MicroRNA-Mediated Restriction of HIV-1 in Resting CD4+ T Cells and Monocytes

Abstract
:1. CD4+ T Cells
1.1. T Cell Activation
1.2. Resting CD4+ T Cells
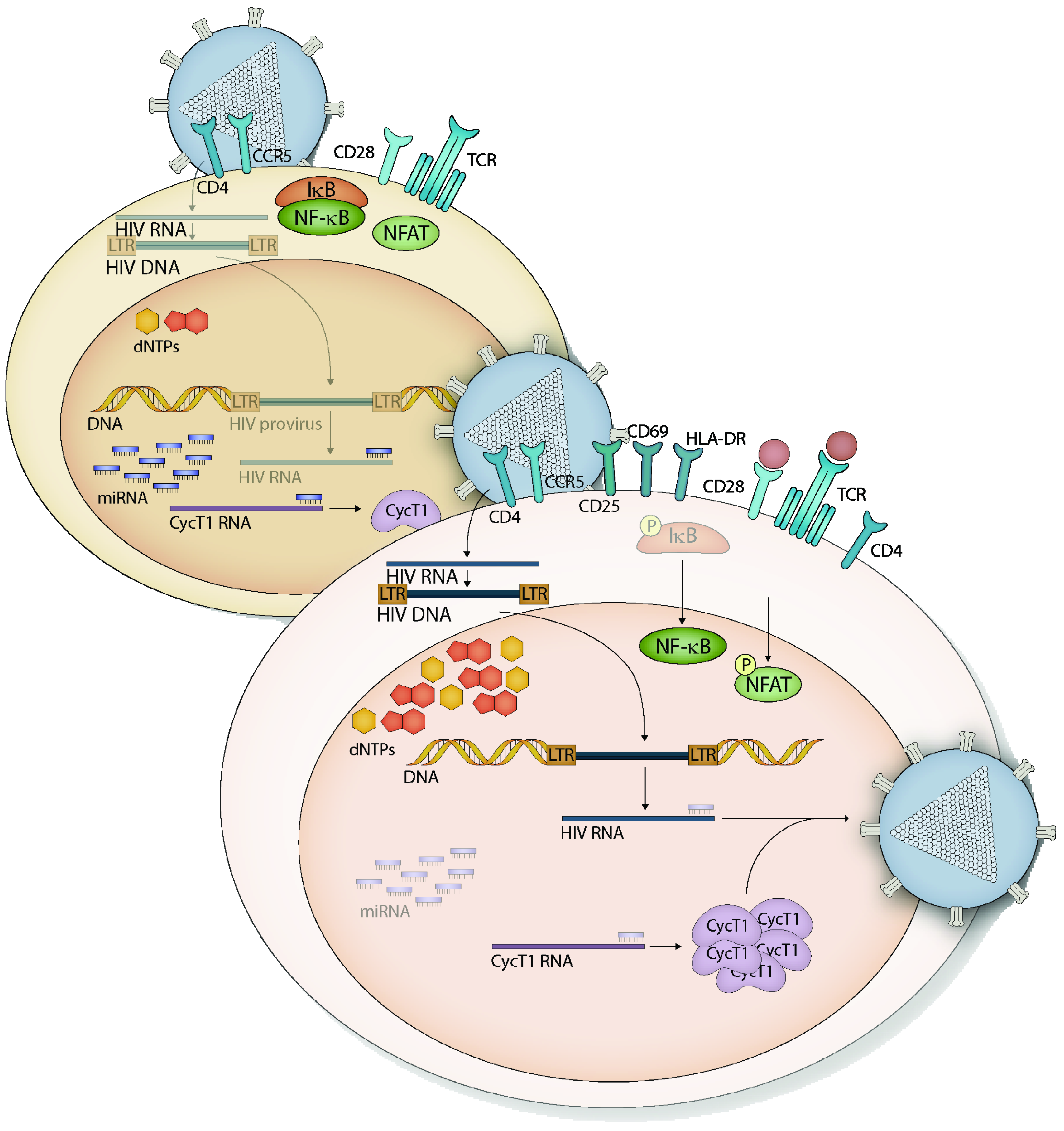
1.3. HIV-1 Restriction in Resting CD4+ T Cells
2. Monocytes and Macrophages
Monocyte and Macrophage Restriction of HIV-1 Replication
3. miRNAs and HIV-1 Replication in Resting CD4+ T Cells and Monocytes
3.1. miRNA Expression Contributes to HIV-1 Restriction

{kind=link}
| CD4+ T cells | HIV-1 RNA target site | References showing downregulation after infection | Other relevant targets | Notes |
|---|---|---|---|---|
| miR-17-92 cluster | N | [82,93] | P/CAF | |
| miR-27 (a, b) | N | 27a: [93,94] | miR-27b: Cyclin T1 | Downregulated by H. saimiri and BLV |
| miR-29 (a, b, c) | Y | [93,94,95] | miR-29b: Cyclin T1 (?) | Targets HIV RNA to RISC and P-bodies [96] |
| miR-150 | Y | [93] | c-Myb, Notch3, Cyclin T1 (?) | Mediates T cell differentiation |
| miR-223 | Y | [93], upregulated in: [95] | Cyclin T1 (?) | Also decreased during monocyte-macrophage differentiation |
| Monocytes | ||||
| miR-198 | N | Upregulated after CEMx174 infection: [94] | Cyclin T1 | Appears to be differentially regulated in CD4+ T cells vs. macrophages |
3.2. Viral Infection Potentiates Changes in miRNAs Highly Expressed in Resting Cells
4. Perspectives
Acknowledgments
Conflict of Interest
References and Notes
- Boomer, J.S.; Green, J.M. An enigmatic tail of CD28 signaling. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Schwartz, R.H. T cell anergy. Annu. Rev. Immunol. 2003, 21, 305–334. [Google Scholar] [CrossRef]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- McKinstry, K.K.; Strutt, T.M.; Swain, S.L. The potential of CD4 T-cell memory. Immunology 2010, 130, 1–9. [Google Scholar] [CrossRef]
- Liao, W.; Lin, J.X.; Leonard, W.J. IL-2 family cytokines: New insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr. Opin. Immunol. 2011, 23, 598–604. [Google Scholar] [CrossRef]
- Sandberg, R.; Neilson, J.R.; Sarma, A.; Sharp, P.A.; Burge, C.B. Proliferating cells express mRNAs with shortened 3' untranslated regions and fewer microRNA target sites. Science 2008, 320, 1643–1647. [Google Scholar] [CrossRef]
- Siliciano, R.F.; Greene, W.C. HIV Latency. Cold Spring Harb. Perspect. Med. 2011, 1. [Google Scholar] [CrossRef]
- Yusuf, I.; Fruman, D.A. Regulation of quiescence in lymphocytes. Trends Immunol. 2003, 24, 380–386. [Google Scholar] [CrossRef]
- Buckley, A.F.; Kuo, C.T.; Leiden, J.M. Transcription factor LKLF is sufficient to program T cell quiescence via a c-Myc-dependent pathway. Nat. Immunol. 2001, 2, 698–704. [Google Scholar] [CrossRef]
- Haaland, R.E.; Yu, W.; Rice, A.P. Identification of LKLF-regulated genes in quiescent CD4+ T lymphocytes. Mol. Immunol. 2005, 42, 627–641. [Google Scholar] [CrossRef]
- Kuo, C.T.; Veselits, M.L.; Leiden, J.M. LKLF: A transcriptional regulator of single-positive T cell quiescence and survival. Science 1997, 277, 1986–1990. [Google Scholar] [CrossRef]
- Liu, J.O. The yins of T cell activation. Sci STKE 2005, 2005. [Google Scholar] [CrossRef]
- Barski, A.; Jothi, R.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Zhao, K. Chromatin poises miRNA- and protein-coding genes for expression. Genome Res. 2009, 19, 1742–1751. [Google Scholar] [CrossRef]
- Smith, A.E.; Chronis, C.; Christodoulakis, M.; Orr, S.J.; Lea, N.C.; Twine, N.A.; Bhinge, A.; Mufti, G.J.; Thomas, N.S. Epigenetics of human T cells during the G0→G1 transition. Genome Res. 2009, 19, 1325–1337. [Google Scholar] [CrossRef]
- Lim, P.S.; Hardy, K.; Bunting, K.L.; Ma, L.; Peng, K.; Chen, X.; Shannon, M.F. Defining the chromatin signature of inducible genes in T cells. Genome Biol. 2009, 10, R107. [Google Scholar] [CrossRef]
- Nechaev, S.; Adelman, K. Pol II waiting in the starting gates: Regulating the transition from transcription initiation into productive elongation. Biochim. Biophys. Acta 2011, 1809, 34–45. [Google Scholar]
- Ramakrishnan, R.; Chiang, K.; Liu, H.; Budhiraja, S.; Donahue, H.; Rice, A.P. Making a short story long: Regulation of P-TEFb and HIV-1 transcriptional elongation in CD4+ T lymphocytes and macrophages. Biology 2012, 94–115. [Google Scholar]
- Ott, M.; Geyer, M.; Zhou, Q. The control of HIV transcription: Keeping RNA polymerase II on track. Cell Host Microbe 2011, 10, 426–435. [Google Scholar] [CrossRef]
- Cohen, A.; Barankiewicz, J.; Lederman, H.M.; Gelfand, E.W. Purine and pyrimidine metabolism in human T lymphocytes. Regulation of deoxyribonucleotide metabolism. J. Biol. Chem. 1983, 258, 12334–12340. [Google Scholar]
- Cohen, A.; Barankiewicz, J.; Lederman, H.M.; Gelfand, E.W. Purine metabolism in human T lymphocytes: Role of the purine nucleoside cycle. Can. J. Biochem. Cell Biol. 1984, 62, 577–583. [Google Scholar] [CrossRef]
- Fairbanks, L.D.; Bofill, M.; Ruckemann, K.; Simmonds, H.A. Importance of ribonucleotide availability to proliferating T-lymphocytes from healthy humans. Disproportionate expansion of pyrimidine pools and contrasting effects of de novo synthesis inhibitors. J. Biol. Chem. 1995, 270, 29682–29689. [Google Scholar]
- Marijnen, Y.M.; de Korte, D.; Haverkort, W.A.; den Breejen, E.J.; van Gennip, A.H.; Roos, D. Studies on the incorporation of precursors into purine and pyrimidine nucleotides via ‘de novo’ and ‘salvage’ pathways in normal lymphocytes and lymphoblastic cell-line cells. Biochim. Biophys. Acta 1989, 1012, 148–155. [Google Scholar]
- Michalek, R.D.; Rathmell, J.C. The metabolic life and times of a T-cell. Immunol. Rev. 2010, 236, 190–202. [Google Scholar] [CrossRef]
- Pearce, E.L. Metabolism in T cell activation and differentiation. Curr. Opin. Immunol. 2010, 22, 314–320. [Google Scholar] [CrossRef]
- Vatakis, D.N.; Bristol, G.; Wilkinson, T.A.; Chow, S.A.; Zack, J.A. Immediate activation fails to rescue efficient human immunodeficiency virus replication in quiescent CD4+ T cells. J. Virol. 2007, 81, 3574–3582. [Google Scholar]
- Spina, C.A.; Guatelli, J.C.; Richman, D.D. Establishment of a stable, inducible form of human immunodeficiency virus type 1 DNA in quiescent CD4 lymphocytes in vitro. J. Virol. 1995, 69, 2977–2988. [Google Scholar]
- Pierson, T.C.; Zhou, Y.; Kieffer, T.L.; Ruff, C.T.; Buck, C.; Siliciano, R.F. Molecular characterization of preintegration latency in human immunodeficiency virus type 1 infection. J. Virol. 2002, 76, 8518–8531. [Google Scholar]
- Swiggard, W.J.; O’Doherty, U.; McGain, D.; Jeyakumar, D.; Malim, M.H. Long HIV type 1 reverse transcripts can accumulate stably within resting CD4+ T cells while short ones are degraded. AIDS Res. Hum. Retroviruses 2004, 20, 285–295. [Google Scholar] [CrossRef]
- Zack, J.A.; Arrigo, S.J.; Weitsman, S.R.; Go, A.S.; Haislip, A.; Chen, I.S. HIV-1 entry into quiescent primary lymphocytes: Molecular analysis reveals a labile, latent viral structure. Cell 1990, 61, 213–222. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, H.; Siliciano, J.D.; Siliciano, R.F. Kinetics of human immunodeficiency virus type 1 decay following entry into resting CD4+ T cells. J. Virol. 2005, 79, 2199–2210. [Google Scholar]
- Korin, Y.D.; Zack, J.A. Nonproductive human immunodeficiency virus type 1 infection in nucleoside-treated G0 lymphocytes. J. Virol. 1999, 73, 6526–6532. [Google Scholar]
- Plesa, G.; Dai, J.; Baytop, C.; Riley, J.L.; June, C.H.; O’Doherty, U. Addition of deoxynucleosides enhances human immunodeficiency virus type 1 integration and 2LTR formation in resting CD4+ T cells. J. Virol. 2007, 81, 13938–13942. [Google Scholar]
- Swiggard, W.J.; Baytop, C.; Yu, J.J.; Dai, J.; Li, C.; Schretzenmair, R.; Theodosopoulos, T.; O’Doherty, U. Human immunodeficiency virus type 1 can establish latent infection in resting CD4+ T cells in the absence of activating stimuli. J. Virol. 2005, 79, 14179–14188. [Google Scholar]
- Dai, J.; Agosto, L.M.; Baytop, C.; Yu, J.J.; Pace, M.J.; Liszewski, M.K.; O’Doherty, U. Human immunodeficiency virus integrates directly into naive resting CD4+ T cells but enters naive cells less efficiently than memory cells. J. Virol. 2009, 83, 4528–4537. [Google Scholar]
- Vatakis, D.N.; Kim, S.; Kim, N.; Chow, S.A.; Zack, J.A. Human immunodeficiency virus integration efficiency and site selection in quiescent CD4+ T cells. J. Virol. 2009, 83, 6222–6233. [Google Scholar] [CrossRef]
- Strebel, K.; Luban, J.; Jeang, K. Human cellular restriction factors that target HIV-1 replication. BMC Med. 2009, 7. [Google Scholar] [CrossRef]
- Ganesh, L.; Burstein, E.; Guha-Niyogi, A.; Louder, M.K.; Mascola, J.R.; Klomp, L.W.; Wijmenga, C.; Duckett, C.S.; Nabel, G.J. The gene product Murr1 restricts HIV-1 replication in resting CD4+ lymphocytes. Nature 2003, 426, 853–857. [Google Scholar]
- Eckstein, D.A.; Penn, M.L.; Korin, Y.D.; Scripture-Adams, D.D.; Zack, J.A.; Kreisberg, J.F.; Roederer, M.; Sherman, M.P.; Chin, P.S.; Goldsmith, M.A. HIV-1 actively replicates in naive CD4(+) T cells residing within human lymphoid tissues. Immunity 2001, 15, 671–682. [Google Scholar] [CrossRef]
- Kinter, A.; Moorthy, A.; Jackson, R.; Fauci, A.S. Productive HIV infection of resting CD4+ T cells: Role of lymphoid tissue microenvironment and effect of immunomodulating agents. AIDS Res. Hum. Retroviruses 2003, 19, 847–856. [Google Scholar] [CrossRef]
- Nishimura, Y.; Brown, C.R.; Mattapallil, J.J.; Igarashi, T.; Buckler-White, A.; Lafont, B.A.; Hirsch, V.M.; Roederer, M.; Martin, M.A. Resting naive CD4+ T cells are massively infected and eliminated by X4-tropic simian-human immunodeficiency viruses in macaques. Proc. Natl. Acad. Sci. USA 2005, 102, 8000–8005. [Google Scholar]
- Li, Q.; Duan, L.; Estes, J.D.; Ma, Z.M.; Rourke, T.; Wang, Y.; Reilly, C.; Carlis, J.; Miller, C.J.; Haase, A.T. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature 2005, 434, 1148–1152. [Google Scholar]
- Finkel, T.H.; Tudor-Williams, G.; Banda, N.K.; Cotton, M.F.; Curiel, T.; Monks, C.; Baba, T.W.; Ruprecht, R.M.; Kupfer, A. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat. Med. 1995, 1, 129–134. [Google Scholar]
- Jekle, A.; Keppler, O.T.; de Clercq, E.; Schols, D.; Weinstein, M.; Goldsmith, M.A. In vivo evolution of human immunodeficiency virus type 1 toward increased pathogenicity through CXCR4-mediated killing of uninfected CD4 T cells. J. Virol. 2003, 77, 5846–5854. [Google Scholar] [CrossRef]
- Doitsh, G.; Cavrois, M.; Lassen, K.G.; Zepeda, O.; Yang, Z.; Santiago, M.L.; Hebbeler, A.M.; Greene, W.C. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 2010, 143, 789–801. [Google Scholar] [CrossRef]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar]
- Chomont, N.; DaFonseca, S.; Vandergeeten, C.; Ancuta, P.; Sekaly, R.P. Maintenance of CD4+ T-cell memory and HIV persistence: Keeping memory, keeping HIV. Curr. Opin. HIV AIDS 2011, 6, 30–36. [Google Scholar] [CrossRef]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages, and dendritic cells. Science 2010, 327, 656–661. [Google Scholar]
- Coleman, C.M.; Wu, L. HIV interactions with monocytes and dendritic cells: Viral latency and reservoirs. Retrovirology 2009, 6. [Google Scholar] [CrossRef]
- Bergamaschi, A.; Pancino, G. Host hindrance to HIV-1 replication in monocytes and macrophages. Retrovirology 2010, 7. [Google Scholar] [CrossRef]
- Naif, H.M.; Li, S.; Alali, M.; Sloane, A.; Wu, L.; Kelly, M.; Lynch, G.; Lloyd, A.; Cunningham, A.L. CCR5 expression correlates with susceptibility of maturing monocytes to human immunodeficiency virus type 1 infection. J. Virol. 1998, 72, 830–836. [Google Scholar]
- Tuttle, D.L.; Harrison, J.K.; Anders, C.; Sleasman, J.W.; Goodenow, M.M. Expression of CCR5 increases during monocyte differentiation and directly mediates macrophage susceptibility to infection by human immunodeficiency virus type 1. J. Virol. 1998, 72, 4962–4969. [Google Scholar]
- Triques, K.; Stevenson, M. Characterization of restrictions to human immunodeficiency virus type 1 infection of monocytes. J. Virol. 2004, 78, 5523–5527. [Google Scholar] [CrossRef]
- Arfi, V.; Riviere, L.; Jarrosson-Wuilleme, L.; Goujon, C.; Rigal, D.; Darlix, J.L.; Cimarelli, A. Characterization of the early steps of infection of primary blood monocytes by human immunodeficiency virus type 1. J. Virol. 2008, 82, 6557–6565. [Google Scholar]
- Cavrois, M.; De Noronha, C.; Greene, W.C. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotechnol. 2002, 20, 1151–1154. [Google Scholar] [CrossRef]
- Sonza, S.; Maerz, A.; Deacon, N.; Meanger, J.; Mills, J.; Crowe, S. Human immunodeficiency virus type 1 replication is blocked prior to reverse transcription and integration in freshly isolated peripheral blood monocytes. J. Virol. 1996, 70, 3863–3869. [Google Scholar]
- O'Brien, W.A.; Namazi, A.; Kalhor, H.; Mao, S.H.; Zack, J.A.; Chen, I.S. Kinetics of human immunodeficiency virus type 1 reverse transcription in blood mononuclear phagocytes are slowed by limitations of nucleotide precursors. J. Virol. 1994, 68, 1258–1263. [Google Scholar]
- Neil, S.; Martin, F.; Ikeda, Y.; Collins, M. Postentry restriction to human immunodeficiency virus-based vector transduction in human monocytes. J. Virol. 2001, 75, 5448–5456. [Google Scholar] [CrossRef]
- Dong, C.; Kwas, C.; Wu, L. Transcriptional restriction of human immunodeficiency virus type 1 gene expression in undifferentiated primary monocytes. J. Virol. 2009, 83, 3518–3527. [Google Scholar] [CrossRef]
- Liou, L.Y.; Herrmann, C.H.; Rice, A.P. Transient induction of cyclin T1 during human macrophage differentiation regulates human immunodeficiency virus type 1 Tat transactivation function. J. Virol. 2002, 76, 10579–10587. [Google Scholar] [CrossRef]
- Liou, L.Y.; Herrmann, C.H.; Rice, A.P. Human immunodeficiency virus type 1 infection induces cyclin T1 expression in macrophages. J. Virol. 2004, 78, 8114–8119. [Google Scholar] [CrossRef]
- Lambotte, O.; Taoufik, Y.; de Goer, M.G.; Wallon, C.; Goujard, C.; Delfraissy, J.F. Detection of infectious HIV in circulating monocytes from patients on prolonged highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2000, 23, 114–119. [Google Scholar]
- Mikovits, J.A.; Lohrey, N.C.; Schulof, R.; Courtless, J.; Ruscetti, F.W. Activation of infectious virus from latent human immunodeficiency virus infection of monocytes in vivo. J. Clin. Invest. 1992, 90, 1486–1491. [Google Scholar] [CrossRef]
- Sonza, S.; Mutimer, H.P.; Oelrichs, R.; Jardine, D.; Harvey, K.; Dunne, A.; Purcell, D.F.; Birch, C.; Crowe, S.M. Monocytes harbour replication-competent, non-latent HIV-1 in patients on highly active antiretroviral therapy. AIDS 2001, 15, 17–22. [Google Scholar] [CrossRef]
- Sonza, S.; Kiernan, R.E.; Maerz, A.L.; Deacon, N.J.; McPhee, D.A.; Crowe, S.M. Accumulation of unintegrated circular viral DNA in monocytes and growth-arrested T cells following infection with HIV-1. J. Leukoc. Biol. 1994, 56, 289–293. [Google Scholar]
- McElrath, M.J.; Steinman, R.M.; Cohn, Z.A. Latent HIV-1 infection in enriched populations of blood monocytes and T cells from seropositive patients. J. Clin. Invest. 1991, 87, 27–30. [Google Scholar] [CrossRef]
- Carter, C.C.; Onafuwa-Nuga, A.; McNamara, L.A.; Riddell, J.t.; Bixby, D.; Savona, M.R.; Collins, K.L. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat. Med. 2010, 16, 446–451. [Google Scholar]
- Durand, C.M.; Ghiaur, G.; Siliciano, J.D.; Rabi, S.A.; Eisele, E.E.; Salgado, M.; Shan, L.; Lai, J.F.; Zhang, H.; Margolick, J.; et al. HIV-1 DNA is detected in bone marrow populations containing CD4+ T cells but is not found in purified CD34+ hematopoietic progenitor cells in most patients on antiretroviral therapy. J. Infect. Dis. 2012, 205, 1014–1018. [Google Scholar] [CrossRef]
- Josefsson, L.; Eriksson, S.; Sinclair, E.; Ho, T.; Killian, M.; Epling, L.; Shao, W.; Lewis, B.; Bacchetti, P.; Loeb, L.; et al. Hematopoietic precursor cells isolated from patients on long-term suppressive HIV therapy did not contain HIV-1 DNA. J. Infect. Dis. 2012, 206, 28–34. [Google Scholar] [CrossRef]
- Chang, J.; Naif, H.M.; Li, S.; Sullivan, J.S.; Randle, C.M.; Cunningham, A.L. Twin studies demonstrate a host cell genetic effect on productive human immunodeficiency virus infection of human monocytes and macrophages in vitro. J. Virol. 1996, 70, 7792–7803. [Google Scholar]
- Eisert, V.; Kreutz, M.; Becker, K.; Konigs, C.; Alex, U.; Rubsamen-Waigmann, H.; Andreesen, R.; von Briesen, H. Analysis of cellular factors influencing the replication of human immunodeficiency virus type I in human macrophages derived from blood of different healthy donors. Virology 2001, 286, 31–44. [Google Scholar] [CrossRef]
- Bol, S.M.; van Remmerden, Y.; Sietzema, J.G.; Kootstra, N.A.; Schuitemaker, H.; van’t Wout, A.B. Donor variation in in vitro HIV-1 susceptibility of monocyte-derived macrophages. Virology 2009, 390, 205–211. [Google Scholar] [CrossRef]
- Herbein, G.; Varin, A. The macrophage in HIV-1 infection: From activation to deactivation? Retrovirology 2010, 7. [Google Scholar] [CrossRef] [Green Version]
- Cassol, E.; Cassetta, L.; Alfano, M.; Poli, G. Macrophage polarization and HIV-1 infection. J. Leukoc. Biol. 2010, 87, 599–608. [Google Scholar] [CrossRef]
- Kaushik, R.; Zhu, X.; Stranska, R.; Wu, Y.; Stevenson, M. A cellular restriction dictates the permissivity of nondividing monocytes/macrophages to lentivirus and gammaretrovirus infection. Cell Host Microbe 2009, 6, 68–80. [Google Scholar] [CrossRef]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef]
- Goldstone, D.C.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.; Rice, G.I.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar]
- Powell, R.D.; Holland, P.J.; Hollis, T.; Perrino, F.W. Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J. Biol. Chem. 2011, 286, 43596–43600. [Google Scholar]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar]
- Triboulet, R.; Mari, B.; Lin, Y.L.; Chable-Bessia, C.; Bennasser, Y.; Lebrigand, K.; Cardinaud, B.; Maurin, T.; Barbry, P.; Baillat, V.; et al. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science 2007, 315, 1579–1582. [Google Scholar] [CrossRef]
- Chable-Bessia, C.; Meziane, O.; Latreille, D.; Triboulet, R.; Zamborlini, A.; Wagschal, A.; Jacquet, J.M.; Reynes, J.; Levy, Y.; Saib, A.; et al. Suppression of HIV-1 replication by microRNA effectors. Retrovirology 2009, 6. [Google Scholar] [CrossRef]
- Wu, H.; Neilson, J.R.; Kumar, P.; Manocha, M.; Shankar, P.; Sharp, P.A.; Manjunath, N. miRNA profiling of naive, effector and memory CD8 T cells. PLoS One 2007, 2, e1020. [Google Scholar] [CrossRef]
- Rossi, R.L.; Rossetti, G.; Wenandy, L.; Curti, S.; Ripamonti, A.; Bonnal, R.J.; Birolo, R.S.; Moro, M.; Crosti, M.C.; Gruarin, P.; et al. Distinct microRNA signatures in human lymphocyte subsets and enforcement of the naive state in CD4+ T cells by the microRNA miR-125b. Nat. Immunol. 2011, 12, 796–803. [Google Scholar] [CrossRef]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef]
- Sung, T.L.; Rice, A.P. miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 2009, 5, e1000263. [Google Scholar] [CrossRef]
- Chiang, K.; Sung, T.L.; Rice, A.P. Regulation of cyclin T1 and HIV-1 replication by microRNAs in resting CD4+ T lymphocytes. J. Virol. 2012, 86, 3244–3252. [Google Scholar] [CrossRef]
- Monticelli, S.; Ansel, K.M.; Xiao, C.; Socci, N.D.; Krichevsky, A.M.; Thai, T.H.; Rajewsky, N.; Marks, D.S.; Sander, C.; Rajewsky, K.; et al. MicroRNA profiling of the murine hematopoietic system. Genome Biol. 2005, 6. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, S.; Mayr, C.; Bartel, D.P.; Lodish, H.F. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc. Natl. Acad. Sci. USA 2007, 104, 7080–7085. [Google Scholar] [CrossRef]
- Xiao, C.; Calado, D.P.; Galler, G.; Thai, T.H.; Patterson, H.C.; Wang, J.; Rajewsky, N.; Bender, T.P.; Rajewsky, K. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell 2007, 131, 146–159. [Google Scholar] [CrossRef]
- Ghisi, M.; Corradin, A.; Basso, K.; Frasson, C.; Serafin, V.; Mukherjee, S.; Mussolin, L.; Ruggero, K.; Bonanno, L.; Guffanti, A.; et al. Modulation of microRNA expression in human T-cell development: Targeting of NOTCH3 by miR-150. Blood 2011, 117, 7053–7062. [Google Scholar] [CrossRef]
- Houzet, L.; Yeung, M.L.; de Lame, V.; Desai, D.; Smith, S.M.; Jeang, K.T. MicroRNA profile changes in human immunodeficiency virus type 1 (HIV-1) seropositive individuals. Retrovirology 2008, 5. [Google Scholar] [CrossRef]
- Hayes, A.M.; Qian, S.; Yu, L.; Boris-Lawrie, K. Tat RNA silencing suppressor activity contributes to perturbation of lymphocyte miRNA by HIV-1. Retrovirology 2011, 8. [Google Scholar] [CrossRef]
- Sun, G.; Li, H.; Wu, X.; Covarrubias, M.; Scherer, L.; Meinking, K.; Luk, B.; Chomchan, P.; Alluin, J.; Gombart, A.F.; Rossi, J.J. Interplay between HIV-1 infection and host microRNAs. Nucleic Acids Res. 2012, 40, 2181–2196. [Google Scholar]
- Nathans, R.; Chu, C.Y.; Serquina, A.K.; Lu, C.C.; Cao, H.; Rana, T.M. Cellular microRNA and P bodies modulate host-HIV-1 interactions. Mol. Cell 2009, 34, 696–709. [Google Scholar] [CrossRef]
- Ghose, R.; Liou, L.Y.; Herrmann, C.H.; Rice, A.P. Induction of TAK (cyclin T1/P-TEFb) in purified resting CD4(+) T lymphocytes by combination of cytokines. J. Virol. 2001, 75, 11336–11343. [Google Scholar] [CrossRef]
- Marshall, R.M.; Salerno, D.; Garriga, J.; Grana, X. Cyclin T1 expression is regulated by multiple signaling pathways and mechanisms during activation of human peripheral blood lymphocytes. J. Immunol. 2005, 175, 6402–6411. [Google Scholar]
- Sung, T.L.; Rice, A.P. Effects of prostratin on Cyclin T1/P-TEFb function and the gene expression profile in primary resting CD4+ T cells. Retrovirology 2006, 3. [Google Scholar] [CrossRef]
- Ahluwalia, J.K.; Khan, S.Z.; Soni, K.; Rawat, P.; Gupta, A.; Hariharan, M.; Scaria, V.; Lalwani, M.; Pillai, B.; Mitra, D.; Brahmachari, S.K. Human cellular microRNA hsa-miR-29a interferes with viral nef protein expression and HIV-1 replication. Retrovirology 2008, 5. [Google Scholar] [CrossRef]
- Hoque, M.; Shamanna, R.A.; Guan, D.; Pe’ery, T.; Mathews, M.B. HIV-1 replication and latency are regulated by translational control of cyclin T1. J. Mol. Biol. 2011, 410, 917–932. [Google Scholar] [CrossRef]
- Chiang, K.; Rice, A.P. Mini ways to stop a virus: MicroRNAs and HIV-1 replication. Future Virol. 2011, 6, 209–221. [Google Scholar] [CrossRef]
- Herschkowitz, J.I.; Fu, X. MicroRNAs add an additional layer to the complexity of cell signaling. Sci. Signal. 2011, 4. [Google Scholar] [CrossRef]
- Chaudhuri, A.A.; So, A.Y.; Sinha, N.; Gibson, W.S.; Taganov, K.D.; O’Connell, R.M.; Baltimore, D. MicroRNA-125b potentiates macrophage activation. J. Immunol. 2011, 187, 5062–5068. [Google Scholar] [CrossRef]
- Wang, X.; Ye, L.; Zhou, Y.; Liu, M.Q.; Zhou, D.J.; Ho, W.Z. Inhibition of anti-HIV microRNA expression: A mechanism for opioid-mediated enhancement of HIV infection of monocytes. Am. J. Pathol. 2011, 178, 41–47. [Google Scholar] [CrossRef]
- Wang, X.; Ye, L.; Hou, W.; Zhou, Y.; Wang, Y.J.; Metzger, D.S.; Ho, W.Z. Cellular microRNA expression correlates with susceptibility of monocytes/macrophages to HIV-1 infection. Blood 2009, 113, 671–674. [Google Scholar] [CrossRef]
- Witwer, K.W.; Watson, A.K.; Blankson, J.N.; Clements, J.E. Relationships of PBMC microRNA expression, plasma viral load, and CD4+ T-cell count in HIV-1-infected elite suppressors and viremic patients. Retrovirology 2012, 9. [Google Scholar] [CrossRef]
- Cazalla, D.; Yario, T.; Steitz, J. Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science 2010, 328, 1563–1566. [Google Scholar] [CrossRef]
- Buck, A.H.; Perot, J.; Chisholm, M.A.; Kumar, D.S.; Tuddenham, L.; Cognat, V.; Marcinowski, L.; Dolken, L.; Pfeffer, S. Post-transcriptional regulation of miR-27 in murine cytomegalovirus infection. RNA 2010, 16, 307–315. [Google Scholar] [CrossRef]
- Libri, V.; Helwak, A.; Miesen, P.; Santhakumar, D.; Borger, J.G.; Kudla, G.; Grey, F.; Tollervey, D.; Buck, A.H. Murine cytomegalovirus encodes a miR-27 inhibitor disguised as a target. Proc. Natl. Acad. Sci. USA 2012, 109, 279–284. [Google Scholar]
- Marcinowski, L.; Tanguy, M.; Krmpotic, A.; Radle, B.; Lisnic, V.J.; Tuddenham, L.; Chane-Woon-Ming, B.; Ruzsics, Z.; Erhard, F.; Benkartek, C.; et al. Degradation of cellular miR-27 by a novel, highly abundant viral transcript is important for efficient virus replication in vivo. PLoS Pathog. 2012, 8, e1002510. [Google Scholar] [CrossRef] [Green Version]
- Kincaid, R.P.; Burke, J.M.; Sullivan, C.S. RNA virus microRNA that mimics a B-cell oncomiR. Proc. Natl. Acad. Sci. USA 2012, 109, 3077–3082. [Google Scholar] [CrossRef]
- Pasquinelli, A.E. MicroRNAs and their targets: Recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012, 13, 271–282. [Google Scholar]
- Ameres, S.L.; Horwich, M.D.; Hung, J.H.; Xu, J.; Ghildiyal, M.; Weng, Z.; Zamore, P.D. Target RNA-directed trimming and tailing of small silencing RNAs. Science 2010, 328, 1534–1539. [Google Scholar] [CrossRef]
- Wyman, S.K.; Knouf, E.C.; Parkin, R.K.; Fritz, B.R.; Lin, D.W.; Dennis, L.M.; Krouse, M.A.; Webster, P.J.; Tewari, M. Post-transcriptional generation of miRNA variants by multiple nucleotidyl transferases contributes to miRNA transcriptome complexity. Genome Res. 2011, 21, 1450–1461. [Google Scholar] [CrossRef]
- Baccarini, A.; Chauhan, H.; Gardner, T.J.; Jayaprakash, A.D.; Sachidanandam, R.; Brown, B.D. Kinetic analysis reveals the fate of a microRNA following target regulation in mammalian cells. Curr. Biol. 2011, 21, 369–376. [Google Scholar] [CrossRef]
- Cloonan, N.; Wani, S.; Xu, Q.; Gu, J.; Lea, K.; Heater, S.; Barbacioru, C.; Steptoe, A.L.; Martin, H.C.; Nourbakhsh, E.; et al. MicroRNAs and their isomiRs function cooperatively to target common biological pathways. Genome Biol. 2011, 12. [Google Scholar] [CrossRef]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 2009, 460, 479–486. [Google Scholar]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef]
- Baek, D.; Villen, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar]
- Selbach, M.; Schwanhausser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008, 455, 58–63. [Google Scholar]
- Hendrickson, D.G.; Hogan, D.J.; McCullough, H.L.; Myers, J.W.; Herschlag, D.; Ferrell, J.E.; Brown, P.O. Concordant regulation of translation and mRNA abundance for hundreds of targets of a human microRNA. PLoS Biol. 2009, 7, e1000238. [Google Scholar] [CrossRef]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, D.; Chen, X.; Li, J.; Li, L.; Bian, Z.; Sun, F.; Lu, J.; Yin, Y.; Cai, X.; et al. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol. Cell 2010, 39, 133–144. [Google Scholar] [CrossRef]
- Muratori, C.; Cavallin, L.E.; Kratzel, K.; Tinari, A.; de Milito, A.; Fais, S.; D’Aloja, P.; Federico, M.; Vullo, V.; Fomina, A.; et al. Massive secretion by T cells is caused by HIV Nef in infected cells and by Nef transfer to bystander cells. Cell Host Microbe 2009, 6, 218–230. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chiang, K.; Rice, A.P. MicroRNA-Mediated Restriction of HIV-1 in Resting CD4+ T Cells and Monocytes. Viruses 2012, 4, 1390-1409. https://doi.org/10.3390/v4091390
Chiang K, Rice AP. MicroRNA-Mediated Restriction of HIV-1 in Resting CD4+ T Cells and Monocytes. Viruses. 2012; 4(9):1390-1409. https://doi.org/10.3390/v4091390
Chicago/Turabian StyleChiang, Karen, and Andrew P. Rice. 2012. "MicroRNA-Mediated Restriction of HIV-1 in Resting CD4+ T Cells and Monocytes" Viruses 4, no. 9: 1390-1409. https://doi.org/10.3390/v4091390