Molecular Mechanisms Underlying Hepatocellular Carcinoma

{kind=link}
{kind=link}
{kind=link}
Abstract
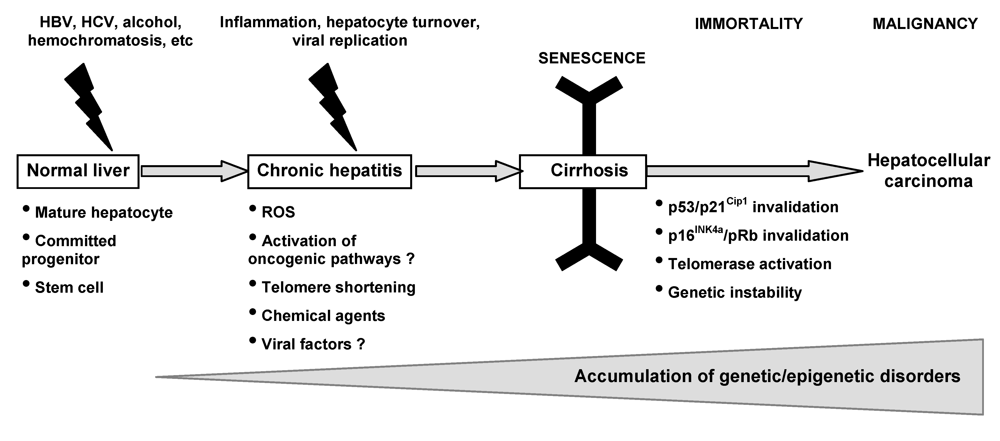
:1. Introduction
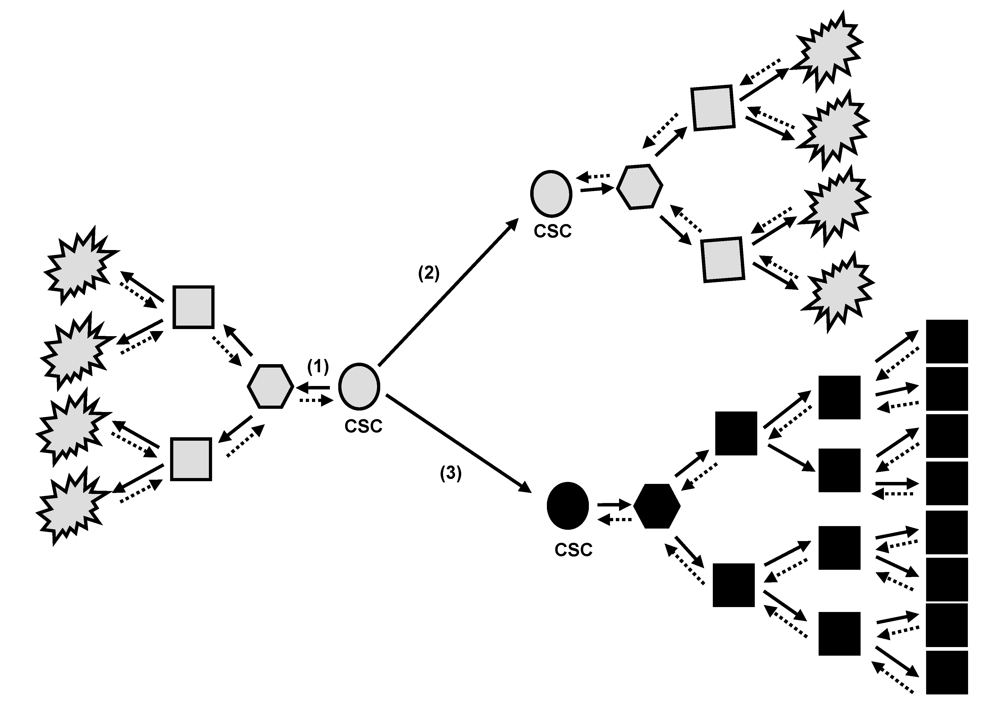
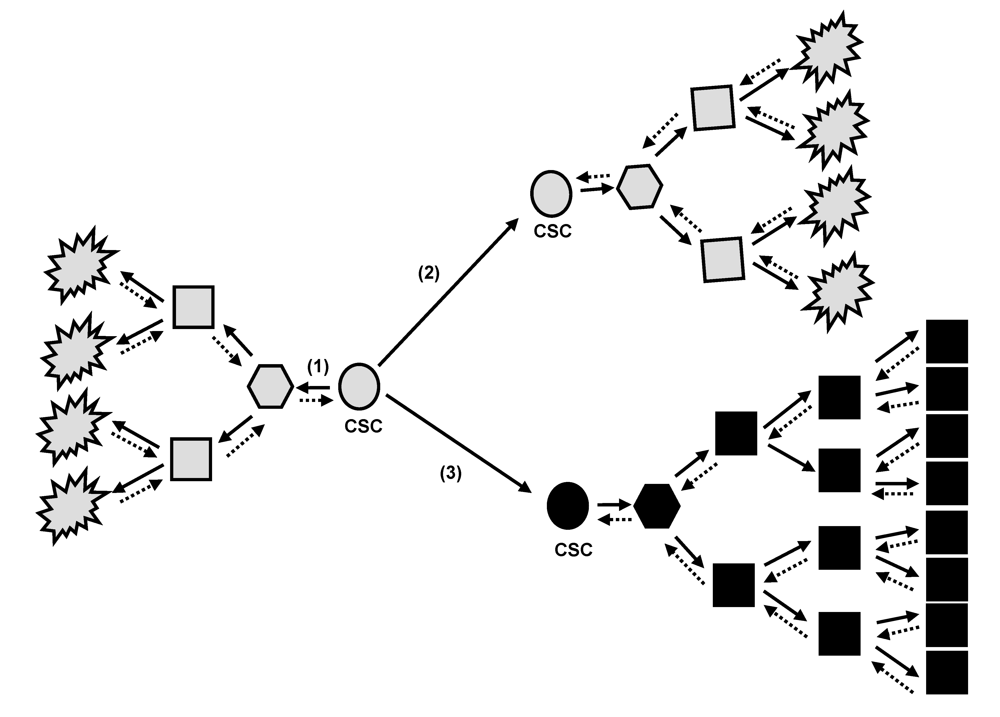
2. Tumor Bulk and Cancer Stem Cell Concept
3. Oncogenic Stress and Cellular Behaviour
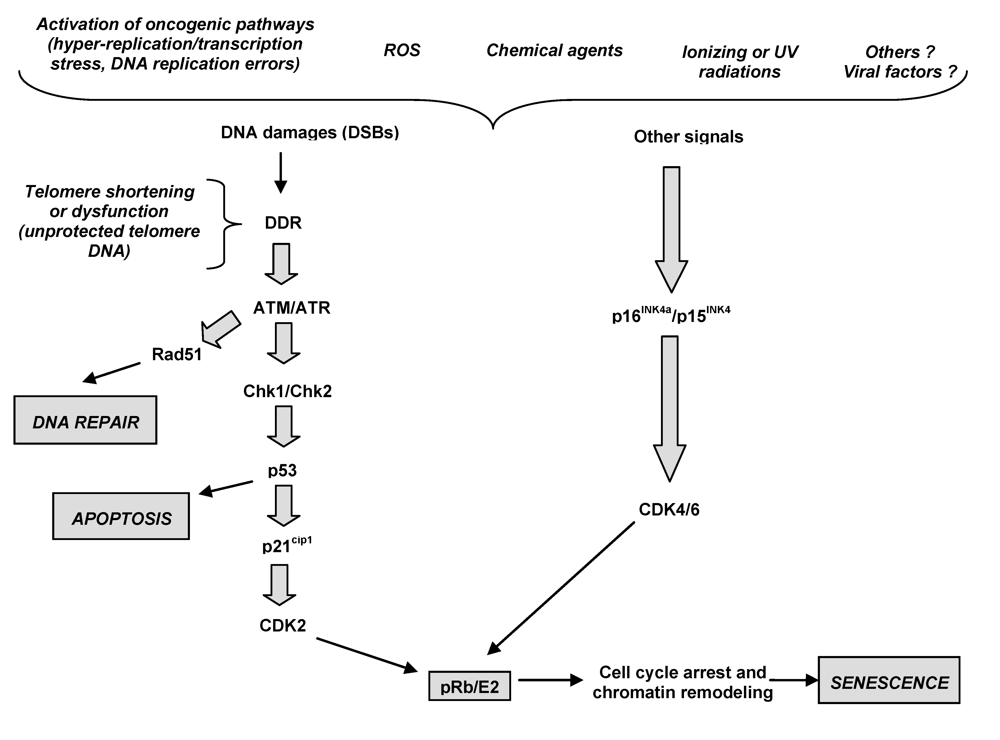
3.1. Senescence pathways as tumor suppressor mechanisms in chronic liver disease
3.2. Senescence-related aberrations in chronic liver disease and hepatocarcinogenesis
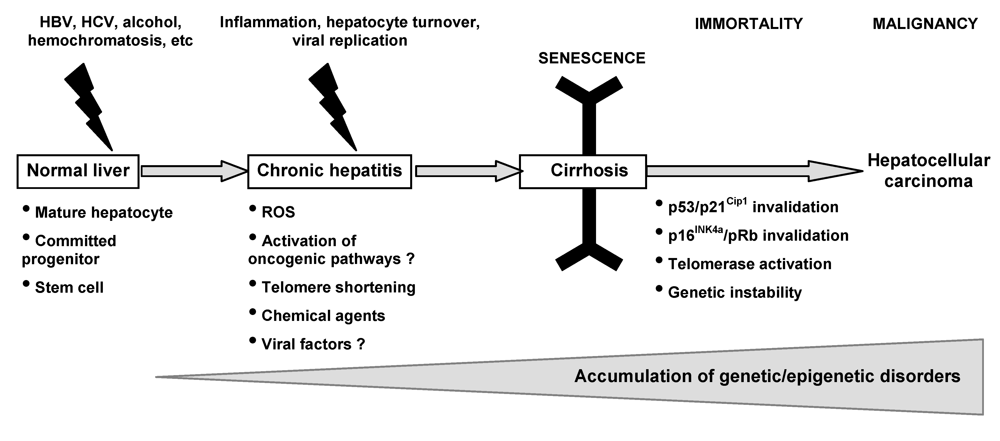
4. Activation of Oncogenic Pathways
4.1. Ras
4.2. c-Myc
4.3. Wnt/β-catenin
5. Liver Inflammation and Hepatocarcinogenesis
6. Antiproliferative and Apoptosis Deficiency and Liver Cell Transformation
7. Conclusion
References
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Estimating the world cancer burden: Globocan 2000. Int. J. Cancer 2001, 94, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Burroughs, A.; Bruix, J. Hepatocellular carcinoma. Lancet 2003, 362, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.; Davila, J.A.; Petersen, N.J.; McGlynn, K.A. The continuing increase in the incidence of hepatocellular carcinoma in the United States: an update. Ann. Intern. Med. 2003, 139, 817–823. [Google Scholar] [PubMed]
- Wang, J.; Chenivesse, X.; Henglein, B.; Brechot, C. Hepatitis B virus integration in a cyclin A gene in a hepatocellular carcinoma. Nature 1990, 343, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Etiemble, J.; Degott, C.; Renard, C.A.; Fourel, G.; Shamoon, B.; Vitvitski-Trépo, L.; Hsu, T.Y.; Tiollais, P.; Babinet, C.; Buendia, M.A. Liver-specific expression and high oncogenic efficiency of a c-myc transgene activated by woodchuck hepatitis virus insertion. Oncogene 1994, 9, 727–737. [Google Scholar] [PubMed]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef]
- Heppner, G.H. Tumor heterogeneity. Cancer Res. 1984, 44, 2259–2265. [Google Scholar] [PubMed]
- Thorgeirsson, S.S.; Grisham, J.W. Molecular pathogenesis of human hepatocellular carcinoma. Nat. Genet. 2002, 31, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Heo, J.; Libbrecht, L.; Chu, I.S.; Kaposi-Novak, P.; Calvisi, D.F.; Mikaelyan, A.; Roberts, L.R.; Demetris, A.J.; Sun, Z.; Nevens, F.; Roskams, T.; Thorgeirsson, S.S. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat. Med. 2006, 12, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Thorgeirsson, S.S. Comparative and integrative functional genomics of HCC. Oncogene 2006, 25, 3801–3809. [Google Scholar] [CrossRef] [PubMed]
- Boyault, S.; Rickman, D.S.; de Reyniès, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Hérault, A.; Saric, J.; Belghiti, J.; Franco, D.; Bioulac-Sage, P.; Laurent-Puig, P.; Zucman-Rossi, J. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007, 45, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Villanueva, A.; Kobayashi, M.; Peix, J.; Chiang, D.Y.; Camargo, A.; Gupta, S.; Moore, J.; Wrobel, M.J.; Lerner, J.; Reich, M.; Chan, J.A.; Glickman, J.N.; Ikeda, K.; Hashimoto, M.; Watanabe, G.; Daidone, M.G.; Roayaie, S.; Schwartz, M.; Thung, S.; Salvesen, H.B.; Gabriel, S.; Mazzaferro, V.; Bruix, J.; Friedman, S.L.; Kumada, H.; Llovet, J.M.; Golub, T.R. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells – perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; Sprick, M.R.; Kemper, K.; Stassi, G.; Medema, J.P. Cancer stem cells - old concepts, new insights. Cell. Death Diff. 2008, 15, 947–958. [Google Scholar] [CrossRef]
- Sell, S.; Pierce, G.B. Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers. Lab. Invest. 1994, 70, 6–22. [Google Scholar] [PubMed]
- Sell, S. Heterogeneity and plasticity of hepatocyte lineage cells. Hepatology 2001, 33, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Sell, S. Cellular origin of hepatocellular carcinoma. Cell. Develop. Biology 2002, 13, 419–424. [Google Scholar] [CrossRef]
- Sell, S. Stem cell origin of cancer and differentiation therapy. Crit. Rev. Oncology Hematology 2004, 52, 1–28. [Google Scholar] [CrossRef]
- Sell, S.; Leffert, H.L. Liver cancer stem cells. J. Clin. Oncol. 2008, 26, 2800–2805. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Ji, J.; Budhu, A.; Forgues, M.; Yang, W.; Wang, H.Y.; Jia, H.; Ye, Q.; Qin, L.X.; Wauthier, E.; Reid, L.M.; Minato, H.; Honda, M.; Kaneko, S.; Tang, Z.Y.; Wang, X.W. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology 2009, 136, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Mallette, F.A.; Ferbeyre, G. The DNA damage signaling pathway connects oncogenic stress to cellular senescence. Cell Cycle 2007, 6, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K. Liver regeneration. J. Cell. Physiol. 2007, 213, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Utoh, R.; Tateno, C.; Yamasaki, C.; Hiraga, N.; Kataoka, M.; Shimada, T.; Chayama, K.; Yoshizato, K. Susceptibility of chimeric mice with livers repopulated by serially subcultured human hepatocytes to hepatitis B virus. Hepatology 2008, 47, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Delhaye, M.; Louis, H.; Degraef, C.; Le Moine, O.; Devière, J.; Gulbis, B.; Jacobovitz, D.; Adler, M.; Galand, P. Relationship between hepatocyte proliferative activity and liver functional reserve in human cirrhosis. Hepatology 1996, 23, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Wege, H.; Le, H.T.; Chui, MS.; Lui, L.; Wu, J.; Giri, G.; Malhi, H.; Sappal, B.S.; Kumaran, V.; Gupta, S.; Zern, M.A. Telomerase reconstitution immortalized human fetal hepatocytes without disrupting their differentiation potential. Gastroenterology 2003, 124, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Paradis, V.; Youssef, N.; Dargere, D.; Ba, N.; Bonvoust, F.; Deschatrette, J.; Bedossa, P. Replicative senescence in normal liver, chronic hepatitis C, and hepatocellular carcinomas. Hum. Pathol. 2001, 32, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Aikata, H.; Takaishi, H.; Kawakami, Y.; Takahashi, S.; Kitamoto, M.; Nakanishi, T.; Nakamura, Y.; Shimamoto, F.; Kajiyama, G.; Ide, T. Telomere reduction in human liver tissues with age and chronic inflammation. Exp. Cell. Res. 2000, 256, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre, M.; Nuciforo, P.G.; Bensimon, A.; Maestro, R.; Pelicci, P.G.; d’Adda di Fagagna, F. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Sarkisian, C.J.; Keister, B.A.; Stairs, D.B.; Boxer, R.B.; Moody, S.E.; Chodosh, L.A. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat. Cell. Biol. 2007, 9, 493–505. [Google Scholar] [CrossRef]
- Pauklin, S.; Kristjuhan, A.; Maimets, T.; Jaks, V. ARF and ATM/ATR cooperate in p53-mediated apoptosis upon oncogenic stress. Biochem. Biophys. Res. Com. 2005, 334, 386–394. [Google Scholar] [CrossRef]
- Zindy, P.; Andrieux, L.; Bonnier, D.; Musso, O.; Langouët, S.; Campion, J.P.; Turlin, B.; Clément, B.; Théret, N. Upregulation of DNA repair genes in active cirrhosis associated with hepatocellular carcinoma. FEBS Lett. 2005, 579, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, R.; Nagashima, M.; Sakamoto, M.; Yamaguchi, N.; Hirohashi, S.; Yokota, J.; Kasai, H. Increased formation of oxidative DNA damage, 8-hydroxydeoxyguanosine, in human livers with chronic hepatitis. Cancer Res. 1994, 54, 3171–3172. [Google Scholar] [PubMed]
- Ohtani, N.; Mann, D.; Hara, E. Cellular senescence: its role in tumor suppression and aging. Cancer Sci. 2009, 5, 792–797. [Google Scholar] [CrossRef]
- Oishi, N.; Shilagardi, K.; Nakamoto, Y.; Honda, M.; Kaneko, S.; Murakami, S. Hepatitis B virus X protein overcomes oncogenic RAS-induced senescence in human immortalized cells. Cancer Sci. 2007, 98, 1540–1548. [Google Scholar] [CrossRef] [PubMed]
- Higashitsuji, H.; Higashitsuji, H.; Itoh, K.; Sakurai, T.; Nagao, T.; Sumitomo, Y.; Masuda, T.; Dawson, S.; Shimada, Y.; Mayer, R.J.; Fujita, J. The oncoprotein gankyrin binds to MDM2/HDM2, enhancing ubiquitylation and degradation of p53. Cancer Cell 2005, 8, 75–87. [Google Scholar] [CrossRef]
- Plentz, R.R.; Park, Y.N.; Lechel, A.; Kim, H.; Nellessen, F.; Langkopf, B.H.; Wilkens, L.; Destro, A.; Fiamengo, B.; Manns, M.P.; Roncalli, M.; Rudolph, K.L. Telomere shortening and inactivation of cell cycle checkpoints characterize human hepatocarcinogenesis. Hepatology 2007, 45, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Roncalli, M.; Bianchi, P.; Bruni, B.; Laghi, L.; Destro, A.; Di Gioia, S.; Gennari, L.; Tommasini, M.; Malesci, A.; Coggi, G. Methylation framework of cell cycle gene inhibitors in cirrhosis and associated hepatocellular carcinoma. Hepatology 2002, 36, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Kalinichenko, V.V.; Major, M.L.; Wang, X.; Petrovic, V.; Kuechle, J.; Yoder, H.M.; Dennewitz, M.B.; Shin, B.; Datta, A.; Raychaudhuri, P.; Costa, R.H. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 2004, 18, 830–850. [Google Scholar] [CrossRef] [PubMed]
- Azechi, H.; Nishida, N.; Fukuda, Y.; Nishimura, T.; Minata, M.; Katsuma, H.; Kuno, M.; Ito, T.; Komeda, T.; Kita, R.; Takahashi, R.; Nakao, K. Disruption of the p16/cyclin D1/retinoblastoma protein pathway in the majority of human hepatocellular carcinomas. Oncology 2001, 60, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Chen, Y.; Wurmbach, E.; Roayaie, S.; Fiel, M.I.; Schwartz, M.; Thung, S.N.; Khitrov, G.; Zhang, W.; Villanueva, A.; Battiston, C.; Mazzaferro, V.; Bruix, J.; Waxman, S.; Friedman, S.L. A molecular signature to discriminate dysplastic nodules from early hepatocellular carcinoma in HCV cirrhosis. Gastroenterology 2006, 131, 1758–1767. [Google Scholar] [CrossRef] [PubMed]
- Farazi, P.A.; Glickman, J.; Jiang, S.; Yu, A.; Rudolph, K.L.; DePinho, R.A. Differential impact of telomere dysfunction on initiation and progression of hepatocellular carcinoma. Cancer Res. 2003, 63, 5021–5027. [Google Scholar] [PubMed]
- Djojosubroto, M.W.; Chin, A.C.; Go, N.; Schaetzlein, S.; Manns, M.P.; Gryaznov, S.; Harley, C.B.; Rudolph, K.L. Telomerase antagonists GRN163 and GRN163L inhibit tumor growth and increase chemosensitivity of human hepatoma. Hepatology 2005, 42, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Lechel, A.; Holstege, H.; Begus, Y.; Schienke, A.; Kamino, K.; Lehmann, U.; Kubicka, S.; Schirmacher, P.; Jonkers, J.; Rudolph, K.L. Telomerase deletion limits progression of p53-mutant hepatocellular carcinoma with short telomeres in chronic liver disease. Gastroenterology 2007, 132, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Saigo, K.; Takashima, H.; Minami, M.; Okanoue, T.; Brechot, C.; Paterlini-Brechot, P. Large scaled analysis of hepatitis B virus (HBV) DNA integration in HBV related hepatocellular carcinomas. Gut 2005, 54, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Luan, F.; Ju, Y.; Shen, H.; Gao, L.; Wang, X.; Liu, S.; Zhang, L.; Sun, W.; Ma, C. In vitro transfection of the hepatitis B virus PreS2 gene into the human hepatocarcinoma cell line HepG2 induces upregulation of human telomerase reverse transcriptase. Biochem. Biophys. Res. Commun. 2007, 355, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Yu, Q.; Subrahmanyam, R.; Difilippantonio, M.J.; Ried, T.; Sen, J.M. β-Catenin expression results in p53-independent DNA damage and oncogene-induced senescence in prelymphomagenic thymocytes in vivo. Mol. Cell. Biol. 2008, 28, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Ansieau, S.; Bastid, J.; Doreau, A.; Morel, A.P.; Bouchet, B.P.; Thomas, C.; Fauvet, F.; Puisieux, I.; Doglioni, C.; Piccinin, S.; Maestro, R.; Voeltzel, T.; Selmi, A.; Valsesia-Wittmann, S.; Caron de Fromentel, C.; Puisieux, A. Induction of EMT by Twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 2008, 14, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. Twisted epithelial–mesenchymal transition blocks senescence. Nat. Cell. Biol. 2008, 10, 1021–1023. [Google Scholar] [CrossRef] [PubMed]
- Buendia, M.A. Genetics of hepatocellular carcinoma. Semin. Cancer Biol. 2000, 10, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Challen, C.; Guo, K.; Collier, J.D.; Cavanagh, D.; Bassendine, M.F. Infrequent point mutations in codons 12 and 61 of ras oncogenes in human hepatocellular carcinomas. J. Hepatol. 1992, 14, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Jadirgar, J.; Nonomura, A.; Patil, J.; Thor, A.; Paronetto, F. Ras oncoprotein p21 expression in hepatocellular carcinoma. J. Exp. Pathol. 1989, 4, 37–46. [Google Scholar] [PubMed]
- Evan, G.I.; Littlewood, T.D. The role of c-myc in cell growth. Curr. Opin. Genet. Dev. 1993, 3, 44–49. [Google Scholar] [CrossRef]
- Hsu, T.; Moroy, T.; Etiemble, J.; Louise, A.; Trepo, C.; Tiollais, P.; Buendia, M.A. Activation of c-myc by woodchuck hepatitis virus insertion in hepatocellular carcinoma. Cell 1988, 55, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Merle, P.; Chevallier, M.; Levy, R.; Maisonnas, M.; Terradillos, O.; Si Ahmed, S.N.; Trepo, C.; Buendia, M.A.; Vitvitski-Trepo, L. Preliminary results of interferon-alpha therapy on woodchuck hepatitis virus-induced hepatocarcinogenesis: possible benefit in female transgenic mice. J. Hepatol. 2001, 34, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Merle, P.; Barraud, L.; Lefrancois, L.; Chevallier, M.; Guerret, S.; Maisonnas, M.; Bordes, I.; Savre-Train, I.; Trepo, C.; Vitvitski-Trepo, L. Long-term high-dose interferon-alpha therapy delays Hepadnavirus-related hepatocarcinogenesis in X/myc transgenic mice. Oncogene 2003, 22, 2762–2771. [Google Scholar] [CrossRef] [PubMed]
- Voravud, N.; Foster, C.S.; Gilbertson, J.A.; Sikora, K.; Waxman, J. Oncogene expression in cholangiocarcinoma and in normal hepatic development. Hum. Pathol. 1989, 12, 1163–1168. [Google Scholar] [PubMed]
- Ebinuma, H.; Saito, H.; Saito, Y.; Wakabayashi, K.; Nakamura, M.; Kurose, I.; Ishii, H. Antisense oligodeoxynucleotide against c-myc mRNA induces differentiation of human hepatocellular carcinoma cells. Int. J. Oncol. 1999, 5, 991–999. [Google Scholar] [PubMed]
- Cheng, J.; Luo, J.; Zhang, X.; Hu, J.; Hui, H.; Wang, C.; Stern, A. Inhibition of cell proliferation in HCC-9204 hepatoma cells by a c-myc specific ribozyme. Cancer Gene Ther. 2000, 3, 407–412. [Google Scholar] [PubMed]
- Himeno, Y.; Fukuda, Y.; Hatanaka, M.; Imura, H. Expression of oncogenes in human liver disease. Liver 1988, 4, 208–212. [Google Scholar] [CrossRef]
- Shen, L.; Fang, J.; Qiu, D.; Zhang, T.; Yang, J.; Chen, S.; Xiao, S. Correlation between DNA methylation and pathological changes in human hepatocellular carcinoma. Hepatogastroenterology 1998, 23, 1753–1759. [Google Scholar]
- Kawate, S.; Kukusato, T.; Ohwada, S.; Watanuki, A.; Morishita, Y. Amplification of c-myc in hepatocellular carcinoma: correlation with clinicopathologic features, proliferative activity and p53 overexpression. Oncology 1999, 57, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Kaposi-Novak, P.; Libbrecht, L.; Woo, Y.G.; Lee, Y.H.; Sears, N.C.; Conner, E.A.; Factor, V.M.; Roskams, T.; Thorgeirsson, S.S. Central role of c-myc during malignant conversion in human hepatocarcinogenesis. Cancer Res. 2009, 69, 2775–2782. [Google Scholar] [CrossRef] [PubMed]
- Poon, T.C.; Wong, N.; Lai, P.B.; Rattray, M.; Johnson, P.J.; Sung, J.J. A tumor progression model for hepatocellular carcinoma: bioinformatic analysis of genomic data. Gastroenterology 2006, 131, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Su, T.S.; Lin, L.H.; Lui, W.Y.; Chang, C.M.; Chou, C.K.; Ting, L.P.; Hu, C.P.; Han, S.H.; P’eng, F.K. Biochem. Biophys. Res. Commun. 1985, 1, 264–268. [Google Scholar] [CrossRef]
- Yuen, M.F.; Wu, P.C.; Lai, V.C.H.; Lau, J.Y.N.; Lai, C.L. Expression of c-Myc, c-Fos, and c-jun in hepatocellular carcinoma. Cancer 2001, 91, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Wei, N.; Deng, X.W. The COP9 signalosome. Annu. Rev. Cell. Dev. Biol. 2003, 19, 261–286. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Kim, J.W.; Gao, P.; Yustein, J. The interplay between MYC and HIF in cancer. Nat. Rev. 2008, 8, 51–56. [Google Scholar] [CrossRef]
- Sears, R.; Leone, G.; DeGregori, J.; Nevins, J.R. Ras enhances Myc protein stability. Mol. Cell. 1999, 3, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Levens, D. Making myc. Curr. Top. Microbiol. Immunol. 2006, 302, 1–32. [Google Scholar] [PubMed]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Kolligs, F.T.; Bommer, G.; Goke, B. Wnt/β-catenin/Tcf signaling: a critical pathway in gastrointestinal tumorigenesis. Digestion 2002, 66, 131–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micsenyi, A.; Tan, X.; Sneddon, T.; Luo, J.H.; Michalopoulos, G.K.; Monga, S.P. β-catenin is temporally regulated during normal liver development. Gastroenterology 2004, 126, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. WNT signaling pathway and stem cell signaling network. Clin. Cancer Res. 2007, 13, 4042–4045. [Google Scholar] [CrossRef]
- de La Coste, A.; Romagnolo, B.; Billuart, P.; Renard, C.A.; Buendia, M.A.; Soubrane, O.; Fabre, M.; Chelly, J.; Beldjord, C.; Kahn, A.; Perret, C. Somatic mutations of the β-catenin gene are frequent in mouse and human hepatocellular carcinoma. Proc Natl Acad Sci USA 1998, 95, 8847–8851. [Google Scholar] [CrossRef] [PubMed]
- Merle, P.; de la Monte, S.; Kim, M.; Herrmann, M.; Tanaka, S.; Von Dem Bussche, A.; Kew, M.C.; Trepo, C.; Wands, J.R. Functional consequences of Frizzled-7 receptor overexpression in human hepatocellular carcinoma. Gastroenterology 2004, 127, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Bengochea, A.; de Souza, M.M.; Lefrancois, L.; Le Roux, E.; Galy, O.; Chemin, I.; Kim, M.; Wands, J.R.; Trepo, C.; Hainaut, P.; Scoazec, J.Y.; Vitvitski, L.; Merle, P. Common dysregulation of Wnt/Frizzled receptor elements in human hepatocellular carcinoma. Br J Cancer 2008, 99, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.C.; Jeng, Y.M.; Mao, T.L.; Chu, J.S.; Lai, P.L.; Peng, S.Y. β-catenin mutations are associated with a subset of low-stage hepatocellular carcinoma negative for hepatitis B virus and with favorable prognosis. Am J Pathol 2000, 157, 763–770. [Google Scholar] [PubMed]
- Devereux, T.R.; Stern, M.C.; Flake, G.P.; Yu, M.C.; Zhang, Z.Q.; London, S.J.; Taylor, J.A. CTNNB1 mutations and beta-catenin protein accumulation in human hepatocellular carcinomas associated with high exposure to aflatoxin B1. Mol. Carcinog. 2001, 31, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Fan, S.T.; Ng, I.O. β-catenin mutation and overexpression in hepatocellular carcinoma: clinicopathologic and prognostic significance. Cancer 2001, 92, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Inagawa, S.; Itabashi, M.; Adachi, S.; Kawamoto, T.; Hori, M.; Shimazaki, J.; Yoshimi, F.; Fukao, K. Expression and prognostic roles of beta-catenin in hepatocellular carcinoma: correlation with tumor progression and postoperative survival. Chin. Cancer Res. 2002, 8, 450–456. [Google Scholar]
- Laurent-Puig, P.; Legoix, P.; Bluteau, O.; Belghiti, J.; Franco, D.; Binot, F.; Mones, G.; Thomas, G.; Bioulac-Sage, P.; Zucman-Rossi, J. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology 2001, 120, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Satoh, S.; Daigo, Y.; Furukawa, Y.; Kato, T.; Miwa, N.; Nishiwaki, T.; Kawasoe, T.; Ishiguro, H.; Fujita, M.; Tokino, T.; Sasaki, Y.; Imaoka, S.; Murata, M.; Shimano, T.; Yamaoka, Y.; Nakamura, Y. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet. 2000, 24, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Fuji, H.; Sankila, A.; Mahler-Araujo, B.M.; Matsuda, M.; Cathomas, G.; Ohgaki, H. β-catenin mutations are frequent in human hepatocellular carcinomas associated with hepatitis C virus infection. Am. J. Pathol. 1999, 155, 1795–1801. [Google Scholar] [PubMed]
- Merle, P.; Kim, M.; Herrmann, M.; Gupte, A.; Lefrançois, L.; Califano, S.; Trepo, C.; Tanaka, S.; Vitvitski, L.; de la Monte, S.; Wands, J.R. Oncogenic role of the Frizzled-7/β-catenin pathway in hepatocellular carcinoma. J. Hepatol. 2005, 43, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, H.C.; Tsedensodnom, O.; Hartley, R.; Lim, Y.S.; Yu, E.; Merle, P.; Wands, J.R. Functional interaction between Wnt3 and Frizzled-7 leads to activation of the Wnt/β-catenin signaling pathway in hepatocellular carcinoma cells. J. Hepatol. 2008, 48, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Longato, L.; de la Monte, S.; Kuzushita, N.; Horimoto, M.; Rogers, A.B.; Slagle, B.L.; Wands, J.R. Overexpression of insulin receptor substrate-1 and hepatitis Bx genes causes premalignant alterations in the liver. Hepatology 2009, 49, 1935–1943. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Zerlanko, B.; Kennedy, A.; Banumathy, G.; Zhang, R.; Adams, P.D. Downregulation of Wnt signaling is an early signal for formation of facultative heterochromatin and onset of cell senescence in primary human cells. Mol. Cell. 2007, 27, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Invest. 2005, 115, 209–218. [Google Scholar] [PubMed]
- Naugler, W.E.; Karin, M. NF-kappaB and cancer-identifying targets and mechanisms. Curr. Opin. Genet. Dev. 2008, 18, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Arsura, M.; Cavin, L.G. Nuclear factor-kappaB and liver carcinogenesis. Cancer Lett. 2005, 229, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Zhang, H.; Yu, J.; Francisco, R.; Dent, P.; Ebert, MP.; Rocken, C.; Farrell, G. Constitutive activation of NF kappaB in human hepatocellular carcinoma: evidence of a cytoprotective role. Hum. Gene Ther. 2006, 17, 280–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, S.; Wang, F.; Venkatraman, M.; Arsura, M. X-linked inhibitor of apoptosis (XIAP) inhibits c-Jun N-terminal kinase 1 (JNK1) activation by transforming growth factor beta1 (TGF-beta1) through ubiquitin-mediated proteosomal degradation of the TGF-beta1-activated kinase 1 (TAK1). J. Biol. Chem. 2005, 280, 38599–38608. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Karin, M. NF-kappaB signaling, liver disease and hepatoprotective agents. Oncogene 2008, 27, 6228–6244. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-kappa B in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Maeda, S.; Chang, L.; Karin, M. Loss of hepatic NF kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc. Natl. Acad. Sci. USA, 2006, 103, 10544–10551. [Google Scholar] [CrossRef]
- Luedde, T.; Beraza, N.; Kotsikoris, V.; van Loo, G.; Nenci, A.; De Vos, R.; Roskams, T.; Trautwein, C.; Pasparakis, M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell 2007, 11, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Schluesener, H.J. Mammalian toll-like receptors: from endogenous ligands to tissue regeneration. Cell. Mol. Life Sci. 2006, 63, 2901–2907. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Kobayashi, T.; Chinen, T.; Takaki, H.; Sanada, T.; Minoda, Y.; Koga, K.; Takaesu, G.; Maehara, Y.; Iida, M.; Yoshimura, A. Deletion of the SOCS3 gene in liver parenchymal cells promotes hepatitis-induced hepatocarcinogenesis. Gastroenterology 2006, 131, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; Magnino, F.; Schmidt, K.; Piguet, A.C.; Lee, J.S.; Semela, D.; St-Pierre, M.V.; Ziemiecki, A.; Cassio, D.; Brenner, C.; Thorgeirsson, S.S.; Dufour, J.F. Hint2, a mitochondrial apoptotic sensitizer down-regulated in hepatocellular carcinoma. Gastroenterology 2006, 130, 2179–2188. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.A.; Zhang, G.M.; Feigenbaum, L.; Zhang, Y.E. Smad3 reduces susceptibility to hepatocarcinoma by sensitizing hepatocytes to apoptosis through downregulation of Bcl-2. Cancer Cell 2006, 9, 445–457. [Google Scholar] [CrossRef]
- Chen, R.H.; Su, Y.H.; Chuang, R.L.; Chang, T.Y. Suppression of transforming growth factor-beta-induced apoptosis through a phosphatidylinositol 3-kinase/Akt-dependent pathway. Oncogene 1998, 17, 1959–1968. [Google Scholar] [PubMed]
- Dennis, P.A.; Rifkin, D.B. Cellular activation of latent transforming growth factor beta requires binding to the cation-independent mannose 6-phosphate/insulin-like growth factor type II receptor. Proc. Natl. Acad. Sci. USA 1991, 88, 580–584. [Google Scholar] [CrossRef]
- Yamada, T.; de Souza, A.T.; Finkelstein, S.; Jirtle, R.L. Loss of the gene encoding mannose 6-phosphate/insulin-like growth factor II receptor is an early event in liver carcinogenesis. Proc. Natl. Acad. Sci. USA 1997, 94, 10351–10355. [Google Scholar] [CrossRef]
- Acquati, F.; Malgaretti, N.; Hauptschein, R.; Rao, P.; Gaidano, G.; Taramelli, R. A 2-Mb YAC contig linking the plasminogen-apoprotein(a) gene family to the insulin-like growth factor 2 receptor (IGF2R) gene on the telomeric region of chromosome 6 (6q26-q27). Genomics 1994, 22, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.H.; Huang, C.C.; Lin, P.R.; Chang, H.W.; Ger, L.P.; Lin, Y.W.; Changchien, C.S.; Lee, C.M.; Tai, M.H. Expression and prognostic role of tumor suppressor gene PTEN/MMAC1/TEP1 in hepatocellular carcinoma. Cancer 2003, 97, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- He, X.C.; Yin, T.; Grindley, J.C.; Tian, Q.; Sato, T.; Tao, W.A.; Dirisina, R.; Porter-Westpfahl, K.S.; Hembree, M.; Johnson, T.; Wiedemann, L.M.; Barrett, T.A.; Hood, L.; Wu, H.; Li, L. PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat. Genet. 2007, 39, 189–198. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Merle, P.; Trepo, C. Molecular Mechanisms Underlying Hepatocellular Carcinoma. Viruses 2009, 1, 852-872. https://doi.org/10.3390/v1030852
Merle P, Trepo C. Molecular Mechanisms Underlying Hepatocellular Carcinoma. Viruses. 2009; 1(3):852-872. https://doi.org/10.3390/v1030852
Chicago/Turabian StyleMerle, Philippe, and Christian Trepo. 2009. "Molecular Mechanisms Underlying Hepatocellular Carcinoma" Viruses 1, no. 3: 852-872. https://doi.org/10.3390/v1030852