Interpreting the Epstein-Barr Virus (EBV) Epigenome Using High-Throughput Data


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Assaying the EBV Epigenome
3. Tour of the EBV Epigenome
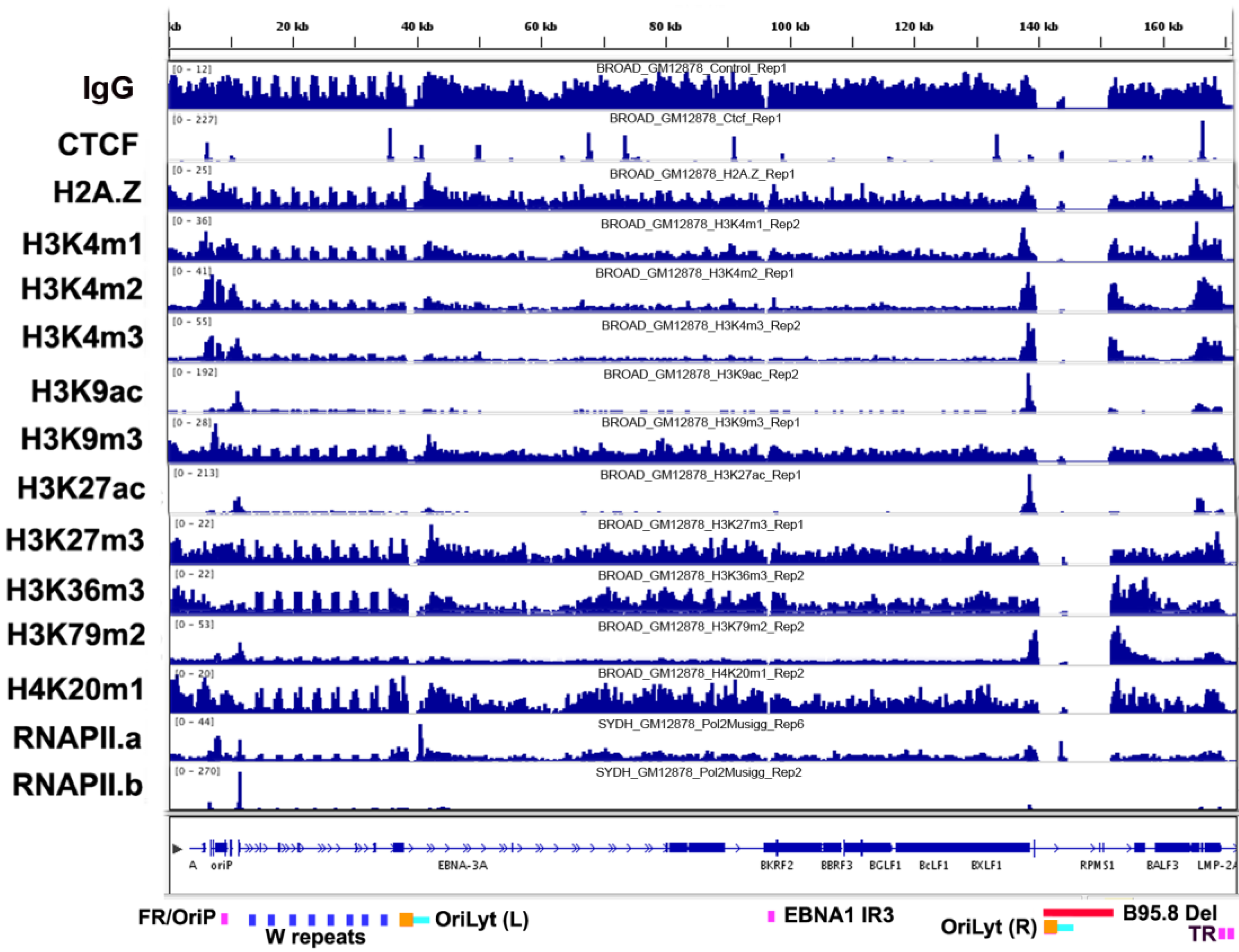
3.1. Overview of Chromatin Structure
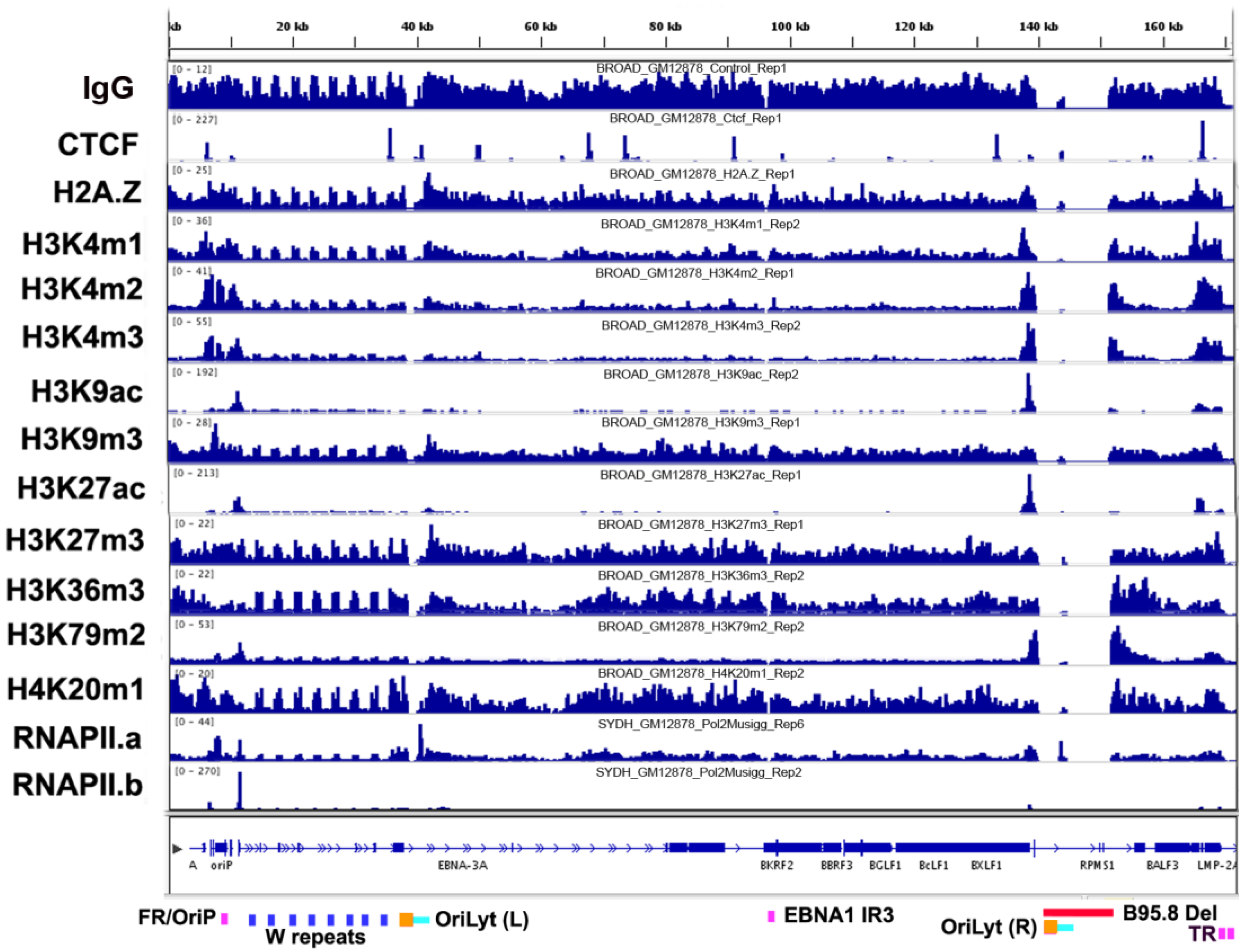
3.2. Histone Modifications
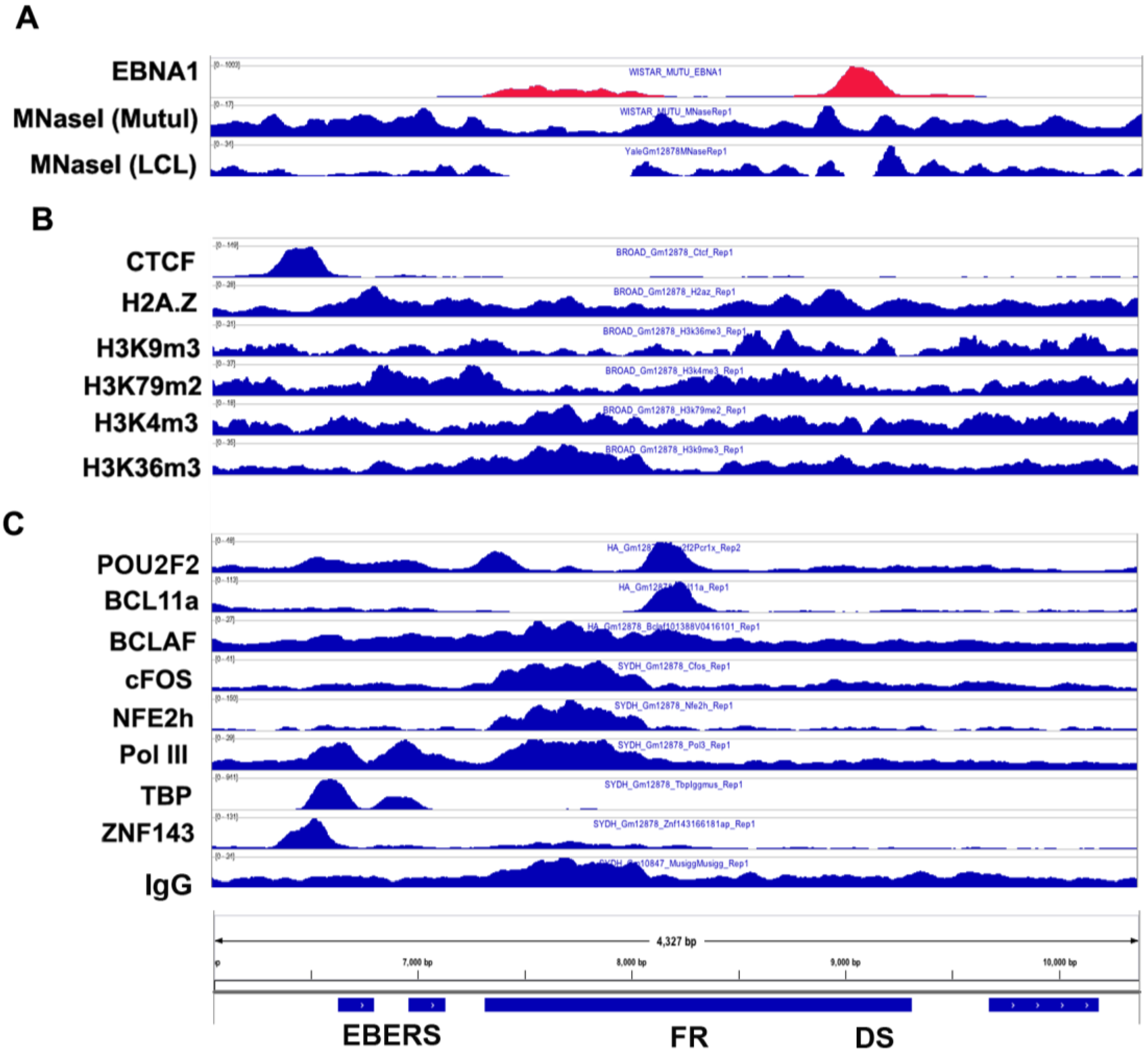
3.3. CTCF and Cohesin Binding Sites
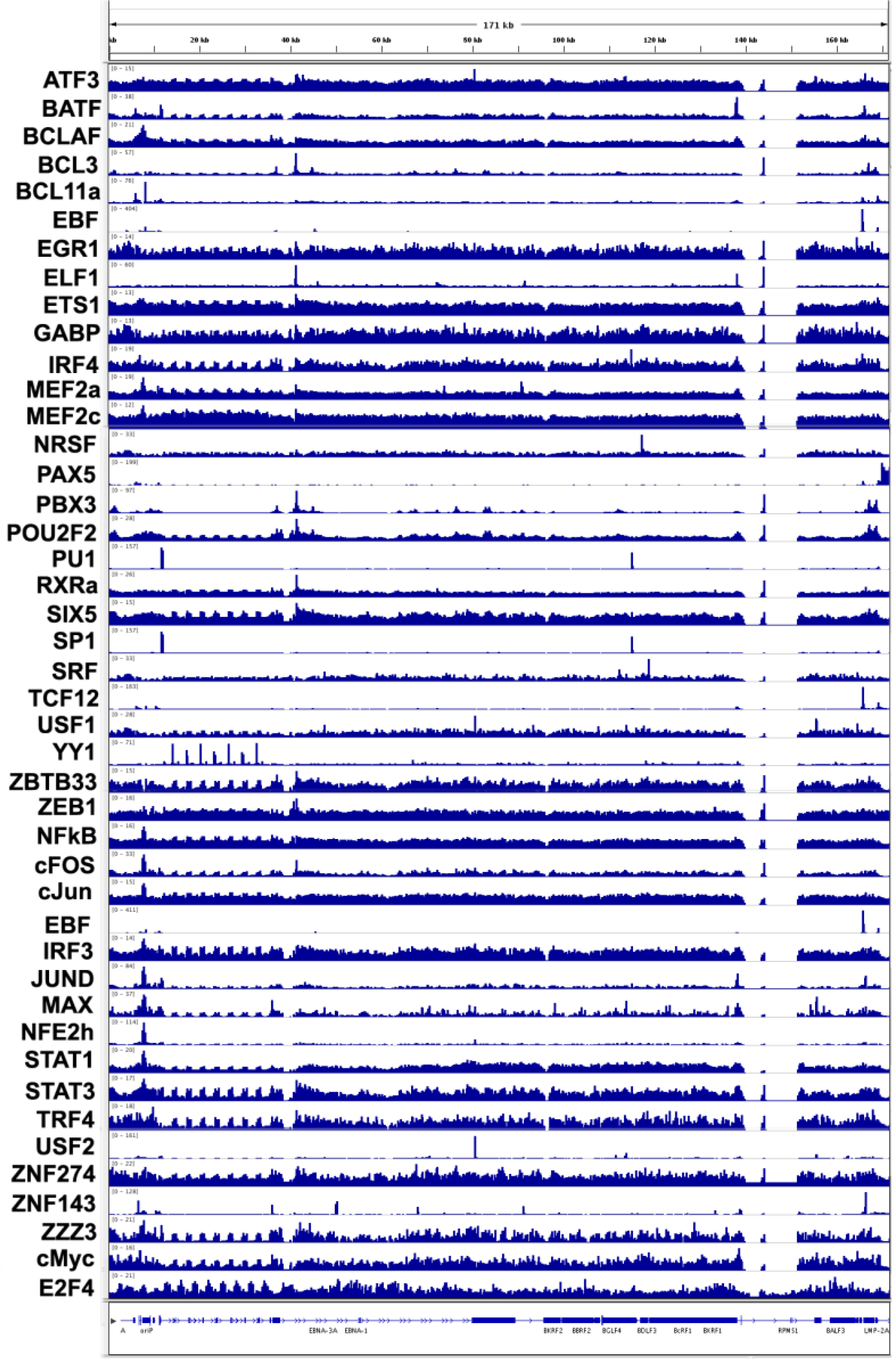
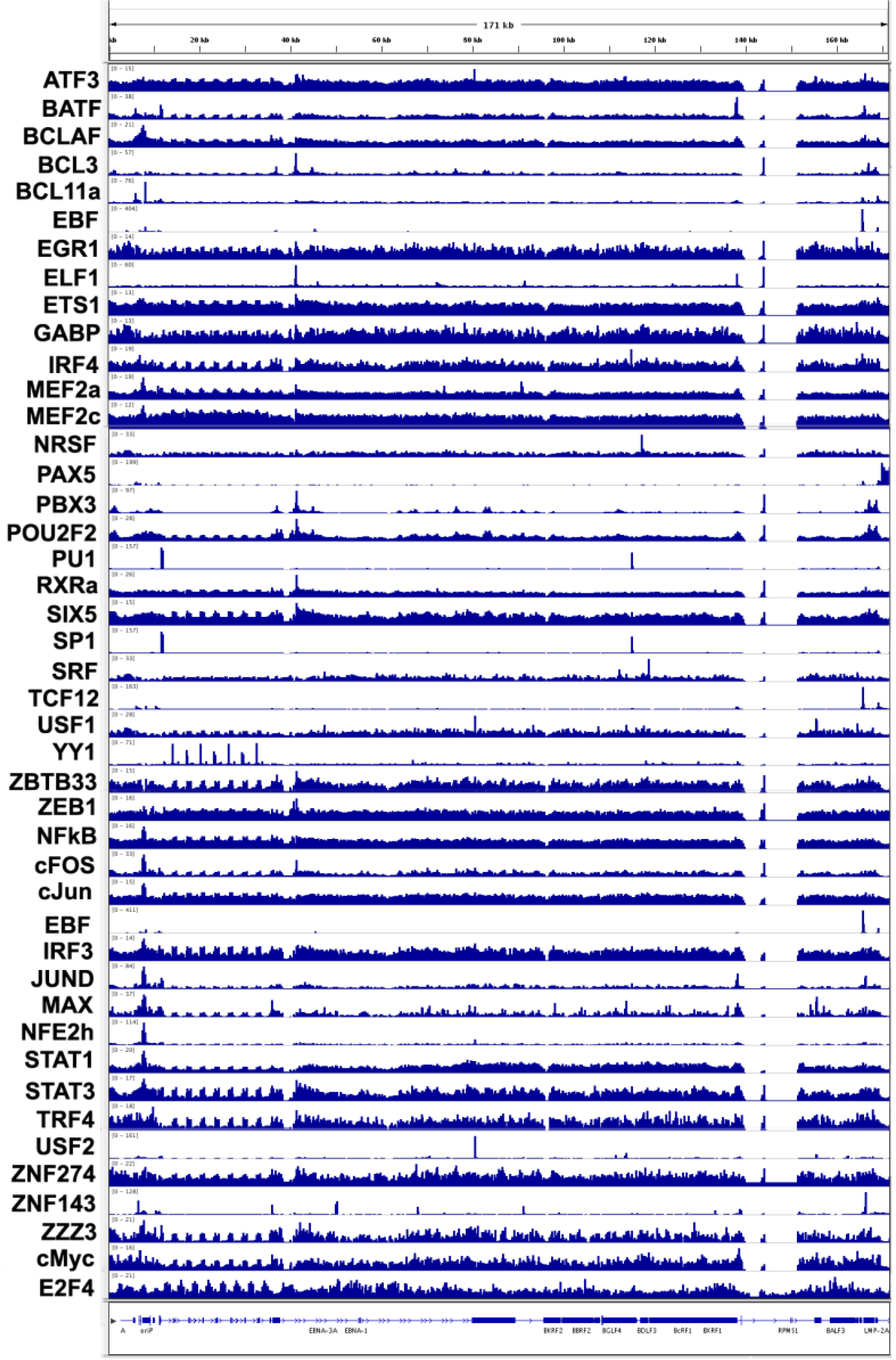
3.4. Transcription Factor Binding Sites
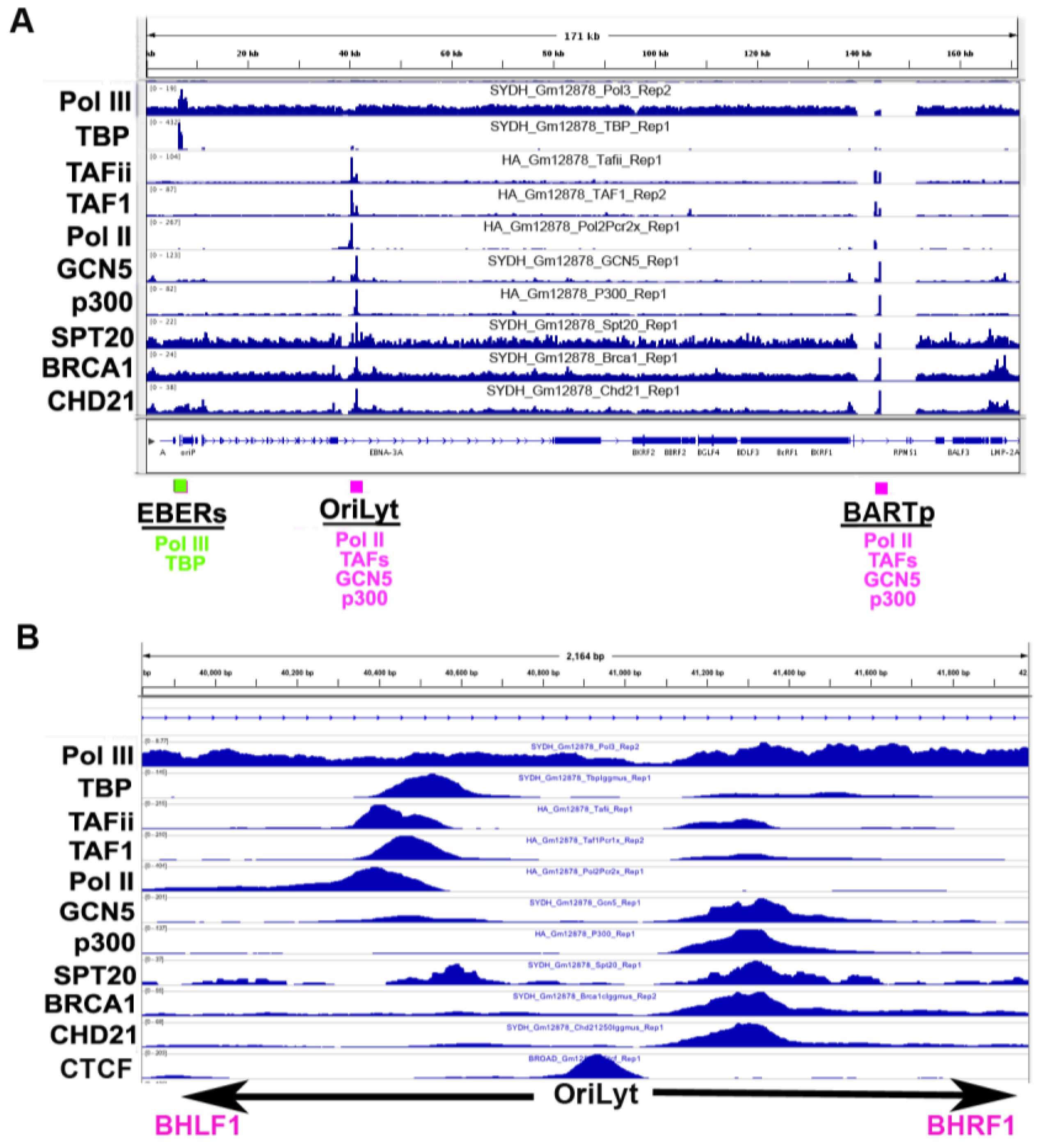
3.5. Co-Activator Binding Sites
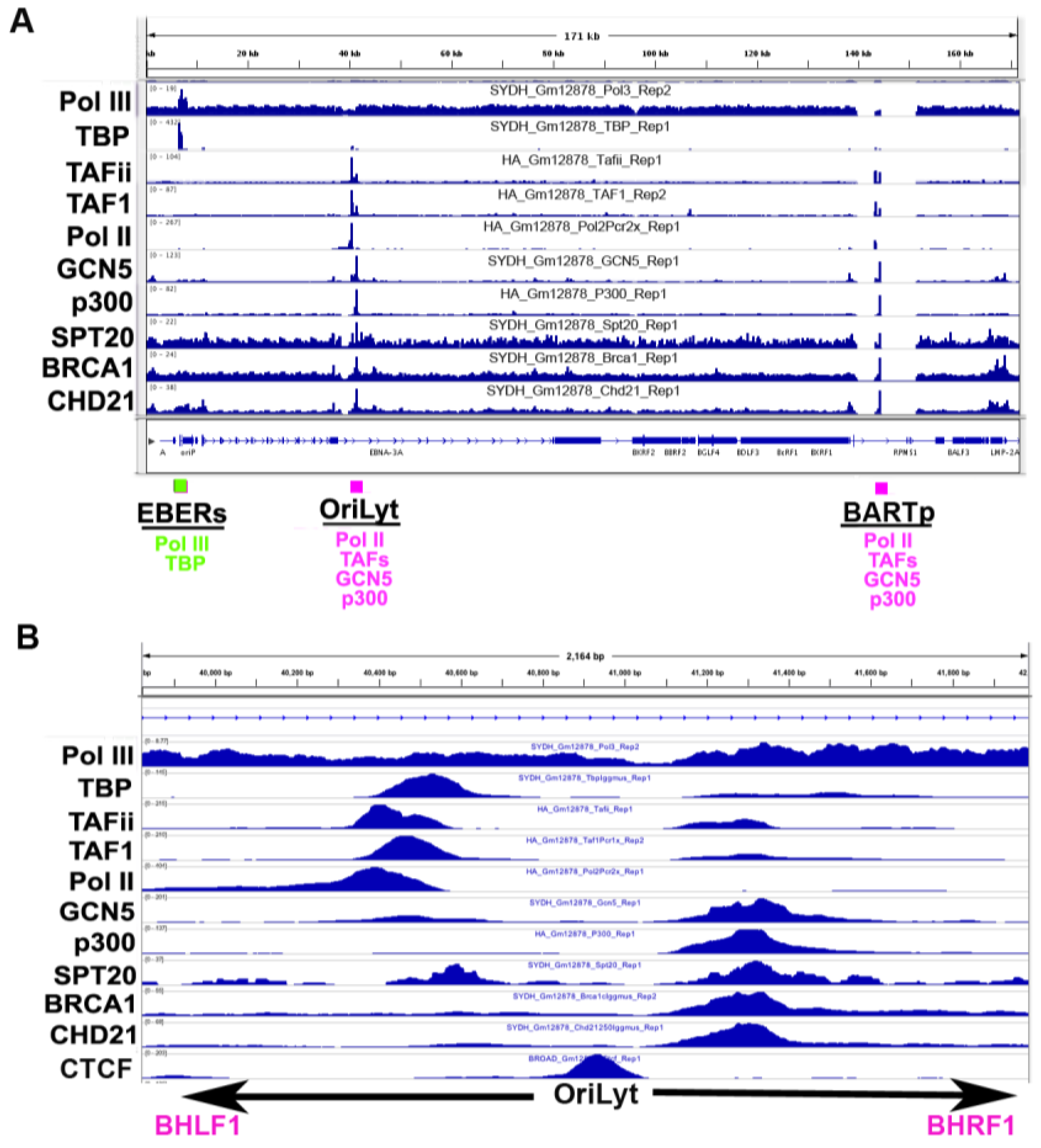
3.6. Origin of Latent Replication
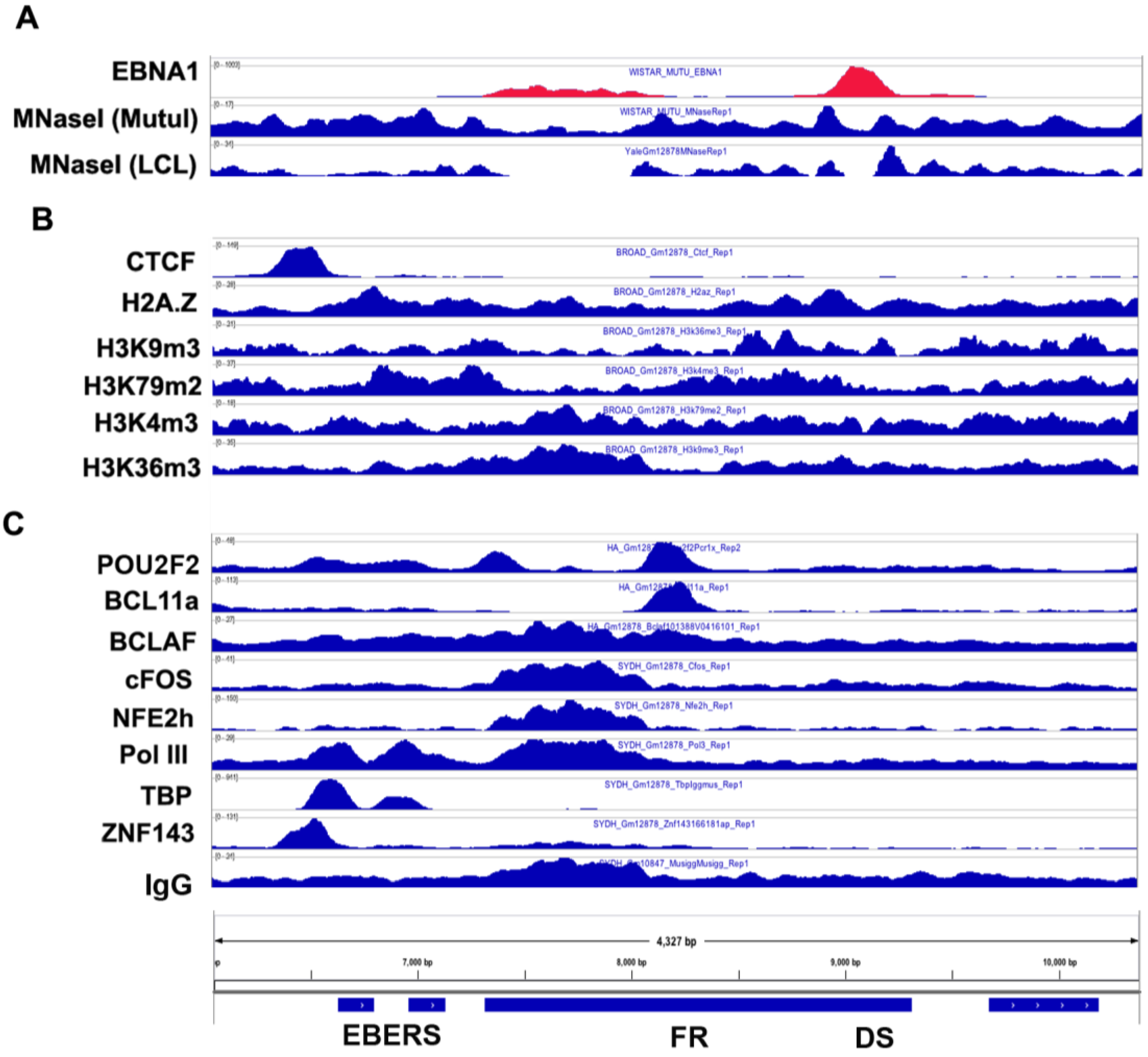
3.7. DNA Methylation
3.8. Negative Results
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Arvey, A.; Tempera, I.; Tsai, K.; Chen, H.S.; Tikhmyanova, N.; Klichinsky, M.; Leslie, C.; Lieberman, P.M. An atlas of the epstein-barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell. Host. Microbe. 2012, 12, 233–245. [Google Scholar] [CrossRef]
- Kieff, E. Epstein-Barr Virus and its replication, 5th ed; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, 2007; p. 2 v, (xix, 3091, 86 p.). [Google Scholar]
- Rickinson, A.B.; Kieff, E. Epstein-Barr Virus, 5th ed; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, 2007; p. 2 v, (xix, 3091, 86 p.). [Google Scholar]
- Rowe, M.; Lear, A.L.; Croom-Carter, D.; Davies, A.H.; Rickinson, A.B. Three pathways of Epstein-Barr virus gene activation from EBNA1-positive latency in B lymphocytes. J. Virol. 1992, 66, 122–131. [Google Scholar]
- Yates, J.L.; Guan, N. Epstein-Barr virus-derived plasmids replicate only once per cell cycle and are not amplified after entry into cells. J. Virol. 1991, 65, 483–488. [Google Scholar]
- Amon, W.; Farrell, P.J. Reactivation of Epstein-Barr virus from latency. Rev. Med. Virol. 2005, 15, 149–156. [Google Scholar] [CrossRef]
- Cai, X.; Schafer, A.; Lu, S.; Bilello, J.P.; Desrosiers, R.C.; Edwards, R.; Raab-Traub, N.; Cullen, B.R. Epstein-Barr virus microRNAs are evolutionarily conserved and differentially expressed. PLoS Pathog. 2006, 2, e23. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinform. 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome. Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Jankelevich, S.; Kolman, J.L.; Bodnar, J.W.; Miller, G. A nuclear matrix attachment region organizes the Epstein-Barr viral plasmid in Raji cells into a single DNA domain. Embo J. 1992, 11, 1165–1176. [Google Scholar]
- Kundaje, A.; Kyriazopoulou-Panagiotopoulou, S.; Libbrecht, M.; Smith, C.L.; Raha, D.; Winters, E.E.; Johnson, S.M.; Snyder, M.; Batzoglou, S.; Sidow, A. Ubiquitous heterogeneity and asymmetry of the chromatin environment at regulatory elements. Genome. Res. 2012, 22, 1735–1747. [Google Scholar] [CrossRef]
- Baer, R.; Bankier, A.T.; Biggin, M.D.; Deininger, P.L.; Farrell, P.J.; Gibson, T.J.; Hatfull, G.; Hudson, G.S.; Satchwell, S.C.; Seguin, C.; et al. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 1984, 310, 207–211. [Google Scholar] [CrossRef]
- Dyson, P.J.; Farrell, P.J. Chromatin structure of Esptein-Barr virus. J. Gen. Virol. 1985, 66, 1931–1940. [Google Scholar] [CrossRef]
- Pepke, S.; Wold, B.; Mortazavi, A. Computation for ChIP-seq and RNA-seq studies. Nat. Methods. 2009, 6, 22–32. [Google Scholar] [CrossRef]
- Shilatifard, A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr. Opin. Cell. Biol. 2008, 20, 341–348. [Google Scholar] [CrossRef]
- Toth, Z.; Maglinte, D.T.; Lee, S.H.; Lee, H.R.; Wong, L.Y.; Brulois, K.F.; Lee, S.; Buckley, J.D.; Laird, P.W.; Marquez, V.E.; Jung, J.U. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 2010, 6, e1001013. [Google Scholar] [CrossRef]
- Prasanth, S.G.; Shen, Z.; Prasanth, K.V.; Stillman, B. Human origin recognition complex is essential for HP1 binding to chromatin and heterochromatin organization. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 15093–15098. [Google Scholar]
- Chakraborty, A.; Shen, Z.; Prasanth, S.G. "ORCanization" on heterochromatin: linking DNA replication initiation to chromatin organization. Epigenetics 2011, 6, 665–670. [Google Scholar] [CrossRef]
- Onder, T.T.; Kara, N.; Cherry, A.; Sinha, A.U.; Zhu, N.; Bernt, K.M.; Cahan, P.; Marcarci, B.O.; Unternaehrer, J.; Gupta, P.B.; Lander, E.S.; Armstrong, S.A.; Daley, G.Q. Chromatin-modifying enzymes as modulators of reprogramming. Nature 2012, 483, 598–602. [Google Scholar] [CrossRef]
- Wakeman, T.P.; Wang, Q.; Feng, J.; Wang, X.F. Bat3 facilitates H3K79 dimethylation by DOT1L and promotes DNA damage-induced 53BP1 foci at G1/G2 cell-cycle phases. EMBO J. 2012, 31, 2169–2181. [Google Scholar] [CrossRef]
- Phillips, J.E.; Corces, V.G. CTCF: master weaver of the genome. Cell 2009, 137, 1194–1211. [Google Scholar] [CrossRef]
- Ohlsson, R.; Lobanenkov, V.; Klenova, E. Does CTCF mediate between nuclear organization and gene expression? Bioessays 2010, 32, 37–50. [Google Scholar] [CrossRef]
- Kang, H.; Wiedmer, A.; Yuan, Y.; Robertson, E.; Lieberman, P.M. Coordination of KSHV latent and lytic gene control by CTCF-cohesin mediated chromosome conformation. PLoS Pathog. 2011, 7, e1002140. [Google Scholar] [CrossRef]
- Kang, H.; Lieberman, P.M. Mechanism of Glycyrrhizic Acid Inhibition of Kaposi's Sarcoma-Associated Herpesvirus: Disruption of CTCF-Cohesin-Mediated RNA Polymerase II Pausing and Sister Chromatid Cohesion. J. Virol. 2011, 85, 11159–11169. [Google Scholar] [CrossRef]
- Tempera, I.; Klichinsky, M.; Lieberman, P.M. EBV latency types adopt alternative chromatin conformations. PLoS Pathog. 2011, 7, e1002180. [Google Scholar] [CrossRef]
- Atchison, M.; Basu, A.; Zaprazna, K.; Papasani, M. Mechanisms of Yin Yang 1 in oncogenesis: the importance of indirect effects. Crit. Rev. Oncog. 2012, 16, 143–161. [Google Scholar]
- Wang, J.; Sugden, B. Origins of bidirectional replication of Epstein-Barr virus: Models for understanding mammalian origins of DNA synthesis. J. Cell. Biochem. 2005, 94, 247–256. [Google Scholar] [CrossRef]
- Lindner, S.E.; Sugden, B. The plasmid replicon of Epstein-Barr virus: mechanistic insights into efficient, licensed, extrachromosomal replication in human cells. Plasmid. 2007, 58, 1–12. [Google Scholar] [CrossRef]
- Zhou, J.; Chau, C.M.; Deng, Z.; Shiekhattar, R.; Spindler, M.P.; Schepers, A.; Lieberman, P.M. Cell cycle regulation of chromatin at an origin of DNA replication. Embo J. 2005, 24, 1406–1417. [Google Scholar] [CrossRef]
- Kalla, M.; Hammerschmidt, W. Human B cells on their route to latent infection--early but transient expression of lytic genes of Epstein-Barr virus. Eur. J. Cell. Biol. 2011, 91, 65–69. [Google Scholar] [CrossRef]
- Tempera, I.; Wiedmer, A.; Dheekollu, J.; Lieberman, P.M. CTCF prevents the epigenetic drift of EBV latency promoter Qp. PLoS Pathog. 2010, 6, e1001048. [Google Scholar] [CrossRef]
- Hughes, D.J.; Marendy, E.M.; Dickerson, C.A.; Yetming, K.D.; Sample, C.E.; Sample, J.T. Contributions of CTCF and DNA methyltransferases DNMT1 and DNMT3B to Epstein-Barr virus restricted latency. J. Virol. 2011, 86, 1034–1045. [Google Scholar]
- Bergbauer, M.; Kalla, M.; Schmeinck, A.; Gobel, C.; Rothbauer, U.; Eck, S.; Benet-Pages, A.; Strom, T.M.; Hammerschmidt, W. CpG-methylation regulates a class of Epstein-Barr virus promoters. PLoS Pathog. 2010, 6, 1001114. [Google Scholar] [CrossRef]
- Ramasubramanyan, S.; Kanhere, A.; Osborn, K.; Flower, K.; Jenner, R.G.; Sinclair, A.J. Genome-Wide Analyses of Zta Binding to the Epstein-Barr Virus Genome Reveals Interactions in both Early and Late Lytic Cycles and an Epigenetic Switch Leading to an Altered Binding Profile. J. Virol. 2012, 86, 12494–12502. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Arvey, A.; Tempera, I.; Lieberman, P.M. Interpreting the Epstein-Barr Virus (EBV) Epigenome Using High-Throughput Data. Viruses 2013, 5, 1042-1054. https://doi.org/10.3390/v5041042
Arvey A, Tempera I, Lieberman PM. Interpreting the Epstein-Barr Virus (EBV) Epigenome Using High-Throughput Data. Viruses. 2013; 5(4):1042-1054. https://doi.org/10.3390/v5041042
Chicago/Turabian StyleArvey, Aaron, Italo Tempera, and Paul M. Lieberman. 2013. "Interpreting the Epstein-Barr Virus (EBV) Epigenome Using High-Throughput Data" Viruses 5, no. 4: 1042-1054. https://doi.org/10.3390/v5041042
APA StyleArvey, A., Tempera, I., & Lieberman, P. M. (2013). Interpreting the Epstein-Barr Virus (EBV) Epigenome Using High-Throughput Data. Viruses, 5(4), 1042-1054. https://doi.org/10.3390/v5041042