Muju Virus, Harbored by Myodes regulus in Korea, Might Represent a Genetic Variant of Puumala Virus, the Prototype Arvicolid Rodent-Borne Hantavirus


Abstract
:1. Introduction
2. Results
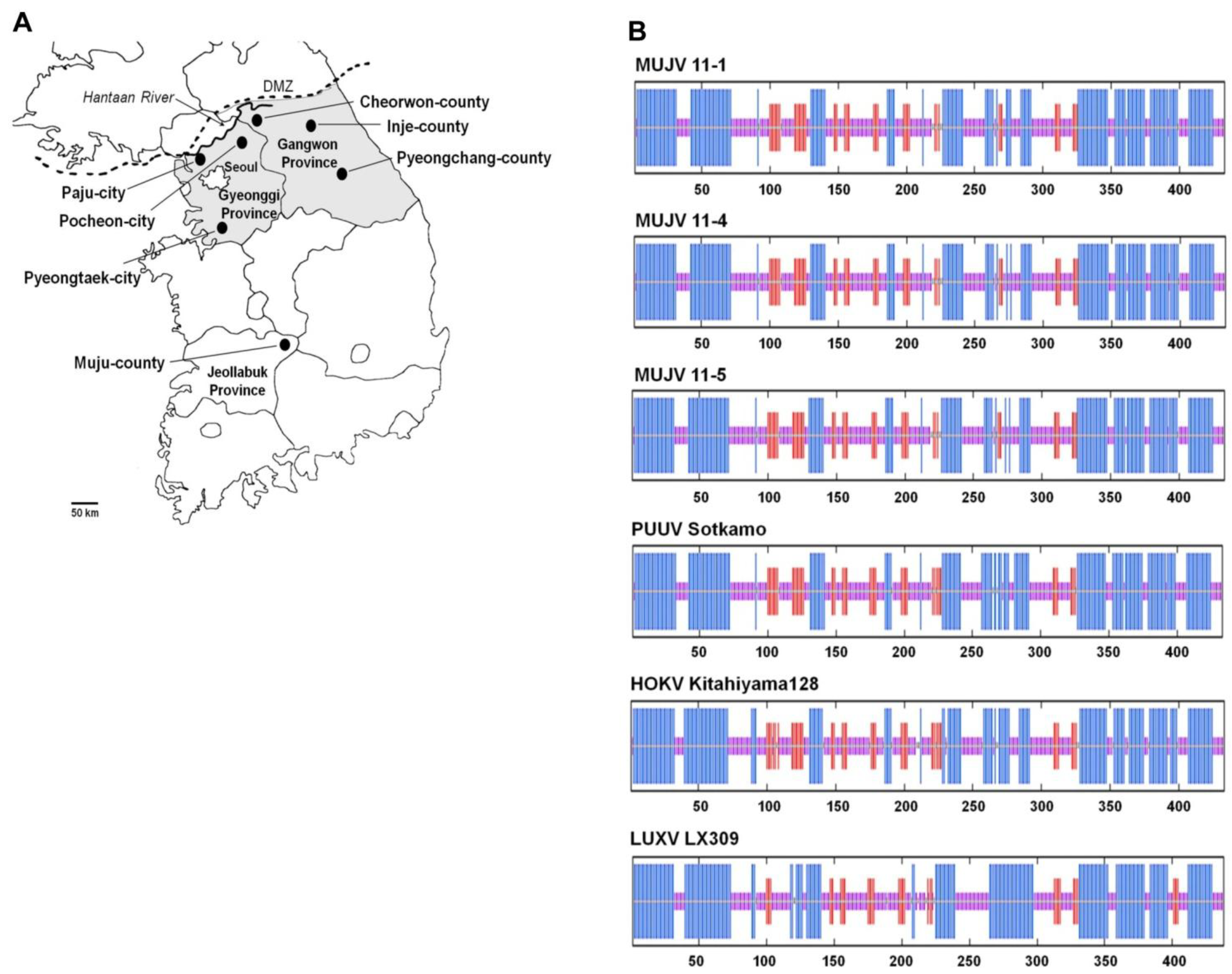

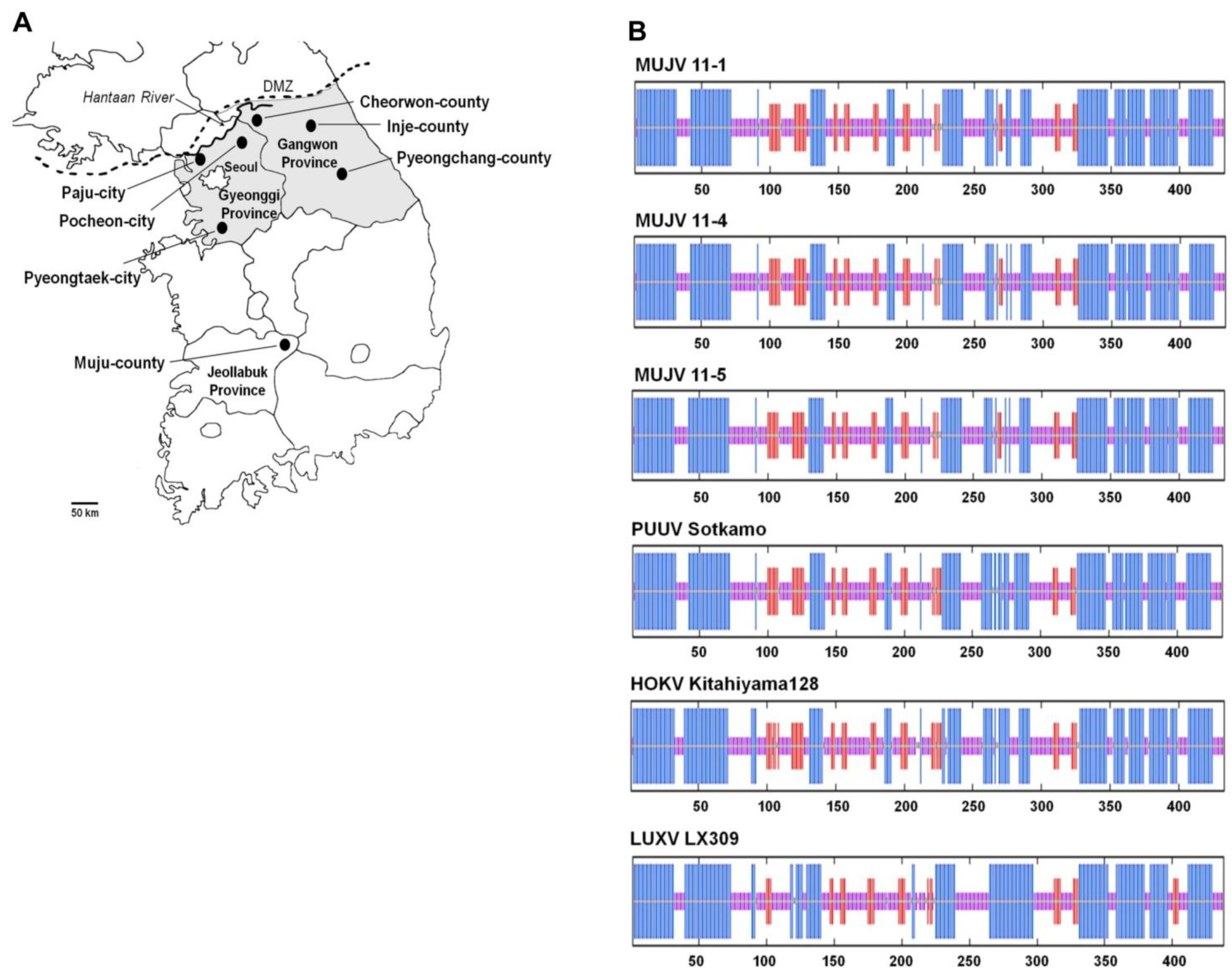{kind=link}
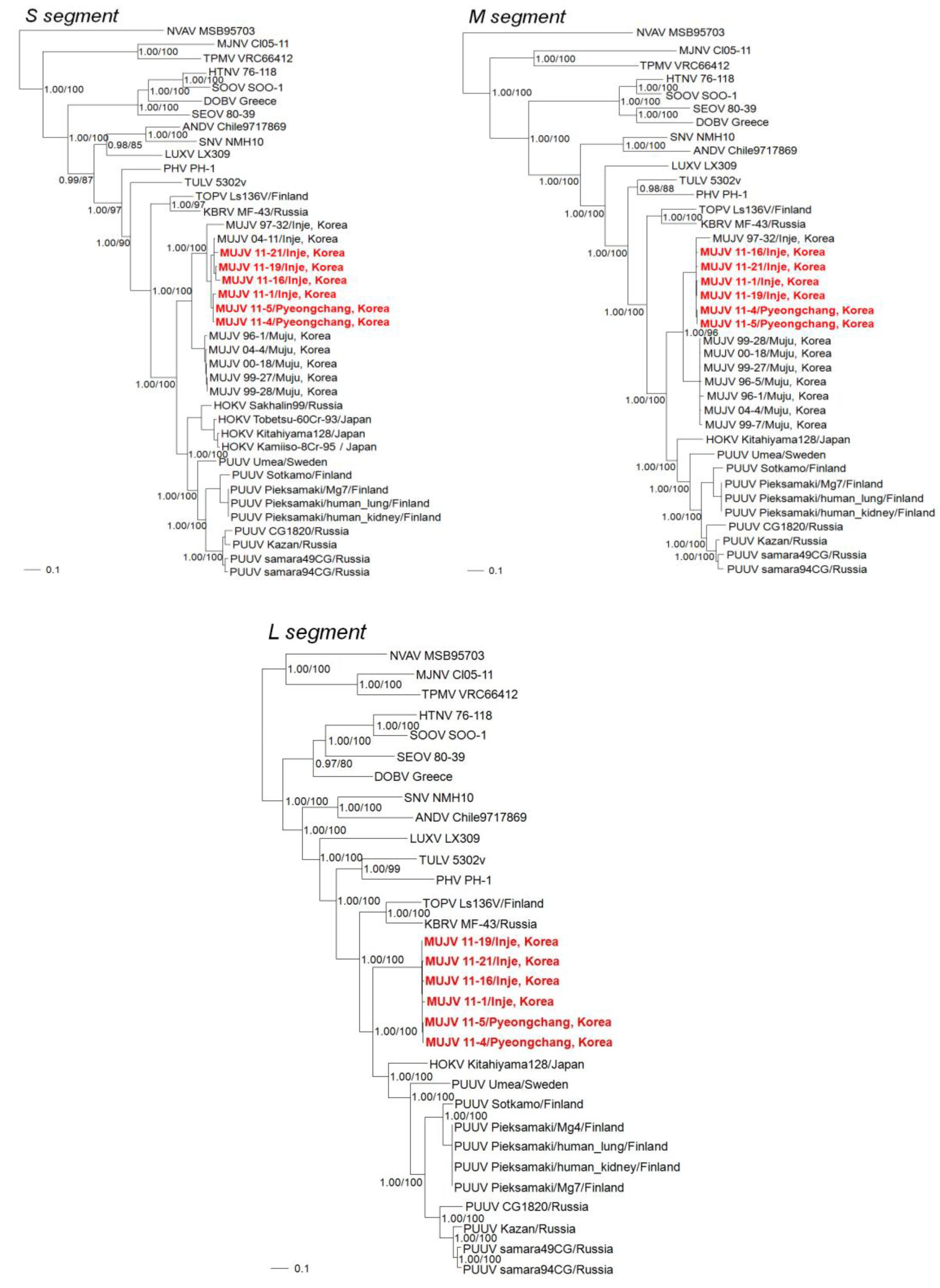
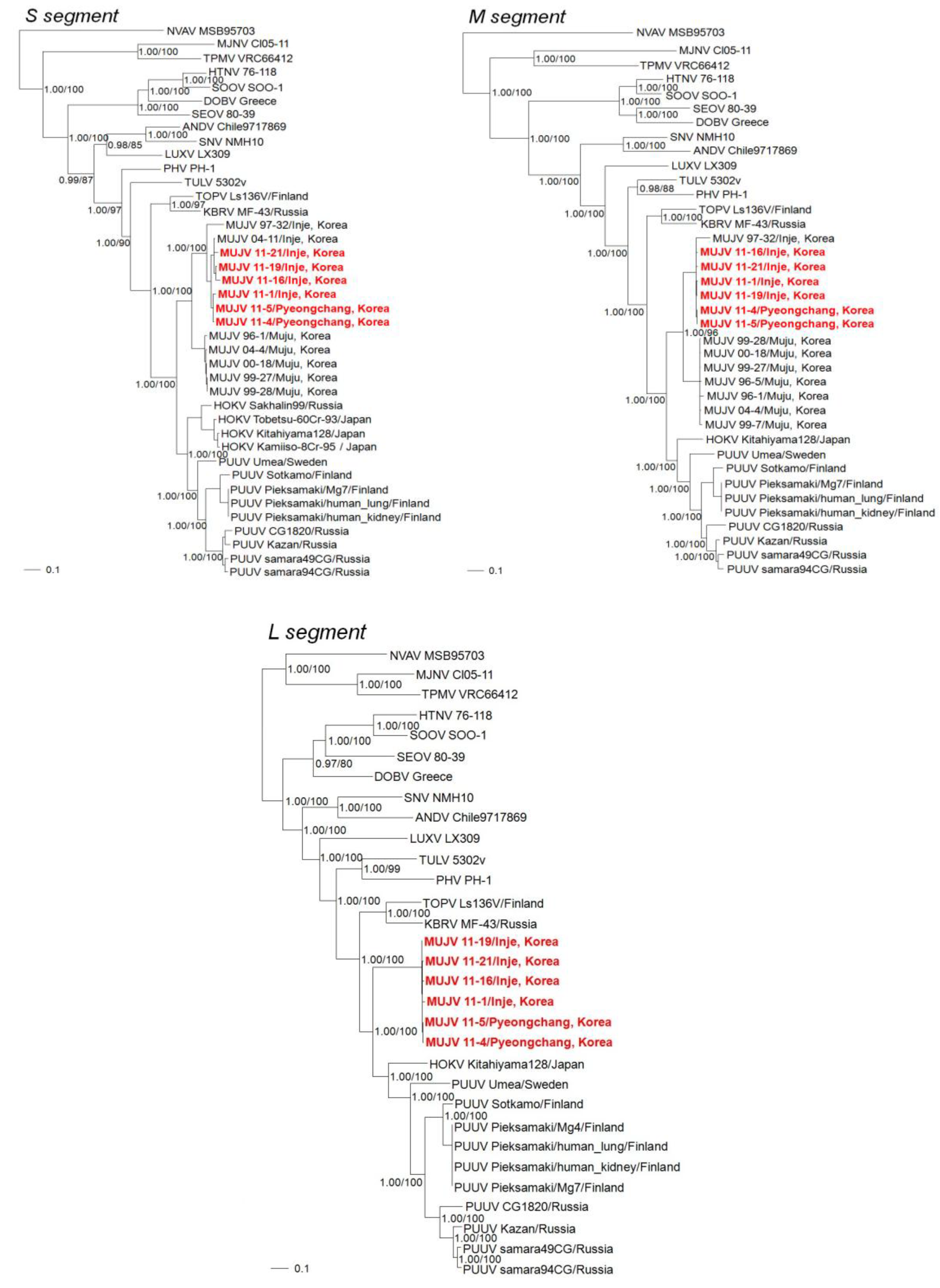{kind=link}
| Virus strain | L segment (nt/aa) | M segment (nt/aa) | S segment (nt/aa) |
|---|---|---|---|
| MUJV 11-1 | 1–6544 (1–2154) JX028271 | 1–3652 (1–1148) JX028272 | 1–1831 (1–433) JX028273 |
| MUJV 11-4 | 1–6544 (1–2154) JX046482 | 1–3652 (1–1148) JX046483 | 1–1831 (1–433) JX046484 |
| MUJV 11-5 | 1~6544 (1–2154) JX046485 | 1–3652 (1–1148) JX046486 | 1–1831 (1–433) JX046487 |
| MUJV 11-16 | 760–4180 (242–1381) JX131287 | 2045–2805 (670–921) JX131288 | 1013–1258 (325–405) JX131289 |
| MUJV 11-19 | 772–4181 (246–1381) JX131290 | 2045–2809 (670–923) JX131291 | 1013–1258 (325–405) JX131292 |
| MUJV 11-21 | 754–4205 (240–1389) JX131293 | 2045–2805 (670–921) JX131294 | 1013–1258 (325–405) JX131295 |
| Virus strain | S segment | M segment | L segment | |||
|---|---|---|---|---|---|---|
| 1299 nt | 433 aa | 3444 nt | 1148 aa | 6462 nt | 2154 aa | |
| MUJV 11-16 | 96.3 | 97.6 | 98.3 | 99.6 | 98.2 | 99.6 |
| MUJV 11-19 | 97.2 | 98.8 | 99.2 | 99.6 | 98.5 | 99.8 |
| MUJV 11-21 | 96.7 | 98.8 | 98.3 | 98.8 | 98.3 | 99.7 |
| MUJV 97-32 | 90.4 | 98.6 | 91.7 | 96.0 | - | - |
| MUJV 04-11 | 96.6 | 98.6 | - | - | - | - |
| MUJV 11-4 | 98.3 | 99.8 | 98.3 | 99.2 | 98.2 | 99.8 |
| MUJV 11-5 | 98.5 | 99.8 | 98.2 | 99.1 | 98.3 | 99.9 |
| MUJV 96-1 | 86.4 | 98.2 | 85.9 | 98.8 | - | - |
| MUJV 96-5 | - | - | 86.7 | 98.8 | - | - |
| MUJV 99-7 | - | - | 86.7 | 100.0 | - | - |
| MUJV 99-27 | 86.8 | 98.8 | 87.1 | 98.8 | - | - |
| MUJV 99-28 | 86.7 | 98.6 | 87.1 | 98.8 | - | - |
| MUJV 00-18 | 86.8 | 98.8 | 86.7 | 98.8 | - | - |
| MUJV 04-4 | 82.7 | 100.0 | 84.2 | 97.5 | - | - |
| HOKV Kitahiyama128 | 82.0 | 95.2 | 78.6 | 92.3 | 79.3 | 93.2 |
| HOKV Kamiiso-8Cr-95 | 81.9 | 95.2 | - | - | - | - |
| HOKV Tobetsu-60Cr-93 | 81.9 | 94.9 | - | - | - | - |
| HOKV Sakhalin99 | 81.4 | 95.2 | - | - | - | - |
| PUUV Sotkamo | 79.4 | 94.2 | 77.6 | 88.9 | 78.7 | 91.6 |
| PUUV Kazan | 79.5 | 93.3 | 77.3 | 90.6 | 78.5 | 92.4 |
| PUUV Umeå | 81.8 | 94.5 | 77.0 | 89.1 | 77.7 | 90.5 |
| PUUV Samara49 | 80.0 | 93.3 | 78.3 | 90.7 | 78.5 | 92.3 |
| PUUV Samara94 | 80.2 | 93.3 | 77.8 | 90.1 | 78.3 | 92.2 |
| PUUV CG1820 | 80.1 | 92.8 | 77.8 | 88.9 | 78.3 | 92.0 |
| PUUV Pieksamaki/Mg7 | 77.3 | 94.5 | 78.4 | 89.7 | 78.7 | 91.9 |
| PUUV Pieksamaki/Mg4 | - | - | - | - | 78.7 | 91.9 |
| PUUV Pieksamaki/hu-lu | 77.3 | 94.5 | 78.4 | 89.7 | 78.7 | 91.9 |
| PUUV Pieksamaki/hu-ki | 77.3 | 94.5 | 78.4 | 89.7 | 78.7 | 91.9 |
| KBRV MF-43 | 77.4 | 87.1 | 74.0 | 84.0 | 76.5 | 90.9 |
| TOPV Ls136V | 77.5 | 87.5 | 74.6 | 84.8 | 77.1 | 89.1 |
| LUXV LX309 | 72.2 | 74.9 | 68.7 | 72.2 | 72.3 | 79.0 |
| PHV PH-1 | 74.8 | 80.1 | 70.9 | 76.4 | 73.4 | 83.4 |
| TULV 5302v | 74.3 | 79.5 | 72.9 | 79.5 | 75.2 | 85.8 |
| HTNV 76-118 | 66.3 | 62.5 | 60.5 | 55.0 | 67.8 | 68.5 |
| SEOV 80-39 | 67.7 | 62.7 | 60.8 | 54.0 | 67.3 | 68.2 |
| SOOV SOO-1 | 66.5 | 61.3 | 60.4 | 54.4 | 68.0 | 68.4 |
| DOBV Greece | 66.6 | 61.1 | 60.1 | 53.8 | 68.3 | 69.4 |
| ANDV Chile9717869 | 72.0 | 74.3 | 65.8 | 66.3 | 71.1 | 76.8 |
| SNV NMH10 | 71.0 | 71.3 | 65.7 | 67.5 | 71.2 | 77.5 |
| MJNV Cl05-11 | 48.0 | 47.0 | 40.7 | 42.1 | 65.4 | 61.9 |
| TPMV VRC66412 | 49.7 | 44.9 | 40.5 | 42.4 | 63.9 | 61.4 |
| NVAV MSB95703 | 59.0 | 53.3 | 56.9 | 44.1 | 65.2 | 61.4 |
3. Discussion
4. Experimental
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Plyusnin, A.; Beatty, B.J.; Elliott, R.M.; Goldbach, R.; Kormelink, R.; Lundkvist, A.; Schmaljohn, C.S.; Tesh, R.B. Bunyaviridae. In Virus Taxonomy: Classification and Nomenclature of Viruses. Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Lefkowitz, E.J., Adams, M.J., Carstens, E.B., Eds.; Elsevier Academic Press: San Diego, CA, USA, 2012; pp. 725–741. [Google Scholar]
- Yanagihara, R.; Gu, S.H.; Arai, S.; Kang, H.J.; Song, J.-W. Hantaviruses: Rediscovery and new beginnings. Virus Res. 2014. [Google Scholar] [CrossRef]
- Fauquet, C.M.; Fargette, D. International Committee on taxonomy of viruses and the 3,142 unassigned species. Virol. J. 2005, 2, e64. [Google Scholar]
- Brummer-Korvenkontio, M.; Vaheri, A.; Hovi, T.; von Bonsdorff, C.H.; Vuorimies, J.; Manni, T.; Penttinen, K.; Oker-Blom, N.; Lähdevirta, J. Nephropathia epidemica: Detection of antigen in bank voles and serologic diagnosis of human infection. J. Infect. Dis. 1980, 141, 131–134. [Google Scholar] [CrossRef]
- Jonsson, C.B.; Figueiredo, L.T.; Vapalahti, O. A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol. Rev. 2010, 23, 412–441. [Google Scholar] [CrossRef]
- Kariwa, H.; Yoshizumi, S.; Arikawa, J.; Yoshimatsu, K.; Takahashi, K.; Takashima, I.; Hashimoto, N. Evidence for the existence of Puumula-related virus among Clethrionomys rufocanus in Hokkaido, Japan. Am. J. Trop. Med. Hyg. 1995, 53, 222–227. [Google Scholar]
- Yashina, L.N.; Danchinova, G.A.; Seregin, S.V.; Khasnatinov, M.A.; Yanagihara, R. Genetic analysis of Hokkaido hantavirus among Myodes rufocanus in the Baikal Lake area. Bull. East. Sib. Sci. Cent. 2013, 4, 147–152. [Google Scholar]
- Song, K.J.; Baek, L.J.; Moon, S.; Ha, S.J.; Kim, S.H.; Park, K.S.; Klein, T.A.; Sames, W.; Kim, H.C.; Lee, J.S.; et al. Muju virus, a novel hantavirus harboured by the arvicolid rodent Myodes regulus in Korea. J. Gen. Virol. 2007, 88, 3121–3129. [Google Scholar] [CrossRef]
- Plyusnin, A.; Vapalahti, O.; Vaheri, A. Hantaviruses: Genome structure, expression and evolution. J. Gen. Virol. 1996, 77, 2677–2687. [Google Scholar] [CrossRef]
- Combet, C.; Blanchet, C.; Geourjon, C.; Deléage, G. NPS@: Network protein sequence analysis. Trends Biochem. Sci. 2000, 25, 147–150. [Google Scholar]
- Piiparinen, H.; Vapalahti, O.; Plyusnin, A.; Vaheri, A.; Lankinen, H. Sequence analysis of the Puumala hantavirus Sotkamo strain L segment. Virus Res. 1997, 51, 1–7. [Google Scholar] [CrossRef]
- Poch, O.; Sauvaget, I.; Delarue, M.; Tordo, N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J. 1989, 8, 3867–3874. [Google Scholar]
- Kang, H.J.; Bennett, S.N.; Sumibcay, L.; Arai, S.; Hope, A.G.; Mocz, G.; Song, J.-W.; Cook, J.A.; Yanagihara, R. Evolutionary insights from a genetically divergent hantavirus harbored by the European common mole (Talpa europaea). PLoS One 2009, 4, e6149. [Google Scholar] [CrossRef]
- Johansson, P.; Olsson, M.; Lindgren, L.; Ahlm, C.; Elgh, F.; Holmstrom, A.; Bucht, G. Complete gene sequence of a human Puumala hantavirus isolate, Puumala Umeå/hu: Sequence comparison and characterisation of encoded gene products. Virus Res. 2004, 105, 147–155. [Google Scholar] [CrossRef]
- Sironen, T.; Plyusnin, A. Genetics and evolution of hantaviruses. In Bunyaviridae: Molecular and Cellular Biology; Plyusnin, A., Elliott, R.M., Eds.; Caister Academic Press: Norfolk, UK, 2011; pp. 61–94. [Google Scholar]
- Maes, P.; Klempa, B.; Clement, J.; Matthijnssens, J.; Gajdusek, D.C.; Krüger, D.H.; van Ranst, M. A proposal for new criteria for the classification of hantaviruses, based on S and M segment protein sequences. Infect. Genet. Evol. 2009, 9, 813–820. [Google Scholar] [CrossRef]
- Yanagihara, R.; Svedmyr, A.; Amyx, H.L.; Lee, P.; Goldgaber, D.; Gajdusek, D.C.; Gibbs, C.J., Jr.; Nyström, K. Isolation and propagation of nephropathia epidemica virus in bank voles. Scand. J. Infect. Dis. 1984, 16, 225–228. [Google Scholar] [CrossRef]
- Traavik, T.; Sommer, A.I.; Mehl, R.; Berdal, B.P.; Stavem, K.; Hunderi, O.H.; Dalrymple, J.M. Nephropathia epidemica in Norway: Antigen and antibodies in rodent reservoirs and antibodies in selected human populations. J. Hyg. (Lond.) 1984, 93, 139–146. [Google Scholar] [CrossRef]
- Plyusnin, A.; Vapalahti, O.; Lankinen, H.; Lehväslaiho, H.; Apekina, N.; Myasnikov, Y.; Kallio-Kokko, H.; Henttonen, H.; Lundkvist, A.; Brummer-Korvenkontio, M. Tula virus: A newly detected hantavirus carried by European common voles. J. Virol. 1994, 68, 7833–7839. [Google Scholar]
- Song, J.-W.; Gligic, A.; Yanagihara, R. Identification of Tula hantavirus in Pitymys subterraneus captured in the Cacak region of Serbia-Yugoslavia. Int. J. Infect. Dis. 2002, 6, 31–36. [Google Scholar] [CrossRef]
- Song, J.-W.; Baek, L.J.; Song, K.-J.; Skrok, A.; Markowski, J.; Bratosiewicz, J.; Kordek, R.; Liberski, P.P.; Yanagihara, R. Characterization of Tula virus from common voles (Microtus arvalis) in Poland: Evidence for geographic-specific phylogenetic clustering. Virus Genes 2004, 29, 239–247. [Google Scholar] [CrossRef]
- Schmidt-Chanasit, J.; Essbauer, S.; Petraityte, R.; Yoshimatsu, K.; Tackmann, K.; Conraths, F.J.; Sasnauskas, K.; Arikawa, J.; Thomas, A.; Pfeffer, M.; et al. Extensive host sharing of central European Tula virus. J. Virol. 2010, 84, 459–474. [Google Scholar] [CrossRef]
- Klempa, B.; Radosa, L.; Krüger, D.H. The broad spectrum of hantaviruses and their hosts in Central Europe. Acta Virol. 2013, 57, 130–137. [Google Scholar] [CrossRef]
- Schlegel, M.; Kindler, E.; Essbauer, S.S.; Wolf, R.; Thiel, J.; Groschup, M.H.; Heckel, G.; Oehme, R.M.; Ulrich, R.G. Tula virus infections in the Eurasian water vole in Central Europe. Vector Borne Zoonotic Dis. 2012, 12, 503–513. [Google Scholar] [CrossRef]
- Firth, C.; Tokarz, R.; Simith, D.B.; Nunes, M.R.; Bhat, M.; Rosa, E.S.; Medeiros, D.B.; Palacios, G.; Vasconcelos, P.F.; Lipkin, W.I. Diversity and distribution of hantaviruses in South America. J. Virol. 2012, 86, 13756–13766. [Google Scholar] [CrossRef]
- Vapalahti, O.; Lundkvist, A.; Fedorov, V.; Conroy, C.J.; Hirvonen, S.; Plyusnina, A.; Nemirov, K.; Fredga, K.; Cook, J.A.; Niemimaa, J.; et al. Isolation and characterization of a hantavirus from Lemmus sibiricus: Evidence for host switch during hantavirus evolution. J. Virol. 1999, 73, 586–5592. [Google Scholar]
- Hörling, J.; Chizhikov, V.; Lundkvist, A.; Jonsson, M.; Ivanov, L.; Dekonenko, A.; Niklasson, B.; Dzagurova, T.; Peters, C.J.; Tkachenko, E.; et al. Khabarovsk virus: A phylogenetically and serologically distinct hantavirus isolated from Microtus fortis trapped in far-east Russia. J. Gen. Virol. 1996, 77, 687–694. [Google Scholar] [CrossRef]
- Zou, Y.; Wang, J.B.; Gaowa, H.S.; Yao, L.S.; Hu, G.W.; Li, M.H.; Chen, H.X.; Plyusnin, A.; Shao, R.; Zhang, Y.Z. Isolation and genetic characterization of hantaviruses carried by Microtus voles in China. J. Med. Virol. 2008, 80, 680–688. [Google Scholar] [CrossRef]
- Nemirov, K.; Vapalahti, O.; Lundkvist, A.; Vasilenko, V.; Golovljova, I.; Plyusnina, A.; Niemimaa, J.; Laakkonen, J.; Henttonen, H.; Vaheri, A.; et al. Isolation and characterization of Dobrava hantavirus carried by the striped field mouse (Apodemus agrarius) in Estonia. J. Gen. Virol. 1999, 80, 371–379. [Google Scholar]
- Klempa, B.; Avsic-Zupanc, T.; Clement, J.; Dzagurova, T.K.; Henttonen, H.; Heyman, P.; Jakab, F.; Krüger, D.H.; Maes, P.; Papa, A.; et al. Complex evolution and epidemiology of Dobrava-Belgrade hantavirus: Definition of genotypes and their characteristics. Arch. Virol. 2013, 158, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Németh, V.; Oldal, M.; Madal, M.; Horváth, G.; Kemenesi, G.; Dallos, B.; Bányal, K.; Jakab, F. Molecular characterization of Dobrava and Kurkino genotypes of Dobrava-Belgrade hantavirus detected in Hungary and Northern Croatia. Virus Genes 2013, 47, 546–549. [Google Scholar] [CrossRef]
- Seto, T.; Tkachenko, E.A.; Morozov, V.G.; Tanikawa, Y.; Kolominov, S.I.; Belov, S.N.; Nakamura, I.; Hashimoto, N.; Kon, Y.; Balakiev, A.E.; et al. An efficient in vivo method for the isolation of Puumala virus in Syrian hamsters and the characterization of the isolates from Russia. J. Virol. Methods 2011, 173, 17–23. [Google Scholar] [CrossRef]
- Sanada, T.; Seto, T.; Ozaki, Y.; Saasa, N.; Yoshimatsu, K.; Arikawa, J.; Yoshii, K.; Kariwa, H. Isolation of Hokkaido virus, genus Hantavirus, using a newly established cell line derived from the kidney of the grey red-backed vole (Myodes rufocanus bedfordiae). J. Gen. Virol. 2012, 93, 2237–2246. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, J.; Yang, X.; Zhou, J.; Yang, W.; Peng, C.; Zhang, H.L.; Shi, Z. A novel hantavirus detected in Yunnan red-backed vole (Eothenomys miletus) in China. J. Gen. Virol. 2011, 92, 1454–1457. [Google Scholar] [CrossRef]
- Parrington, M.A.; Lee, P.W.; Kang, C.Y. Molecular characterization of the Prospect Hill virus M RNA segment: A comparison with the M RNA segments of other hantaviruses. J. Gen. Virol. 1991, 72, 1845–1854. [Google Scholar] [CrossRef]
- Plyusnin, A.; Cheng, Y.; Vapalahti, O.; Pejcoch, M.; Unar, J.; Jelinkova, Z.; Lehväslaiho, H.; Lundkvist, A.; Vaheri, A. Genetic variation in Tula hantaviruses: Sequence analysis of the S and M segments of strains from Central Europe. Virus Res. 1995, 39, 237–250. [Google Scholar] [CrossRef]
- Chizhikov, V.E.; Spiropoulou, C.F.; Morzunov, S.P.; Monroe, M.C.; Peters, C.J.; Nichol, S.T. Complete genetic characterization and analysis of isolation of Sin Nombre virus. J. Virol. 1995, 69, 8132–8136. [Google Scholar]
- Schmaljohn, C.S. Nucleotide sequence of the L genome segment of Hantaan virus. Nucleic Acids Res. 1990, 18, 6728. [Google Scholar] [CrossRef]
- Nemirov, K.; Vapalahti, O.; Papa, A.; Plyusnina, A.; Lundkvist, A.; Antoniadis, A.; Vaheri, A.; Plyusnin, A. Genetic characterization of new Dobrava hantavirus isolate from Greece. J. Virol. 2003, 77, 8793–8800. [Google Scholar] [CrossRef]
- Baek, L.J.; Kariwa, H.; Lokugamage, K.; Yoshimatsu, K.; Arikawa, J.; Takashima, I.; Kang, J.I.; Moon, S.S.; Chung, S.Y.; Kim, E.J.; et al. Soochong virus: An antigenically and genetically distinct hantavirus isolated from Apodemus peninsulae in Korea. J. Med. Virol. 2006, 78, 290–297. [Google Scholar]
- Antic, D.; Lim, B.U.; Kang, C.Y. Nucleotide sequence and coding capacity of the large (L) genomic RNA segment of Seoul 80–39 virus, a member of the Hantavirus genus. Virus Res. 1991, 19, 59–65. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods); Version 4; Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML Web-Servers. Syst. Biol. 2008, 75, 758–771. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. MODELTEST: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, J.G.; Gu, S.H.; Baek, L.J.; Shin, O.S.; Park, K.S.; Kim, H.-C.; Klein, T.A.; Yanagihara, R.; Song, J.-W. Muju Virus, Harbored by Myodes regulus in Korea, Might Represent a Genetic Variant of Puumala Virus, the Prototype Arvicolid Rodent-Borne Hantavirus. Viruses 2014, 6, 1701-1714. https://doi.org/10.3390/v6041701
Lee JG, Gu SH, Baek LJ, Shin OS, Park KS, Kim H-C, Klein TA, Yanagihara R, Song J-W. Muju Virus, Harbored by Myodes regulus in Korea, Might Represent a Genetic Variant of Puumala Virus, the Prototype Arvicolid Rodent-Borne Hantavirus. Viruses. 2014; 6(4):1701-1714. https://doi.org/10.3390/v6041701
Chicago/Turabian StyleLee, Jin Goo, Se Hun Gu, Luck Ju Baek, Ok Sarah Shin, Kwang Sook Park, Heung-Chul Kim, Terry A. Klein, Richard Yanagihara, and Jin-Won Song. 2014. "Muju Virus, Harbored by Myodes regulus in Korea, Might Represent a Genetic Variant of Puumala Virus, the Prototype Arvicolid Rodent-Borne Hantavirus" Viruses 6, no. 4: 1701-1714. https://doi.org/10.3390/v6041701