Chromatin, Non-Coding RNAs, and the Expression of HIV

{kind=link}
Abstract
:1. Introduction
2. Regulation of HIV-1 Chromatin and Latency
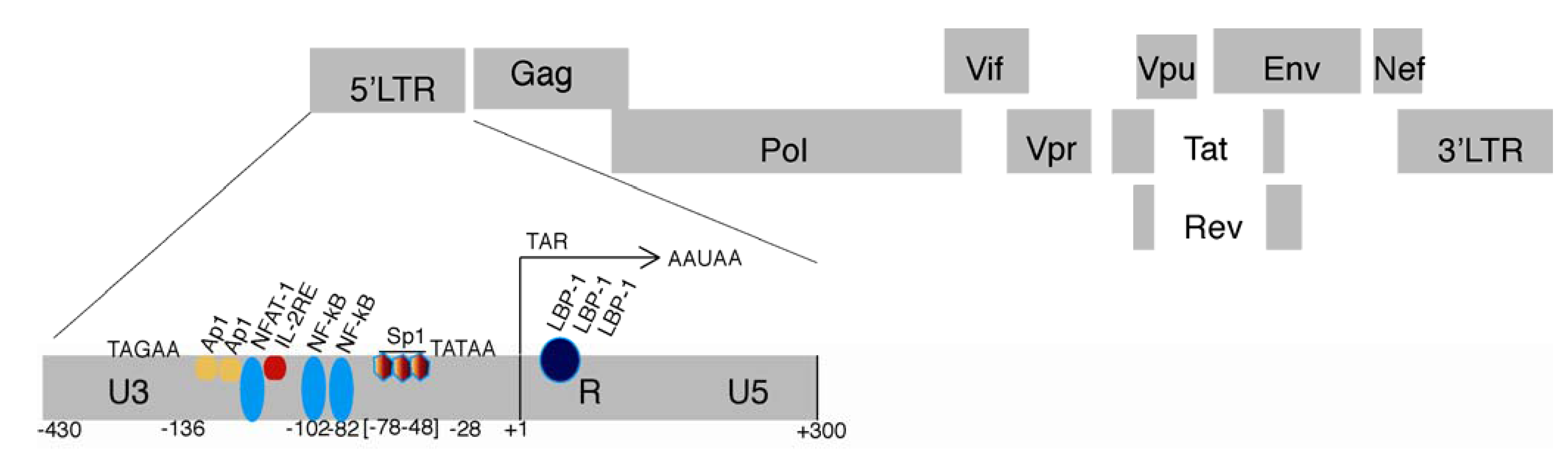
2.1. The HIV LTR
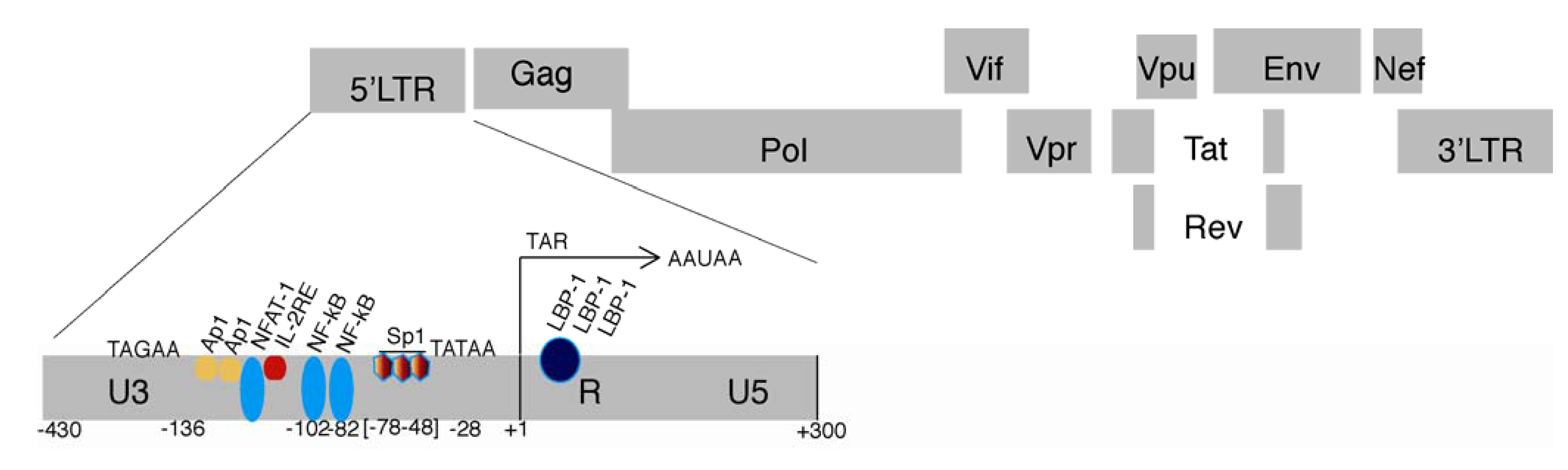
2.2. HIV Chromatin Regulation
3. NcRNAs and Their Role in Genetic Regulation
3.1. LncRNAs, Xist and PTENpg1
3.2. MicroRNAs (miRNAs) and Small Interfering RNAs (siRNAs); The Role of Dicer and Drosha
3.3. Mechanisms of RNA-Directed Epigenetic Modification
4. HIV and Non-Coding RNAs
4.1. HIV and asRNAs
4.2. HIV and miRNAs
5. Final Remarks and Future Work
Acknowledgments
Conflict of Interest
References and Notes
- Pereira, L.A.; Bentley, K.; Peeters, A.; Churchill, M.J.; Deacon, N.J. A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Res. 2000, 28, 663–668. [Google Scholar] [CrossRef]
- Jones, K.A.; Peterlin, B.M. Control of RNA initiation and elongation at the HIV-1 promoter. Annu. Rev. Biochem. 1994, 63, 717–743. [Google Scholar] [CrossRef]
- Jones, K.A.; Kadonaga, J.T.; Luciw, P.A.; Tjian, R. Activation of the AIDS retrovirus promoter by the cellular transcription factor, Sp1. Science 1986, 232, 755–759. [Google Scholar]
- Rittner, K.; Churcher, M.J.; Gait, M.J.; Karn, J. The human immunodeficiency virus long terminal repeat includes a specialised initiator element which is required for Tat-responsive transcription. J. Mol. Biol. 1995, 248, 562–580. [Google Scholar]
- Majello, B.; de Luca, P.; Hagen, G.; Suske, G.; Lania, L. Different members of the Sp1 multigene family exert opposite transcriptional regulation of the long terminal repeat of HIV-1. Nucleic Acids Res. 1994, 22, 4914–4921. [Google Scholar] [CrossRef]
- Graeble, M.A.; Churcher, M.J.; Lowe, A.D.; Gait, M.J.; Karn, J. Human immunodeficiency virus type 1 transactivator protein, Tat, stimulates transcriptional read-through of distal terminator sequences in vitro. Proc. Natl. Acad. Sci. USA 1993, 90, 6184–6188. [Google Scholar]
- Wei, P.; Garber, M.E.; Fang, S.-M.; Fischer, W.H.; Jones, K.A. A Novel CDK9-Associated C-Type Cyclin Interacts Directly with HIV-1 Tat and Mediates Its High-Affinity, Loop-Specific Binding to TAR RNA. Cell 1998, 92, 451–462. [Google Scholar] [CrossRef]
- Brigati, C.; Giacca, M.; Noonan, D.M.; Albini, A. HIV Tat, its TARgets and the control of viral gene expression. FEMS Microbiol. Lett. 2003, 220, 57–65. [Google Scholar]
- Col, E.; Caron, C.; Seigneurin-Berny, D.; Gracia, J.; Favier, A.; Khochbin, S. The histone acetyltransferase, hGCN5, interacts with and acetylates the HIV transactivator, Tat. J. Biol. Chem. 2001, 276, 28179–28184. [Google Scholar]
- Deng, L.; de la Fuente, C.; Fu, P.; Wang, L.; Donnelly, R.; Wade, J.D.; Lambert, P.; Li, H.; Lee, C.G.; Kashanchi, F. Acetylation of HIV-1 Tat by CBP/P300 increases transcription of integrated HIV-1 genome and enhances binding to core histones. Virology 2000, 277, 278–295. [Google Scholar]
- Kiernan, R.E.; Vanhulle, C.; Schiltz, L.; Adam, E.; Xiao, H.; Maudoux, F.; Calomme, C.; Burny, A.; Nakatani, Y.; Jeang, K.T.; et al. HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J. 1999, 18, 6106–6118. [Google Scholar]
- Ott, M.; Schnolzer, M.; Garnica, J.; Fischle, W.; Emiliani, S.; Rackwitz, H.R.; Verdin, E. Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Curr. Biol. 1999, 9, 1489–1492. [Google Scholar] [CrossRef]
- Rabbi, M.F.; Saifuddin, M.; Gu, D.S.; Kagnoff, M.F.; Roebuck, K.A. U5 region of the human immunodeficiency virus type 1 long terminal repeat contains TRE-like cAMP-responsive elements that bind both AP-1 and CREB/ATF proteins. Virology 1997, 233, 235–245. [Google Scholar] [CrossRef]
- Roebuck, K.A.; Gu, D.S.; Kagnoff, M.F. Activating protein-1 cooperates with phorbol ester activation signals to increase HIV-1 expression. AIDS 1996, 10, 819–826. [Google Scholar]
- Luger, K.; Richmond, T.J. The histone tails of the nucleosome. Curr. Opin. Genet. Dev. 1998, 8, 140–146. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- He, G.; Ylisastigui, L.; Margolis, D.M. The regulation of HIV-1 gene expression: The emerging role of chromatin. DNA Cell Biol. 2002, 21, 697–705. [Google Scholar] [CrossRef]
- Pumfery, A.; Deng, L.; Maddukuri, A.; de la Fuente, C.; Li, H.; Wade, J.D.; Lambert, P.; Kumar, A.; Kashanchi, F. Chromatin remodeling and modification during HIV-1 Tat-activated transcription. Curr. HIV Res. 2003, 1, 343–362. [Google Scholar]
- Verdin, E. DNase I-hypersensitive sites are associated with both long terminal repeats and with the intragenic enhancer of integrated human immunodeficiency virus type 1. J. Virol. 1991, 65, 6790–6799. [Google Scholar]
- Van Lint, C.; Emiliani, S.; Ott, M.; Verdin, E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996, 15, 1112–1120. [Google Scholar]
- Coull, J.J.; Romerio, F.; Sun, J.M.; Volker, J.L.; Galvin, K.M.; Davie, J.R.; Shi, Y.; Hansen, U.; Margolis, D.M. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J. Virol. 2000, 74, 6790–6799. [Google Scholar] [CrossRef]
- Jiang, G.; Espeseth, A.; Hazuda, D.J.; Margolis, D.M. c-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. J. Virol. 2007, 81, 10914–10923. [Google Scholar] [CrossRef]
- Imai, K.; Okamoto, T. Transcriptional repression of human immunodeficiency virus type 1 by AP-4. J. Biol. Chem. 2006, 281, 12495–12505. [Google Scholar] [CrossRef]
- Williams, S.A.; Chen, L.F.; Kwon, H.; Ruiz-Jarabo, C.M.; Verdin, E.; Greene, W.C. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006, 25, 139–149. [Google Scholar] [CrossRef]
- Coull, J.J.; He, G.; Melander, C.; Rucker, V.C.; Dervan, P.B.; Margolis, D.M. Targeted derepression of the Human Immunodeficiency Virus type 1 long terminal repeat by pyrrole-imidazole polyamides. J. Virol. 2002, 76, 12349–12354. [Google Scholar]
- Kauder, S.E.; Bosque, A.; Lindqvist, A.; Planelles, V.; Verdin, E. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 2009, 5, e1000495. [Google Scholar] [CrossRef]
- Bednarik, D.P.; Cook, J.A.; Pitha, P.M. Inactivation of the HIV LTR by DNA CpG methylation: Evidence for a role in latency. EMBO J. 1990, 9, 1157–1164. [Google Scholar]
- Bednarik, D.P.; Duckett, C.; Kim, S.U.; Perez, V.L.; Griffis, K.; Guenthner, P.C.; Folks, T.M. DNA CpG methylation inhibits binding of NF-kappa B proteins to the HIV-1 long terminal repeat cognate DNA motifs. New Biol. 1991, 3, 969–976. [Google Scholar]
- Hong, L.; Schroth, G.P.; Matthews, H.R.; Yau, P.; Bradbury, E.M. Studies of the DNA binding properties of histone H4 amino terminus. Thermal denaturation studies reveal that acetylation markedly reduces the binding constant of the H4 “tail” to DNA. J. Biol. Chem. 1993, 268, 305–314. [Google Scholar]
- Marzio, G.; Tyagi, M.; Gutierrez, M.I.; Giacca, M. HIV-1 tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc. Natl. Acad. Sci. USA 1998, 95, 13519–13524. [Google Scholar]
- Hottiger, M.O.; Nabel, G.J. Interaction of human immunodeficiency virus type 1 Tat with the transcriptional coactivators p300 and CREB binding protein. J. Virol. 1998, 72, 8252–8256. [Google Scholar]
- Lusic, M.; Marcello, A.; Cereseto, A.; Giacca, M. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 2003, 22, 6550–6561. [Google Scholar] [CrossRef]
- Narlikar, G.J.; Fan, H.Y.; Kingston, R.E. Cooperation between complexes that regulate chromatin structure and transcription. Cell 2002, 108, 475–487. [Google Scholar] [CrossRef]
- Cosma, M.P. Ordered recruitment: Gene-specific mechanism of transcription activation. Mol. Cell 2002, 10, 227–236. [Google Scholar] [CrossRef]
- Panning, B.; Dausman, J.; Jaenisch, R. X chromosome inactivation is mediated by Xist RNA stabilization. Cell 1997, 90, 907–916. [Google Scholar] [CrossRef]
- Gartler, S.M.; Goldman, M.A. X-Chromosome inactivation. In eLS; John Wiley & Sons, Ltd.: New York, NY, USA, 2001. [Google Scholar]
- Brown, C.J.; Ballabio, A.; Rupert, J.L.; Lafreniere, R.G.; Grompe, M.; Tonlorenzi, R.; Willard, H.F. A gene from the region of the Human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 1991, 349, 38–44. [Google Scholar]
- Penny, G.D.; Kay, G.F.; Sheardown, S.A.; Rastan, S.; Brockdorff, N. Requirement for Xist in X chromosome inactivation. Nature 1996, 379, 131–137. [Google Scholar] [CrossRef]
- Simon, J.A.; Kingston, R.E. Mechanisms of Polycomb gene silencing: Knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009, 10, 697–708. [Google Scholar]
- Plath, K.; Fang, J.; Mlynarczyk-Evans, S.K.; Cao, R.; Worringer, K.A.; Wang, H.; de la Cruz, C.C.; Otte, A.P.; Panning, B.; Zhang, Y. Role of histone H3 lysine 27 methylation in X inactivation. Science 2003, 300, 131–135. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.J.; Lee, J.T. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 2008, 322, 750–756. [Google Scholar] [CrossRef]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; St Laurent, G., 3rd; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretas e. Nat. Med. 2008, 14, 723–730. [Google Scholar] [CrossRef]
- Tufarelli, C.; Stanley, J.A.; Garrick, D.; Sharpe, J.A.; Ayyub, H.; Wood, W.G.; Higgs, D.R. Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat. Genet. 2003, 34, 157–165. [Google Scholar]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef]
- Johnsson, P.; Ackley, A.; Vidarsdottir, L.; Lui, W.; Corcoran, M.; Grandér, D.; Morris, K.V. A pseudogene long non-coding RNA network regulates PTEN transcription and translation in human cells. 2013, in press. [Google Scholar]
- Kawasaki, H.; Wadhwa, R.; Taira, K. World of small RNAs: From ribozymes to siRNA and miRNA. Differentiation 2004, 72, 58–64. [Google Scholar]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Radmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar]
- Sontheimer, E.J. Assembly and function of RNA silencing complexes. Nat. Rev. Mol. Cell Biol. 2005, 6, 127–138. [Google Scholar]
- Conrad, R.; Barrier, M.; Ford, L.P. Role of miRNA and miRNA processing factors in development and disease. Birth Defects Res. C Embryo Today 2006, 78, 107–117. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of Novel genes coding for small expressed RNAs. Science 2001, 294, 853–857. [Google Scholar]
- Lau, N.C.; Lim, L.P.; Weinstein, E.G.; Bartel, D.P. An abundant class of tine RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001, 294, 858–862. [Google Scholar] [CrossRef]
- Mourelatos, Z.; Dostie, J.; Paushkin, S.; Sharma, A.; Charroux, B.; Abel, L.; Rappsilber, J.; Mann, M.; Dreyfuss, G. miRNPs: A novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev. 2002, 16, 720–728. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar]
- Pfeffer, S.; Zavolan, M.; Grasser, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of virus-encoded microRNAs. Science 2004, 304, 734–736. [Google Scholar]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar]
- Sullivan, C.S.; Sung, C.K.; Pack, C.D.; Grundhoff, A.; Lukacher, A.E.; Benjamin, T.L.; Ganem, D. Murine Polyomavirus encodes a microRNA that cleaves early RNA transcripts but is not essential for experimental infection. Virology 2009, 387, 157–167. [Google Scholar]
- Singh, J.; Singh, C.P.; Bhavani, A.; Nagaraju, J. Discovering microRNAs from Bombyx mori nucleopolyhedrosis virus. Virology 2010, 407, 120–128. [Google Scholar] [CrossRef]
- Lee, J.T. Epigenetic regulation by long noncoding RNAs. Science 2012, 338, 1435–1439. [Google Scholar]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar]
- Han, J.; Kim, D.; Morris, K.V. Promoter-associated RNA is required for RNA-directed transcriptional gene silencing in human cells. Proc. Natl. Acad. Sci. USA 2007, 104, 12422–12427. [Google Scholar] [CrossRef]
- Beisel, C.; Paro, R. Silencing chromatin: Comparing modes and mechanisms. Nat. Rev. Genet. 2011, 12, 123–135. [Google Scholar]
- Mesnard, J.M.; Barbeau, B.; Devaux, C. HBZ, a new important player in the mystery of adult T-cell leukemia. Blood 2006, 108, 3979–3982. [Google Scholar] [CrossRef]
- Gaudray, G.; Gachon, F.; Basbous, J.; Biard-Piechaczyk, M.; Devaux, C.; Mesnard, J.M. The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J. Virol. 2002, 76, 12813–12822. [Google Scholar]
- Larocca, D.; Chao, L.A.; Seto, M.H.; Brunck, T.K. Human T-cell leukemia virus minus strand transcription in infected T-cells. Biochem. Biophys. Res. Commun. 1989, 163, 1006–1013. [Google Scholar]
- Miller, R.H. Human immunodeficiency virus may encode a novel protein on the genomic DNA plus strand. Science 1988, 239, 1420–1422. [Google Scholar]
- Michael, N.L.; Vahey, M.T.; d'Arcy, L.; Ehrenberg, P.K.; Mosca, J.D.; Rappaport, J.; Redfield, R.R. Negative-strand RNA transcripts are produced in human immunodeficiency virus type 1-infected cells and patients by a novel promoter downregulated by Tat. J. Virol. 1994, 68, 979–987. [Google Scholar]
- Torresilla, C.; Larocque, E.; Landry, S.; Halin, M.; Coulombe, Y.; Masson, J.Y.; Mesnard, J.M.; Barbeau, B. Detection of the HIV-1 minus strand-encoded Antisense Protein and its association with autophagy. J. Virol. 2013, 87, 5089–5105. [Google Scholar] [CrossRef]
- Tagieva, N.E.; Vaquero, C. Expression of naturally occurring antisense RNA inhibits human immunodeficiency virus type 1 heterologous strain replication. J. Gen. Virol. 1997, 78, 2503–2511. [Google Scholar]
- Landry, S.; Halin, M.; Lefort, S.; Audet, B.; Vaquero, C.; Mesnard, J.M.; Barbeau, B. Detection, characterization and regulation of antisense transcripts in HIV-1. Retrovirology 2007, 4, 71. [Google Scholar]
- Bentley, K.; Deacon, N.; Sonza, S.; Zeichner, S.; Churchill, M. Mutational analysis of the HIV-1 LTR as a promoter of negative sense transcription. Arch. Virol. 2004, 149, 2277–2294. [Google Scholar] [CrossRef]
- Peeters, A.; Lambert, P.F.; Deacon, N.J. A fourth Sp1 site in the human immunodeficiency virus type 1 long terminal repeat is essential for negative-sense transcription. J. Virol. 1996, 70, 6665–6672. [Google Scholar]
- Kobayashi-Ishihara, M.; Yamagishi, M.; Hara, T.; Matsuda, Y.; Takahashi, R.; Miyake, A.; Nakano, K.; Yamochi, T.; Ishida, T.; Watanabe, T. HIV-1-encoded antisense RNA suppresses viral replication for a prolonged period. Retrovirology 2012, 9, 38. [Google Scholar]
- Grewal, S.I.; Moazed, D. Heterochromatin and epigenetic control of gene expression. Science 2003, 301, 798–802. [Google Scholar] [CrossRef]
- Hsieh, J.; Fire, A. Recognition and silencing of repeated DNA. Annu. Rev. Genet. 2000, 34, 187–204. [Google Scholar] [CrossRef]
- Morris, K.V. The emerging role of RNA in the regulation of gene transcription in human cells. Semin. Cell Dev. Biol. 2011, 22, 351–358. [Google Scholar] [CrossRef]
- Narayanan, A.; Kehn-Hall, K.; Bailey, C.; Kashanchi, F. Analysis of the roles of HIV-derived microRNAs. Expert Opin. Biol. Ther. 2011, 11, 17–29. [Google Scholar] [CrossRef]
- Jeang, K.T.; Chang, Y.; Berkhout, B.; Hammarskjold, M.L.; Rekosh, D. Regulation of HIV expression: Mechanisms of action of Tat and Rev. AIDS 1991, 5, S3–S14. [Google Scholar] [CrossRef]
- Bennasser, Y.; Le, S.Y.; Yeung, M.L.; Jeang, K.T. HIV-1 encoded candidate micro-RNAs and their cellular targets. Retrovirology 2004, 1, 43. [Google Scholar] [CrossRef]
- Klase, Z.; Kale, P.; Winograd, R.; Gupta, M.V.; Heydarian, M.; Berro, R.; McCaffrey, T.; Kashanchi, F. HIV-1 TAR element is processed by Dicer to yield a viral micro-RNA involved in chromatin remodeling of the viral LTR. BMC Mol. Biol. 2007, 8, 63. [Google Scholar] [CrossRef]
- Bennasser, Y.; Yeung, M.L.; Jeang, K.T. HIV-1 TAR RNA subverts RNA interference in transfected cells through sequestration of TAR RNA-binding protein, TRBP. J. Biol. Chem. 2006, 281, 27674–27678. [Google Scholar] [CrossRef]
- Purzycka, K.J.; Adamiak, R.W. The HIV-2 TAR RNA domain as a potential source of viral-encoded miRNA. A reconnaissance study. Nucleic Acids Symp. Ser. (Oxf.) 2008. [Google Scholar] [CrossRef]
- Couturier, J.P.; Root-Bernstein, R.S. HIV may produce inhibitory microRNAs (miRNAs) that block production of CD28, CD4 and some interleukins. J. Theor. Biol. 2005, 235, 169–184. [Google Scholar]
- Schopman, N.C.; Willemsen, M.; Liu, Y.P.; Bradley, T.; van Kampen, A.; Baas, F.; Berkhout, B.; Haasnoot, J. Deep sequencing of virus-infected cells reveals HIV-encoded small RNAs. Nucleic Acids Res. 2012, 40, 414–427. [Google Scholar] [CrossRef]
- Bennasser, Y.; Le, S.Y.; Benkirane, M.; Jeang, K.T. Evidence that HIV-1 encodes an siRNA and a suppressor of RNA silencing. Immunity 2005, 22, 607–619. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Groen, J.N.; Morris, K.V. Chromatin, Non-Coding RNAs, and the Expression of HIV. Viruses 2013, 5, 1633-1645. https://doi.org/10.3390/v5071633
Groen JN, Morris KV. Chromatin, Non-Coding RNAs, and the Expression of HIV. Viruses. 2013; 5(7):1633-1645. https://doi.org/10.3390/v5071633
Chicago/Turabian StyleGroen, Jessica N., and Kevin V. Morris. 2013. "Chromatin, Non-Coding RNAs, and the Expression of HIV" Viruses 5, no. 7: 1633-1645. https://doi.org/10.3390/v5071633
APA StyleGroen, J. N., & Morris, K. V. (2013). Chromatin, Non-Coding RNAs, and the Expression of HIV. Viruses, 5(7), 1633-1645. https://doi.org/10.3390/v5071633