Phosphoinositides in the Hepatitis C Virus Life Cycle

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
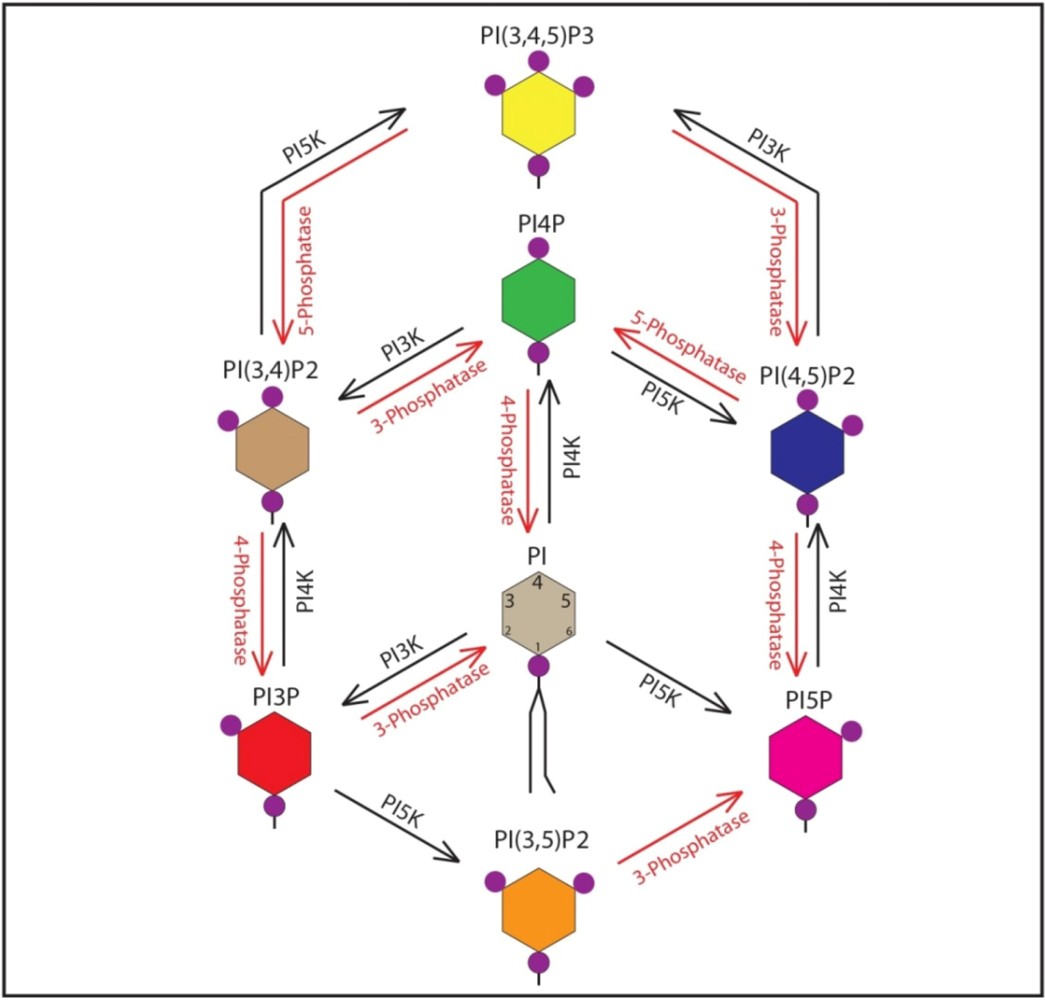
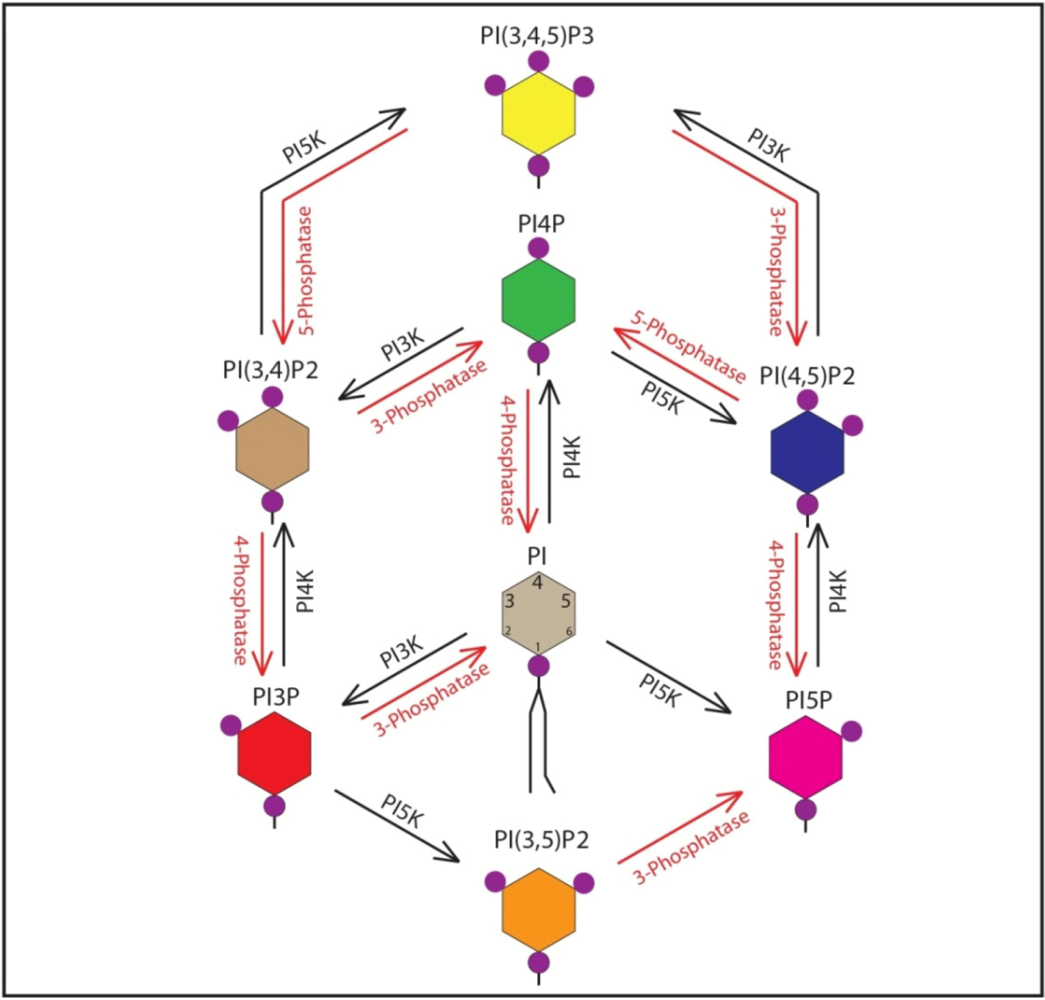
2. Cellular Functions of Phosphoinositides
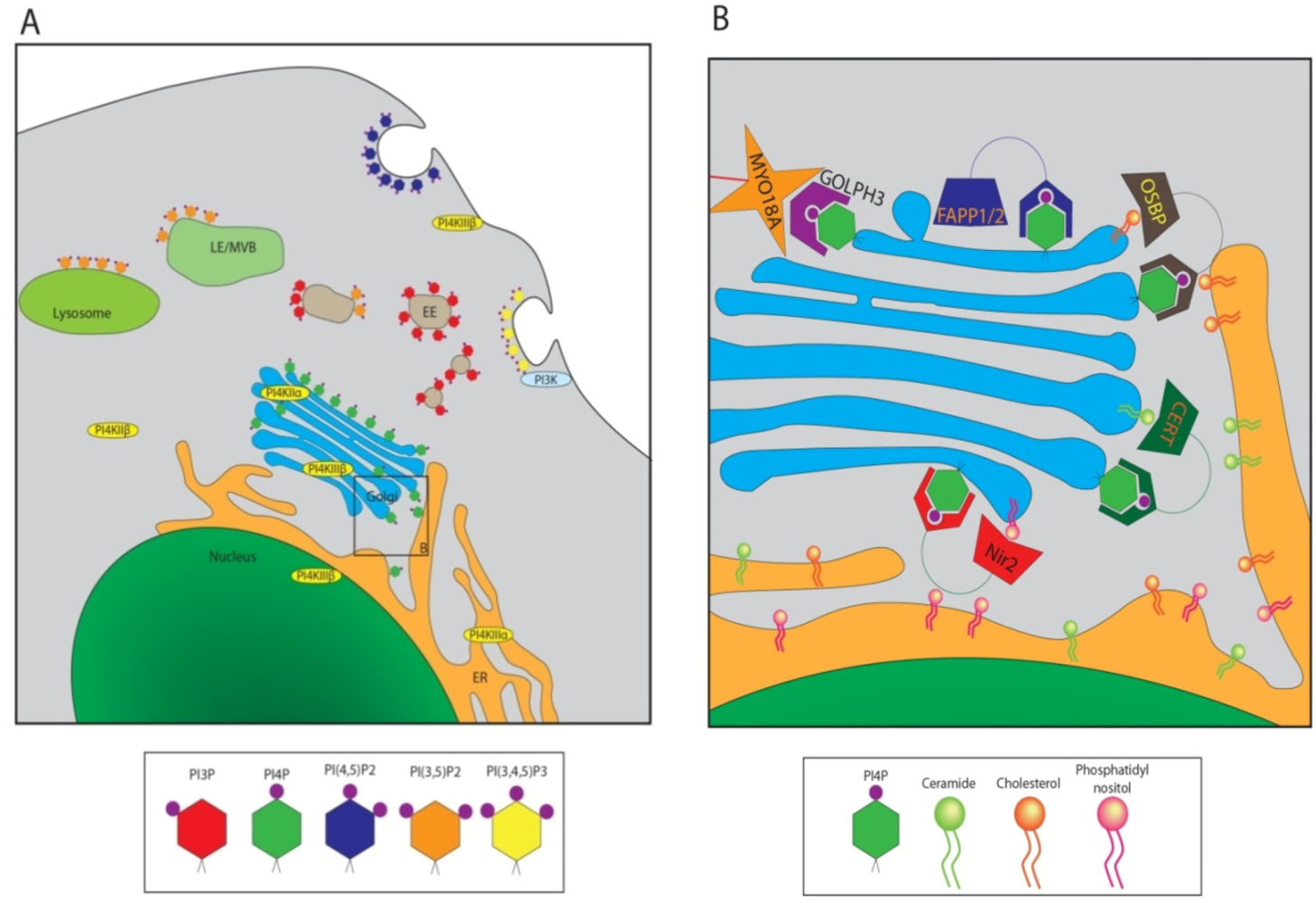
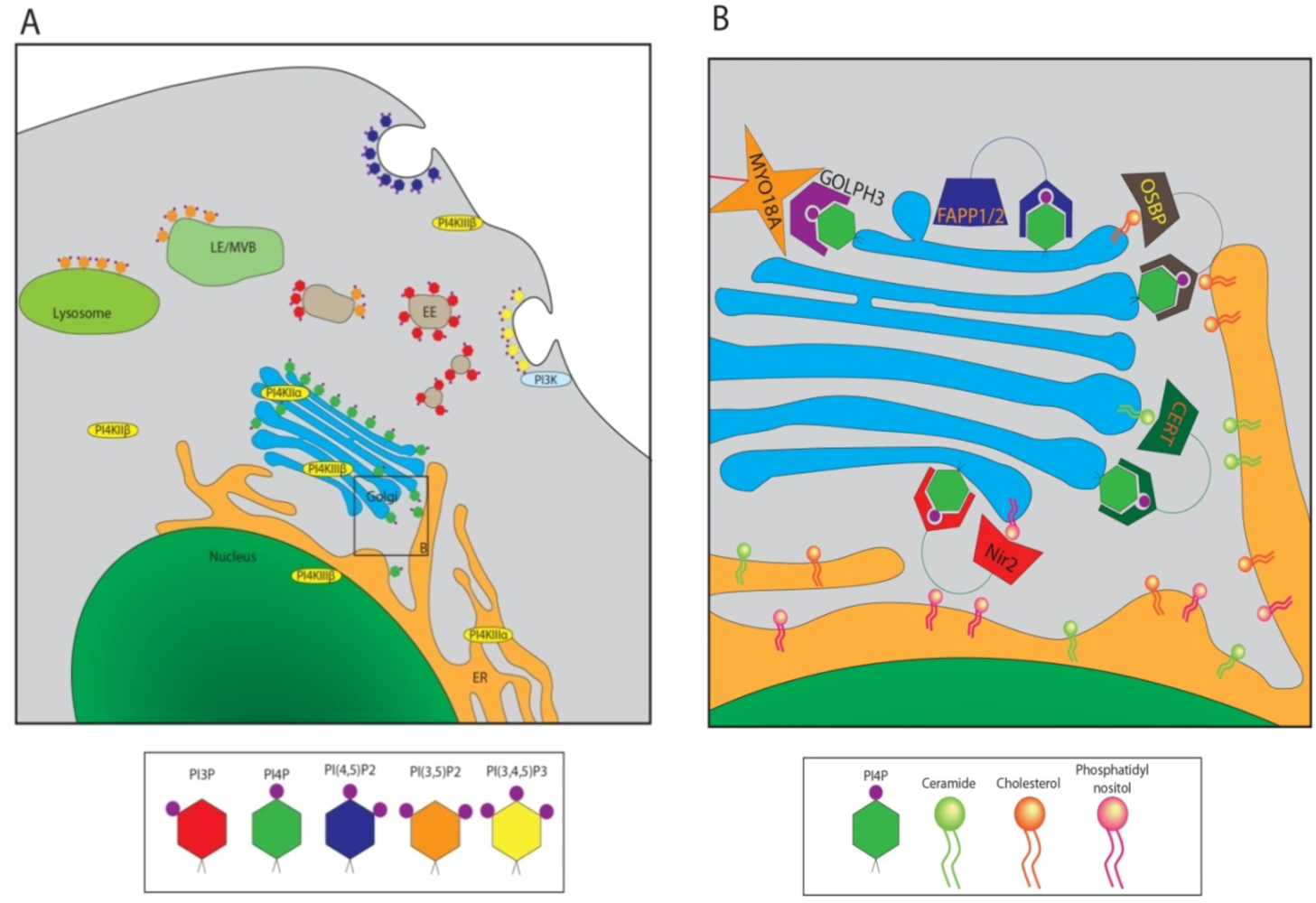
3. PI4P Significance in HCV Replication
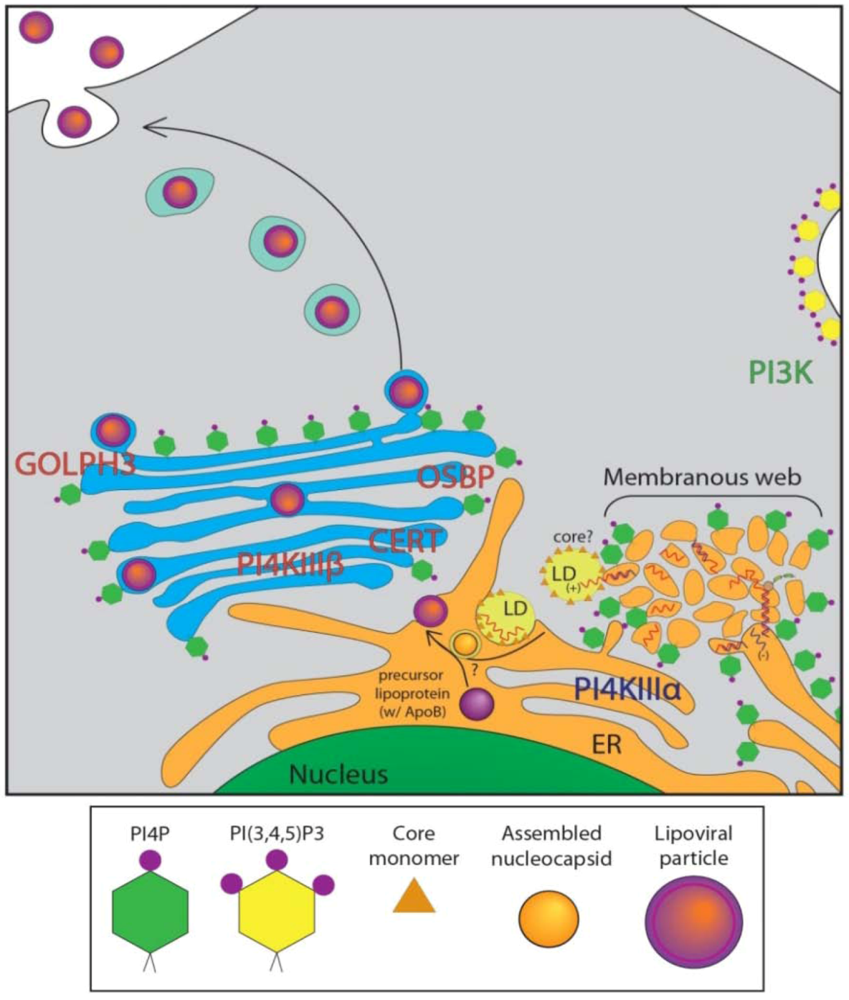
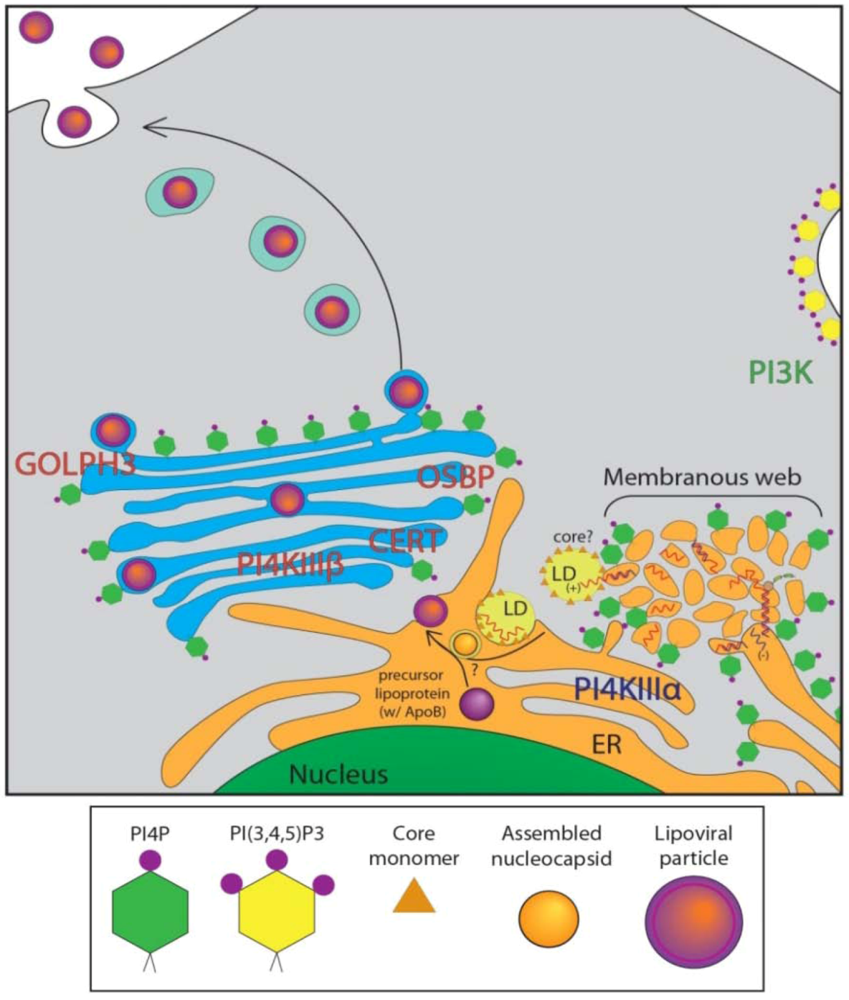
4. Role of Golgi-localized PI4P in HCV Secretion
5. HCV and PI3K Activation
6. Targeting PI4Ks for HCV Therapeutics
7. Concluding Remarks
Acknowledgments
Conflict of Interest
References and Notes
- Shepard, C.W.; Finelli, L.; Alter, M.J. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 2005, 5, 558–567. [Google Scholar] [CrossRef]
- Pawlotsky, J.M. Pathophysiology of hepatitis C virus infection and related liver disease. Trends Microbiol. 2004, 12, 96–102. [Google Scholar] [CrossRef]
- Tellinghuisen, T.L.; Evans, M.J.; von Hahn, T.; You, S.; Rice, C.M. Studying hepatitis C virus: Making the best of a bad virus. J. Virol. 2007, 81, 8853–8867. [Google Scholar] [CrossRef]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef]
- McLauchlan, J. Lipid droplets and hepatitis C virus infection. Biochim. Biophys. Acta 2009, 1791, 552–559. [Google Scholar] [CrossRef]
- Syed, G.H.; Amako, Y.; Siddiqui, A. Hepatitis C virus hijacks host lipid metabolism. Trends Endocrinol. Metab. 2010, 21, 33–40. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Penin, F.; Lohmann, V.; Andre, P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011, 19, 95–103. [Google Scholar] [CrossRef]
- Siddiqi, S.A. VLDL exits from the endoplasmic reticulum in a specialized vesicle, the VLDL transport vesicle, in rat primary hepatocytes. Biochem. J. 2008, 413, 333–342. [Google Scholar] [CrossRef]
- Gusarova, V.; Seo, J.; Sullivan, M.L.; Watkins, S.C.; Brodsky, J.L.; Fisher, E.A. Golgi-associated maturation of very low density lipoproteins involves conformational changes in apolipoprotein B, but is not dependent on apolipoprotein E. J. Biol. Chem. 2007, 282, 19453–19462. [Google Scholar]
- Kapadia, S.B.; Chisari, F.V. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. USA 2005, 102, 2561–2566. [Google Scholar]
- Gastaminza, P.; Kapadia, S.B.; Chisari, F.V. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J. Virol. 2006, 80, 11074–11081. [Google Scholar] [CrossRef]
- Chang, K.S.; Jiang, J.; Cai, Z.; Luo, G. Human apolipoprotein E is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 2007, 81, 13783–13793. [Google Scholar]
- Li, Q.; Brass, A.L.; Ng, A.; Hu, Z.; Xavier, R.J.; Liang, T.J.; Elledge, S.J. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl. Acad. Sci. USA 2009, 106, 16410–16415. [Google Scholar]
- Tai, A.W.; Benita, Y.; Peng, L.F.; Kim, S.S.; Sakamoto, N.; Xavier, R.J.; Chung, R.T. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 2009, 5, 298–307. [Google Scholar] [CrossRef]
- Vaillancourt, F.H.; Pilote, L.; Cartier, M.; Lippens, J.; Liuzzi, M.; Bethell, R.C.; Cordingley, M.G.; Kukolj, G. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology 2009, 387, 5–10. [Google Scholar] [CrossRef]
- Supekova, L.; Supek, F.; Lee, J.; Chen, S.; Gray, N.; Pezacki, J.P.; Schlapbach, A.; Schultz, P.G. Identification of human kinases involved in hepatitis C virus replication by small interference RNA library screening. J. Biol. Chem. 2008, 283, 29–36. [Google Scholar]
- Berger, K.L.; Cooper, J.D.; Heaton, N.S.; Yoon, R.; Oakland, T.E.; Jordan, T.X.; Mateu, G.; Grakoui, A.; Randall, G. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 7577–7582. [Google Scholar]
- De Matteis, M.A.; Di Campli, A.; Godi, A. The role of the phosphoinositides at the Golgi complex. Biochim. Biophys. Acta 2005, 1744, 396–405. [Google Scholar] [CrossRef]
- Sasaki, T.; Takasuga, S.; Sasaki, J.; Kofuji, S.; Eguchi, S.; Yamazaki, M.; Suzuki, A. Mammalian phosphoinositide kinases and phosphatases. Prog. Lipid Res. 2009, 48, 307–343. [Google Scholar] [CrossRef]
- Di Paolo, G.; De Camilli, P. Phosphoinositides in cell regulation and membrane dynamics. Nature 2006, 443, 651–657. [Google Scholar] [CrossRef]
- De Matteis, M.A.; D'Angelo, G. The role of the phosphoinositides at the Golgi complex. Biochem. Soc. Symp. 2007, 74, 107–116. [Google Scholar] [CrossRef]
- D'Angelo, G.; Vicinanza, M.; Di Campli, A.; De Matteis, M.A. The multiple roles of PtdIns(4)P—Not just the precursor of PtdIns(4,5)P2. J. Cell Sci. 2008, 121, 1955–1963. [Google Scholar] [CrossRef]
- Balla, A.; Balla, T. Phosphatidylinositol 4-kinases: Old enzymes with emerging functions. Trends Cell Biol. 2006, 16, 351–361. [Google Scholar] [CrossRef]
- Reiss, S.; Rebhan, I.; Backes, P.; Romero-Brey, I.; Erfle, H.; Matula, P.; Kaderali, L.; Poenisch, M.; Blankenburg, H.; Hiet, M.S.; et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 2011, 9, 32–45. [Google Scholar] [CrossRef]
- Hsu, N.Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef]
- Bishe, B.; Syed, G.H.; Field, S.J.; Siddiqui, A. Role of phosphatidylinositol 4-phosphate (PI4P) and its binding protein GOLPH3 in hepatitis C virus secretion. J. Biol. Chem. 2012, 287, 27637–27647. [Google Scholar]
- Graham, T.R.; Burd, C.G. Coordination of Golgi functions by phosphatidylinositol 4-kinases. Trends Cell Biol. 2011, 21, 113–121. [Google Scholar] [CrossRef]
- Wenk, M.R.; De Camilli, P. Protein-lipid interactions and phosphoinositide metabolism in membrane traffic: Insights from vesicle recycling in nerve terminals. Proc. Natl. Acad. Sci. USA 2004, 101, 8262–8269. [Google Scholar] [CrossRef]
- Behnia, R.; Munro, S. Organelle identity and the signposts for membrane traffic. Nature 2005, 438, 597–604. [Google Scholar] [CrossRef]
- Ngo, M.H.; Colbourne, T.R.; Ridgway, N.D. Functional implications of sterol transport by the oxysterol-binding protein gene family. Biochem. J. 2010, 429, 13–24. [Google Scholar] [CrossRef]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef]
- Mayinger, P. Signaling at the Golgi. CSH Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Roth, M.G. Phosphoinositides in constitutive membrane traffic. Physiol. Rev. 2004, 84, 699–730. [Google Scholar] [CrossRef]
- Lemmon, M.A. Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 2008, 9, 99–111. [Google Scholar] [CrossRef]
- Weixel, K.M.; Blumental-Perry, A.; Watkins, S.C.; Aridor, M.; Weisz, O.A. Distinct Golgi populations of phosphatidylinositol 4-phosphate regulated by phosphatidylinositol 4-kinases. J. Biol. Chem. 2005, 280, 10501–10508. [Google Scholar]
- Farhan, H.; Weiss, M.; Tani, K.; Kaufman, R.J.; Hauri, H.P. Adaptation of endoplasmic reticulum exit sites to acute and chronic increases in cargo load. EMBO J. 2008, 27, 2043–2054. [Google Scholar] [CrossRef]
- Blumental-Perry, A.; Haney, C.J.; Weixel, K.M.; Watkins, S.C.; Weisz, O.A.; Aridor, M. Phosphatidylinositol 4-phosphate formation at ER exit sites regulates ER export. Dev. Cell 2006, 11, 671–682. [Google Scholar] [CrossRef]
- Balla, A.; Tuymetova, G.; Tsiomenko, A.; Varnai, P.; Balla, T. A plasma membrane pool of phosphatidylinositol 4-phosphate is generated by phosphatidylinositol 4-kinase type-III alpha: Studies with the PH domains of the oxysterol binding protein and FAPP1. Mol. Biol. Cell 2005, 16, 1282–1295. [Google Scholar] [CrossRef]
- Hammond, G.R.; Fischer, M.J.; Anderson, K.E.; Holdich, J.; Koteci, A.; Balla, T.; Irvine, R.F. PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science 2012, 337, 727–730. [Google Scholar]
- Rohde, H.M.; Cheong, F.Y.; Konrad, G.; Paiha, K.; Mayinger, P.; Boehmelt, G. The human phosphatidylinositol phosphatase SAC1 interacts with the coatomer I complex. J. Biol. Chem. 2003, 278, 52689–52699. [Google Scholar]
- Dippold, H.C.; Ng, M.M.; Farber-Katz, S.E.; Lee, S.K.; Kerr, M.L.; Peterman, M.C.; Sim, R.; Wiharto, P.A.; Galbraith, K.A.; Madhavarapu, S.; et al. GOLPH3 bridges phosphatidylinositol-4- phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell 2009, 139, 337–351. [Google Scholar] [CrossRef]
- Peretti, D.; Dahan, N.; Shimoni, E.; Hirschberg, K.; Lev, S. Coordinated lipid transfer between the endoplasmic reticulum and the Golgi complex requires the VAP proteins and is essential for Golgi-mediated transport. Mol. Biol. Cell 2008, 19, 3871–3884. [Google Scholar] [CrossRef]
- Godi, A.; di Campli, A.; Konstantakopoulos, A.; di Tullio, G.; Alessi, D.R.; Kular, G.S.; Daniele, T.; Marra, P.; Lucocq, J.M.; de Matteis, M.A. FAPPS control Golgi-to-cell-surface membrane traffic by binding to ARF and PtdIns(4)P. Nat. Cell Biol. 2004, 6, 393–404. [Google Scholar] [CrossRef]
- Hanada, K.; Kumagai, K.; Yasuda, S.; Miura, Y.; Kawano, M.; Fukasawa, M.; Nishijima, M. Molecular machinery for non-vesicular trafficking of ceramide. Nature 2003, 426, 803–809. [Google Scholar] [CrossRef]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef]
- Salonen, A.; Ahola, T.; Kaariainen, L. Viral RNA replication in association with cellular membranes. Curr. Top. Microbiol. Immunol. 2005, 285, 139–173. [Google Scholar]
- Deng, Y.; Almsherqi, Z.A.; Ng, M.M.; Kohlwein, S.D. Do viruses subvert cholesterol homeostasis to induce host cubic membranes? Trends Cell Biol. 2010, 20, 371–379. [Google Scholar] [CrossRef]
- den Boon, J.A.; Ahlquist, P. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Ann. Rev. Microbiol. 2010, 64, 241–256. [Google Scholar] [CrossRef]
- Sasaki, J.; Ishikawa, K.; Arita, M.; Taniguchi, K. ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J. 2012, 31, 754–766. [Google Scholar]
- Greninger, A.L.; Knudsen, G.M.; Betegon, M.; Burlingame, A.L.; Derisi, J.L. The 3A protein from multiple picornaviruses utilizes the golgi adaptor protein ACBD3 to recruit PI4KIIIbeta. J. Virol. 2012, 86, 3605–3616. [Google Scholar] [CrossRef]
- Arita, M.; Kojima, H.; Nagano, T.; Okabe, T.; Wakita, T.; Shimizu, H. Phosphatidylinositol 4-kinase III beta is a target of enviroxime-like compounds for antipoliovirus activity. J. Virol. 2011, 85, 2364–2372. [Google Scholar] [CrossRef]
- Trotard, M.; Lepere-Douard, C.; Regeard, M.; Piquet-Pellorce, C.; Lavillette, D.; Cosset, F.L.; Gripon, P.; le Seyec, J. Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. FASEB J. 2009, 23, 3780–3789. [Google Scholar] [CrossRef]
- Borawski, J.; Troke, P.; Puyang, X.; Gibaja, V.; Zhao, S.; Mickanin, C.; Leighton-Davies, J.; Wilson, C.J.; Myer, V.; Cornellataracido, I.; et al. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J. Virol. 2009, 83, 10058–10074. [Google Scholar]
- Tai, A.W.; Salloum, S. The role of the phosphatidylinositol 4-kinase PI4KA in hepatitis C virus-induced host membrane rearrangement. PloS One 2011, 6, e26300. [Google Scholar]
- Berger, K.L.; Kelly, S.M.; Jordan, T.X.; Tartell, M.A.; Randall, G. Hepatitis C virus stimulates the phosphatidylinositol 4-kinase III alpha-dependent phosphatidylinositol 4-phosphate production that is essential for its replication. J. Virol. 2011, 85, 8870–8883. [Google Scholar]
- Lim, Y.S.; Hwang, S.B. Hepatitis C virus NS5A protein interacts with phosphatidylinositol 4-kinase type IIIalpha and regulates viral propagation. J. Biol. Chem. 2011, 286, 11290–11298. [Google Scholar]
- Bianco, A.; Reghellin, V.; Donnici, L.; Fenu, S.; Alvarez, R.; Baruffa, C.; Peri, F.; Pagani, M.; Abrignani, S.; Neddermann, P.; et al. Metabolism of phosphatidylinositol 4-kinase IIIalpha-dependent PI4P is subverted by HCV and is targeted by a 4-anilino quinazoline with antiviral activity. PLoS Pathog. 2012, 8, e1002576. [Google Scholar] [CrossRef]
- de Graaf, P.; Zwart, W.T.; van Dijken, R.A.; Deneka, M.; Schulz, T.K.; Geijsen, N.; Coffer, P.J.; Gadella, B.M.; Verkleij, A.J.; van der Sluijs, P.; et al. Phosphatidylinositol 4-kinasebeta is critical for functional association of rab11 with the Golgi complex. Mol. Biol. Cell 2004, 15, 2038–2047. [Google Scholar] [CrossRef]
- Zhang, L.; Hong, Z.; Lin, W.; Shao, R.X.; Goto, K.; Hsu, V.W.; Chung, R.T. ARF1 and GBF1 generate a PI4P-enriched environment supportive of hepatitis C virus replication. PloS One 2012, 7, e32135. [Google Scholar]
- Haynes, L.P.; Thomas, G.M.; Burgoyne, R.D. Interaction of neuronal calcium sensor-1 and ADP-ribosylation factor 1 allows bidirectional control of phosphatidylinositol 4-kinase beta and trans-Golgi network-plasma membrane traffic. J. Biol. Chem. 2005, 280, 6047–6054. [Google Scholar]
- Coller, K.E.; Heaton, N.S.; Berger, K.L.; Cooper, J.D.; Saunders, J.L.; Randall, G. Molecular determinants and dynamics of hepatitis C virus secretion. PLoS Pathog. 2012, 8, e1002466. [Google Scholar] [CrossRef]
- Gosert, R.; Egger, D.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Bienz, K.; Moradpour, D. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 2003, 77, 5487–5492. [Google Scholar]
- Egger, D.; Wolk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar]
- Ferraris, P.; Blanchard, E.; Roingeard, P. Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J. Gen. Virol. 2010, 91, 2230–2237. [Google Scholar] [CrossRef] [Green Version]
- Berger, K.L.; Randall, G. Potential roles for cellular cofactors in hepatitis C virus replication complex formation. Commun. Integr. Biol. 2009, 2, 471–473. [Google Scholar] [CrossRef]
- Amemiya, F.; Maekawa, S.; Itakura, Y.; Kanayama, A.; Matsui, A.; Takano, S.; Yamaguchi, T.; Itakura, J.; Kitamura, T.; Inoue, T.; et al. Targeting lipid metabolism in the treatment of hepatitis C virus infection. J. Infect. Dis. 2008, 197, 361–370. [Google Scholar] [CrossRef]
- Sakamoto, H.; Okamoto, K.; Aoki, M.; Kato, H.; Katsume, A.; Ohta, A.; Tsukuda, T.; Shimma, N.; Aoki, Y.; Arisawa, M.; et al. Host sphingolipid biosynthesis as a target for hepatitis C virus therapy. Nature Chem. Biol. 2005, 1, 333–337. [Google Scholar] [CrossRef]
- Amako, Y.; Sarkeshik, A.; Hotta, H.; Yates, J., 3rd.; Siddiqui, A. Role of oxysterol binding protein in hepatitis C virus infection. J. Virol. 2009, 83, 9237–9246. [Google Scholar]
- Aizaki, H.; Morikawa, K.; Fukasawa, M.; Hara, H.; Inoue, Y.; Tani, H.; Saito, K.; Nishijima, M.; Hanada, K.; Matsuura, Y.; et al. Critical role of virion-associated cholesterol and sphingolipid in hepatitis C virus infection. J. Virol. 2008, 82, 5715–5724. [Google Scholar] [CrossRef]
- Sagan, S.M.; Rouleau, Y.; Leggiadro, C.; Supekova, L.; Schultz, P.G.; Su, A.I.; Pezacki, J.P. The influence of cholesterol and lipid metabolism on host cell structure and hepatitis C virus replication. Biochem. Cell Biol. 2006, 84, 67–79. [Google Scholar] [CrossRef]
- McLauchlan, J. Hepatitis C virus: Viral proteins on the move. Biochem. Soc. Trans. 2009, 37, 986–990. [Google Scholar] [CrossRef]
- Merz, A.; Long, G.; Hiet, M.S.; Brugger, B.; Chlanda, P.; Andre, P.; Wieland, F.; Krijnse-Locker, J.; Bartenschlager, R. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 2011, 286, 3018–3032. [Google Scholar]
- He, J.; Scott, J.L.; Heroux, A.; Roy, S.; Lenoir, M.; Overduin, M.; Stahelin, R.V.; Kutateladze, T.G. Molecular basis of phosphatidylinositol 4-phosphate and ARF1 GTPase recognition by the FAPP1 pleckstrin homology (PH) domain. J. Biol. Chem. 2011, 286, 18650–18657. [Google Scholar]
- Zoncu, R.; Perera, R.M.; Sebastian, R.; Nakatsu, F.; Chen, H.; Balla, T.; Ayala, G.; Toomre, D.; De Camilli, P.V. Loss of endocytic clathrin-coated pits upon acute depletion of phosphatidylinositol 4,5-bisphosphate. Proc. Natl. Acad. Sci. USA 2007, 104, 3793–3798. [Google Scholar]
- Shimizu, Y.; Hishiki, T.; Ujino, S.; Sugiyama, K.; Funami, K.; Shimotohno, K. Lipoprotein component associated with hepatitis C virus is essential for virus infectivity. Curr. Opin. Virol. 2011, 1, 19–26. [Google Scholar] [CrossRef]
- He, Y.; Nakao, H.; Tan, S.L.; Polyak, S.J.; Neddermann, P.; Vijaysri, S.; Jacobs, B.L.; Katze, M.G. Subversion of cell signaling pathways by hepatitis C virus nonstructural 5A protein via interaction with GRB2 and P85 phosphatidylinositol 3-kinase. J. Virol. 2002, 76, 9207–9217. [Google Scholar]
- Street, A.; Macdonald, A.; Crowder, K.; Harris, M. The hepatitis C virus NS5A protein activates a phosphoinositide 3-kinase-dependent survival signaling cascade. J. Biol. Chem. 2004, 279, 12232–12241. [Google Scholar]
- Mannova, P.; Fang, R.; Wang, H.; Deng, B.; McIntosh, M.W.; Hanash, S.M.; Beretta, L. Modification of host lipid raft proteome upon hepatitis C virus replication. Mol. Cell. Proteomics 2006, 5, 2319–2325. [Google Scholar] [CrossRef]
- Ishida, H.; Li, K.; Yi, M.; Lemon, S.M. p21-activated kinase 1 is activated through the mammalian target of rapamycin/p70 S6 kinase pathway and regulates the replication of hepatitis C virus in human hepatoma cells. J. Biol. Chem. 2007, 282, 11836–11848. [Google Scholar]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Waris, G.; Siddiqui, A. Hepatitis C virus stimulates the expression of cyclooxygenase-2 via oxidative stress: Role of prostaglandin E2 in RNA replication. J. Virol. 2005, 79, 9725–9734. [Google Scholar] [CrossRef]
- Street, A.; Macdonald, A.; McCormick, C.; Harris, M. Hepatitis C virus NS5A-mediated activation of phosphoinositide 3-kinase results in stabilization of cellular beta-catenin and stimulation of beta-catenin-responsive transcription. J. Virol. 2005, 79, 5006–5016. [Google Scholar]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar]
- Wymann, M.P.; Marone, R. Phosphoinositide 3-kinase in disease: Timing, location, and scaffolding. Curr. Opin. Cell Biol. 2005, 17, 141–149. [Google Scholar]
- Milward, A.; Mankouri, J.; Harris, M. Hepatitis C virus NS5A protein interacts with beta-catenin and stimulates its transcriptional activity in a phosphoinositide-3 kinase-dependent fashion. J. Gen. Virol. 2010, 91, 373–381. [Google Scholar] [CrossRef]
- Li, S.; Brown, M.S.; Goldstein, J.L. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3441–3446. [Google Scholar] [CrossRef]
- Waris, G.; Felmlee, D.J.; Negro, F.; Siddiqui, A. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J. Virol. 2007, 81, 8122–8130. [Google Scholar] [CrossRef]
- Park, C.Y.; Jun, H.J.; Wakita, T.; Cheong, J.H.; Hwang, S.B. Hepatitis C virus nonstructural 4B protein modulates sterol regulatory element-binding protein signaling via the AKT pathway. J. Biol. Chem. 2009, 284, 9237–9246. [Google Scholar]
- Shetty, S.; Eckhardt, E.R.; Post, S.R.; van der Westhuyzen, D.R. Phosphatidylinositol-3-kinase regulates scavenger receptor class B type I subcellular localization and selective lipid uptake in hepatocytes. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2125–2131. [Google Scholar] [CrossRef]
- Bartosch, B.; Vitelli, A.; Granier, C.; Goujon, C.; Dubuisson, J.; Pascale, S.; Scarselli, E.; Cortese, R.; Nicosia, A.; Cosset, F.L. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 2003, 278, 41624–41630. [Google Scholar]
- Grove, J.; Huby, T.; Stamataki, Z.; Vanwolleghem, T.; Meuleman, P.; Farquhar, M.; Schwarz, A.; Moreau, M.; Owen, J.S.; Leroux-Roels, G. et al. Scavenger receptor BI and BII expression levels modulate hepatitis C virus infectivity. J. Virol. 2007, 81, 3162–3169. [Google Scholar]
- Carnero, A.; Blanco-Aparicio, C.; Renner, O.; Link, W.; Leal, J.F. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr. Cancer Drug Targets 2008, 8, 187–198. [Google Scholar]
- Clement, S.; Peyrou, M.; Sanchez-Pareja, A.; Bourgoin, L.; Ramadori, P.; Suter, D.; Vinciguerra, M.; Guilloux, K.; Pascarella, S.; Rubbia-Brandt, L.; et al. Down-regulation of phosphatase and tensin homolog by hepatitis C virus core 3a in hepatocytes triggers the formation of large lipid droplets. Hepatology 2011, 54, 38–49. [Google Scholar]
- Rahman, M.A.; Kohno, H.; Nagasue, N. Cox-2—A target for preventing hepatic carcinoma? Expert Opin. Ther. Targets 2002, 6, 483–490. [Google Scholar]
- Gao, M.; Nettles, R.E.; Belema, M.; Snyder, L.B.; Nguyen, V.N.; Fridell, R.A.; Serrano-Wu, M.H.; Langley, D.R.; Sun, J.H.; O'Boyle, D.R., 2nd.; et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 2010, 465, 96–100. [Google Scholar] [CrossRef]
- Delang, L.; Vliegen, I.; Froeyen, M.; Neyts, J. Comparative study of the genetic barriers and pathways towards resistance of selective inhibitors of hepatitis C virus replication. Antimicrob. Agents Chemother. 2011, 55, 4103–4113. [Google Scholar] [CrossRef]
- Balla, A.; Kim, Y.J.; Varnai, P.; Szentpetery, Z.; Knight, Z.; Shokat, K.M.; Balla, T. Maintenance of hormone-sensitive phosphoinositide pools in the plasma membrane requires phosphatidylinositol 4-kinase IIIalpha. Mol. Biol. Cell 2008, 19, 711–721. [Google Scholar]
- Knight, Z.A.; Shokat, K.M. Features of selective kinase inhibitors. Chem. Biol. 2005, 12, 621–637. [Google Scholar] [CrossRef]
- Heinz, B.A.; Vance, L.M. The antiviral compound enviroxime targets the 3A coding region of rhinovirus and poliovirus. J. Virol. 1995, 69, 4189–4197. [Google Scholar]
- Lamarche, M.J.; Borawski, J.; Bose, A.; Capacci-Daniel, C.; Colvin, R.; Dennehy, M.; Ding, J.; Dobler, M.; Drumm, J.; Gaither, L.A.; et al. Anti-HCV activity and toxicity of PI4KIIIbeta inhibitors. Antimicrob. Agents Chemother. 2012, 56, 5149–5156. [Google Scholar]
- Vaillancourt, F.H.; Brault, M.; Pilote, L.; Uyttersprot, N.; Gaillard, E.T.; Stoltz, J.H.; Knight, B.L.; Pantages, L.; McFarland, M.; Breitfelder, S.; et al. Evaluation of phosphatidylinositol-4-kinase IIIalpha as a hepatitis C virus drug target. J. Virol. 2012, 86, 11595–11607. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bishé, B.; Syed, G.; Siddiqui, A. Phosphoinositides in the Hepatitis C Virus Life Cycle. Viruses 2012, 4, 2340-2358. https://doi.org/10.3390/v4102340
Bishé B, Syed G, Siddiqui A. Phosphoinositides in the Hepatitis C Virus Life Cycle. Viruses. 2012; 4(10):2340-2358. https://doi.org/10.3390/v4102340
Chicago/Turabian StyleBishé, Bryan, Gulam Syed, and Aleem Siddiqui. 2012. "Phosphoinositides in the Hepatitis C Virus Life Cycle" Viruses 4, no. 10: 2340-2358. https://doi.org/10.3390/v4102340