
Protein/Peptide Aggregation and Amyloidosis on Biointerfaces


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
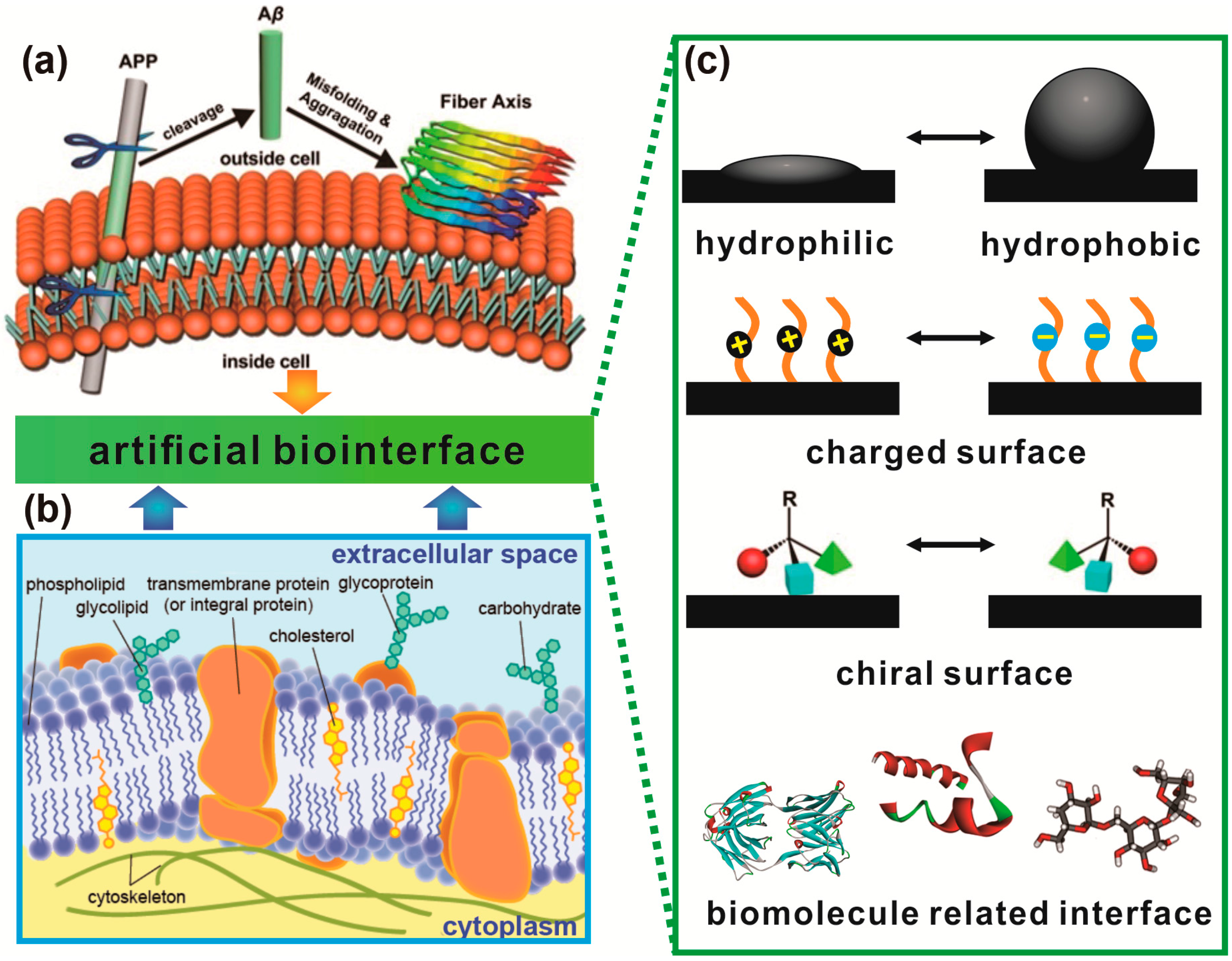
1. Introduction
1.1. Role of Protein/Peptide Aggregation in Daily Life
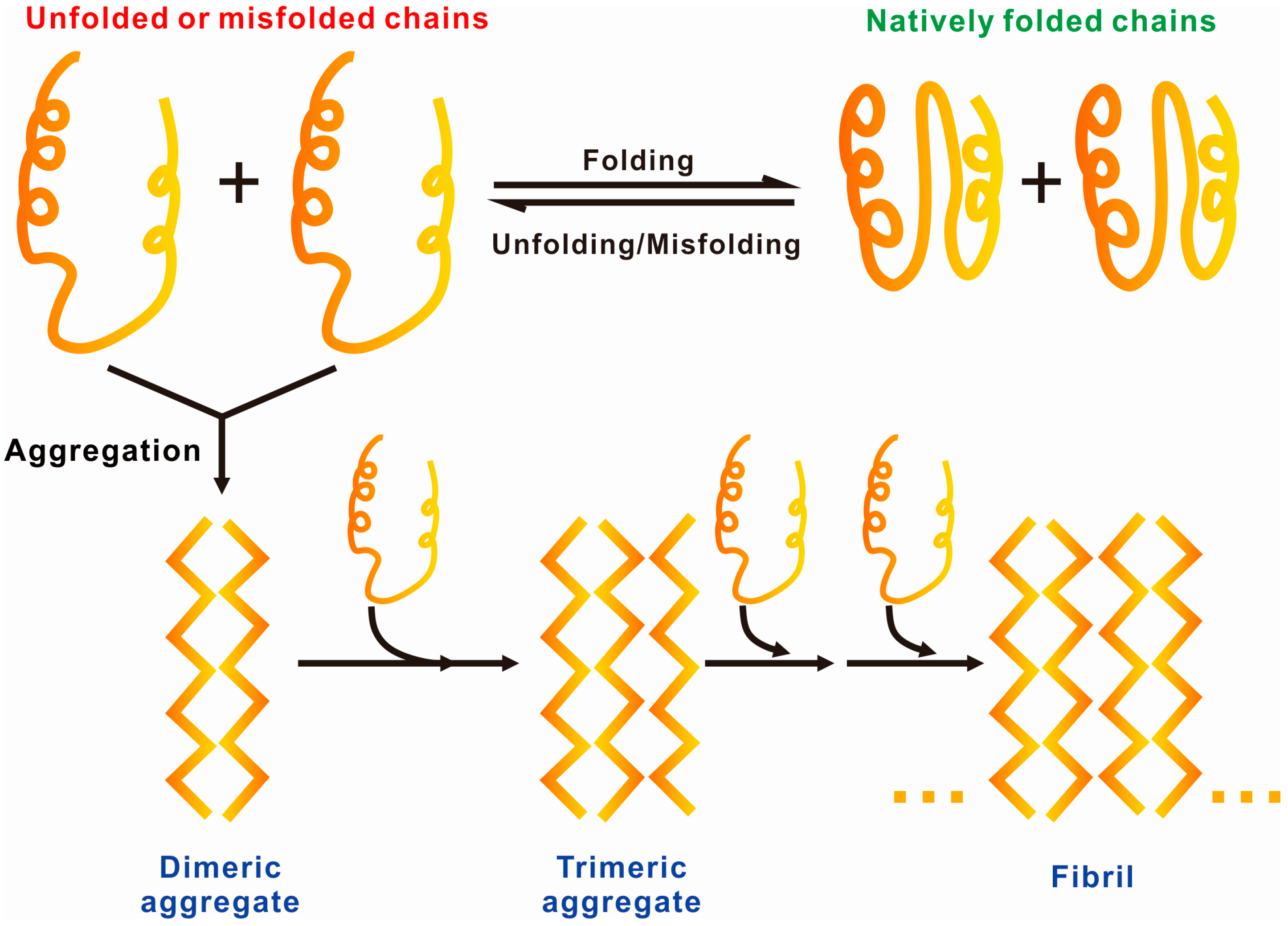
1.2. General Driving Forces of Protein/Peptide Aggregation
1.3. Protein Aggregation Modulated by Biological Membranes
2. Hydrophobic–Hydrophilic Interfaces Triggered Accumulation of Amyloid Protein
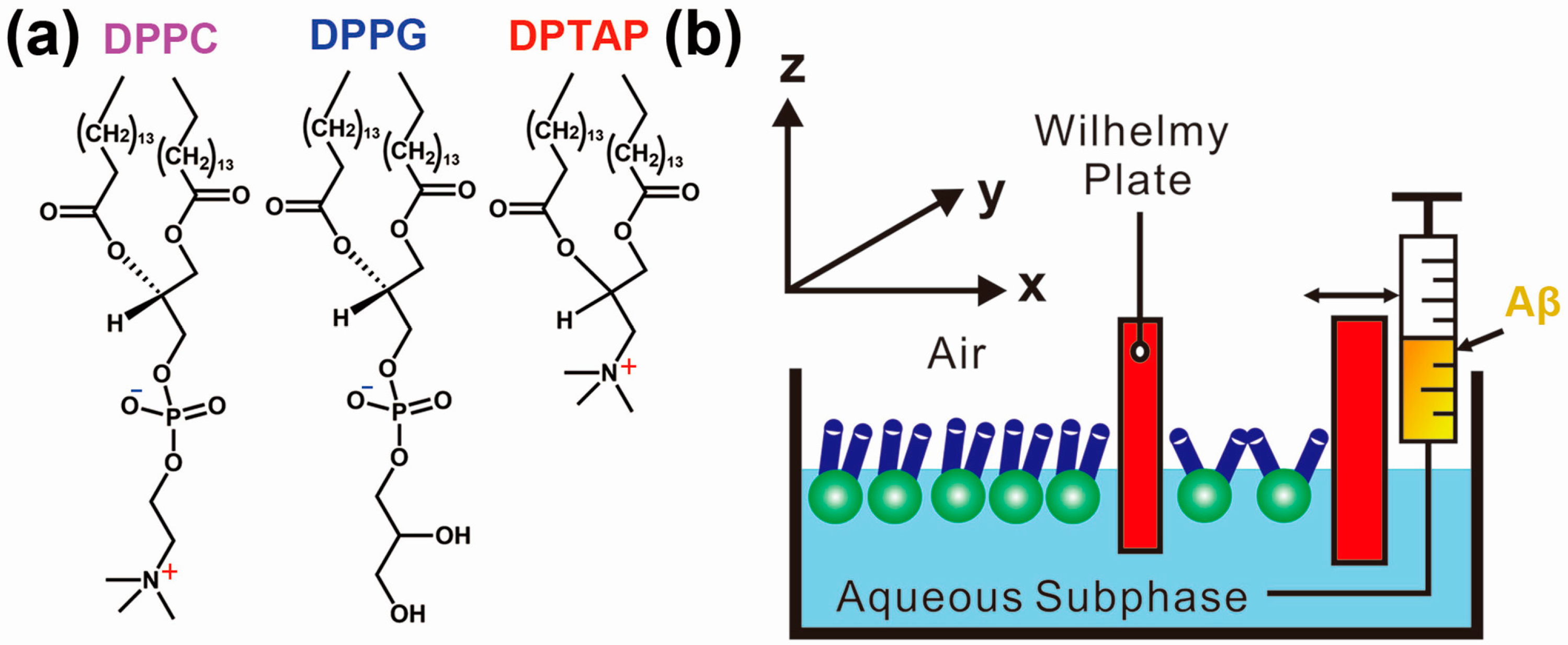
2.1. Effect of the Air–Water Interface on Protein Aggregation
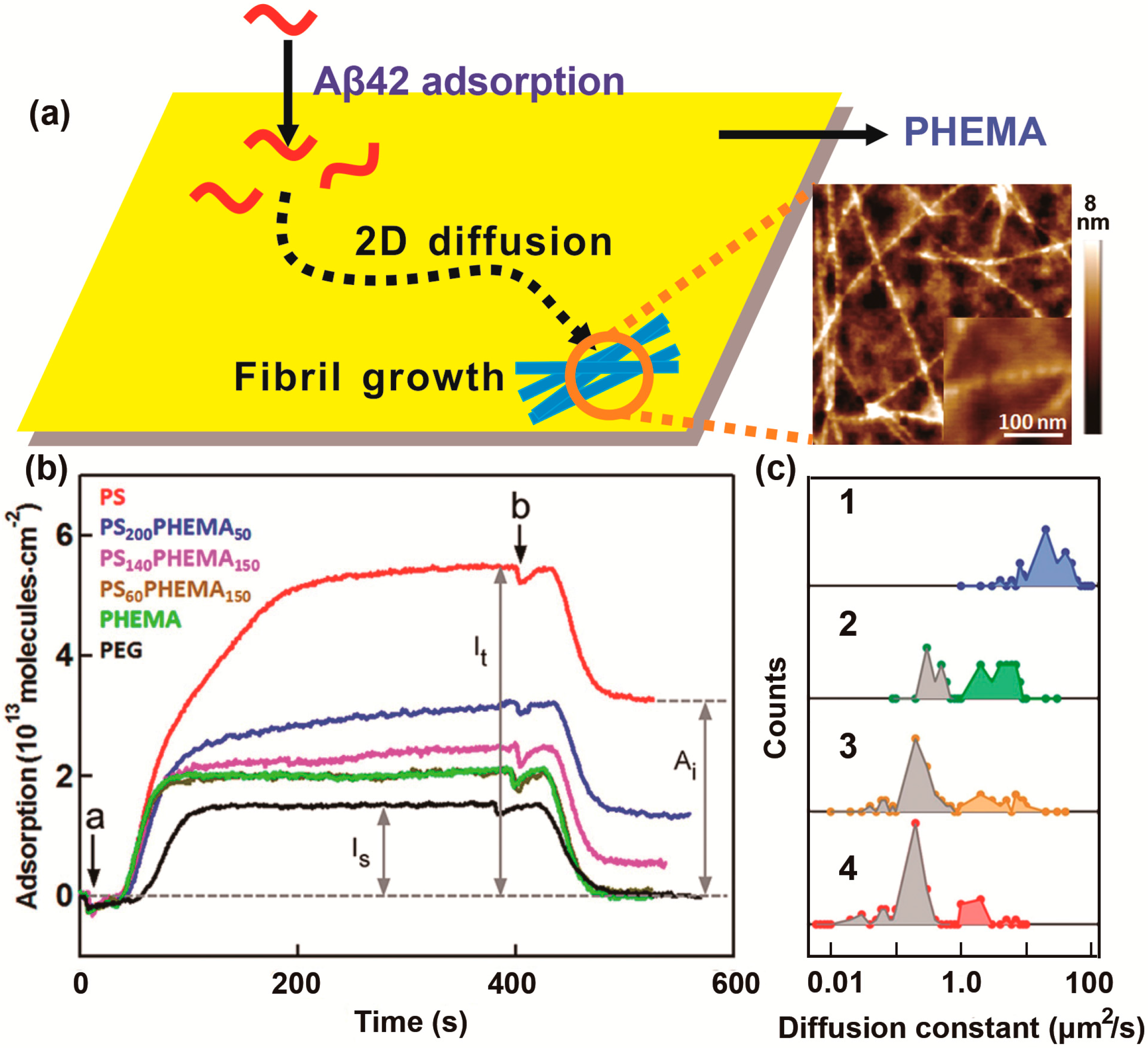
2.2. Effect of the Hydrophobic Surface on Protein Aggregation
2.3. Dominated Effect of Hydrophobic–Hydrophilic Interface on Protein Aggregation in the Presence of Metal Ions
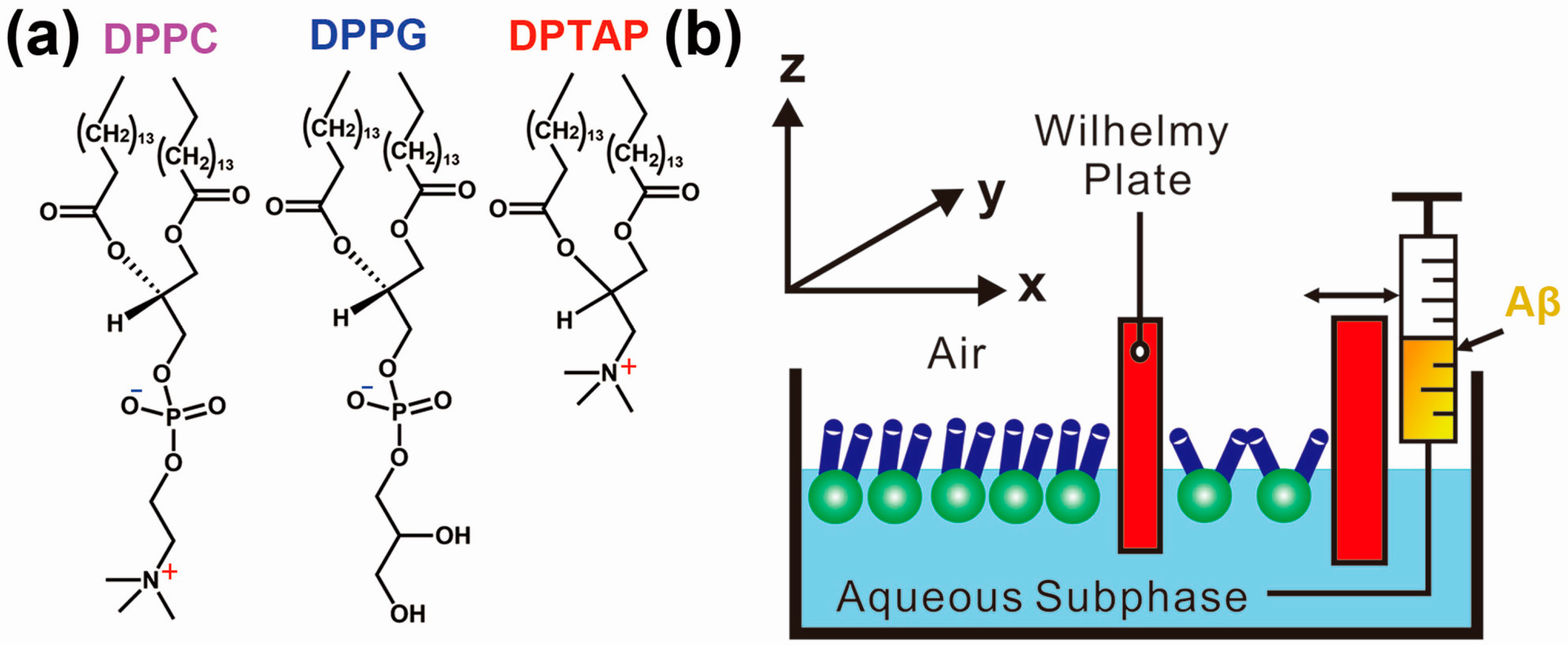
3. Protein Adsorption and Aggregation Influenced by Surface Charge
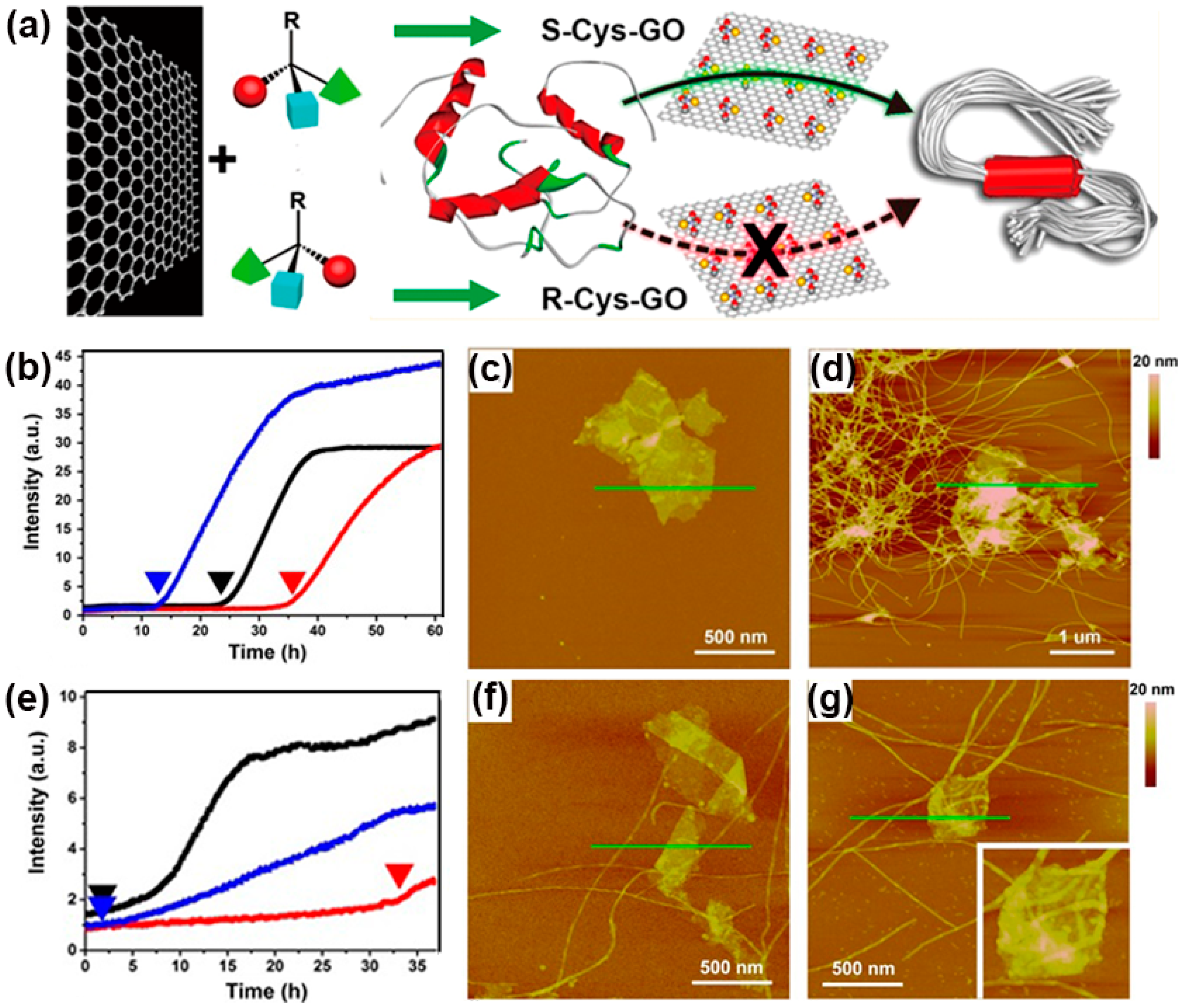
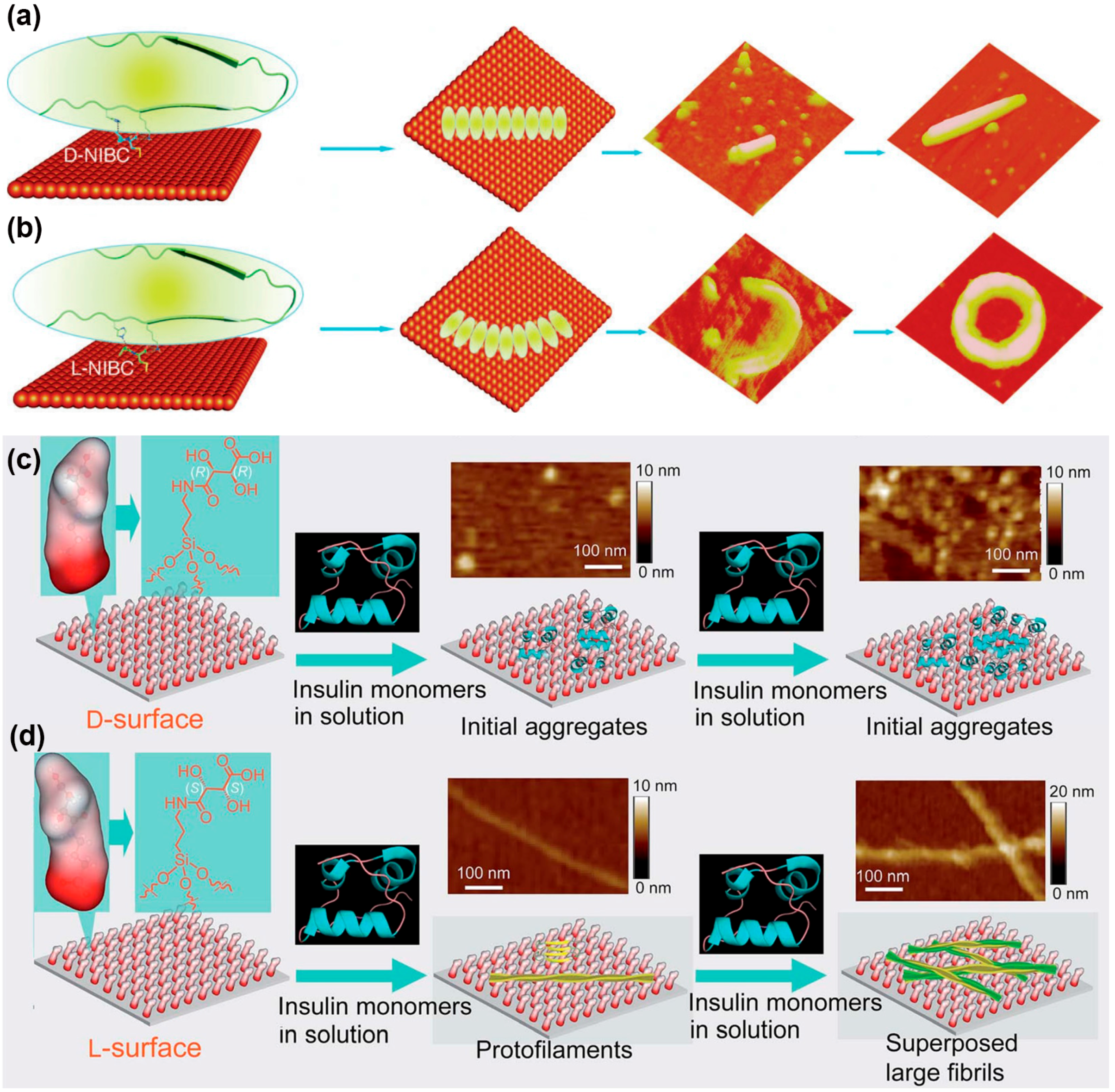
4. Protein Aggregation Modulated by Chiral Interface
5. Protein Aggregation Modulated by Biomolecules and Their Interfaces
6. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tyers, M.; Mann, M. From genomics to proteomics. Nature 2003, 42, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, A.; Thornton, J.M. Understanding nature’s catalytic toolkit. Trends Biochem. Sci. 2005, 30, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Oda, N.; Levin, J.D.; Spoonde, A.Y.; Frank, E.G.; Levine, A.S.; Woodgate, R.; Ackerman, E.J. Arrested DNA replication in Xenopus and release by Escherichia coli mutagenesis proteins. Science 1996, 272, 1644–1646. [Google Scholar] [CrossRef] [PubMed]
- Whisstock, J.C.; Lesk, A.M. Prediction of protein function from protein sequence and structure. Q. Rev. Biophys. 2003, 36, 307–340. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Kobayashi, Y. Mechanism of protein folding. Proteins Struct. Funct. Bioinf. 2000, 41, 288–298. [Google Scholar]
- Stefani, M. Protein folding and misfolding on surfaces. Int. J. Mol. Sci. 2009, 9, 2515–2542. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradović, Z. Intrinsic disorder and protein function. Biochemistry 2002, 9, 6573–6582. [Google Scholar] [CrossRef]
- Tyedmers, J.; Mogk, A.; Bukau, B. Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 2010, 11, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J.; Pinheiro, T.J.T. Danger—Misfolding proteins. Nature 2002, 416, 483–484. [Google Scholar] [CrossRef] [PubMed]
- Mezzenga, R.; Fischer, P. The self-assembly, aggregation and phase transitions of food protein systems in one, two and three dimensions. Rep. Prog. Phys. 2013, 76, 46601–46643. [Google Scholar] [CrossRef] [PubMed]
- Sidelmann, J.J.; Gram, J.; Jespersen, J.; Kluft, C. Fibrin clot formation and lysis: Basic mechanisms. Semin. Thromb. Hemost. 2000, 26, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Alsberg, E.; Feinstein, E.; Joy, M.P.; Prentiss, M.; Ingber, D.E. Magnetically-guided self-assembly of fibrin matrices with ordered nano-scale structure for tissue engineering. Tissue Eng. 2006, 12, 3247–3256. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Benedek, G.B. Observation of protein diffusivity in intact human and bovine lenses with application to cataract. Investig. Ophthalmol. Vis. Sci. 1975, 14, 449–456. [Google Scholar]
- Siezen, R.J.; Fisch, M.R.; Slingsby, C.; Benedek, G.B. Opacification of gamma-crystallin solutions from calf lens in relation to cold cat aract formation. Proc. Natl. Acad. Sci. USA 1985, 82, 1701–1705. [Google Scholar] [CrossRef] [PubMed]
- Harrington, D.J.; Adachi, K.; Royer, W.E. The high resolution crystal structure of deoxyhemoglobin S. J. Mol. Biol. 1997, 272, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Konstantinides, S.; Schafer, K.; Koschnick, S.; Loskutoff, D. Leptin-dependent platelet aggregation and arterial thrombosis suggests a mechanism for atherothrombotic disease in obesity. J. Clin. Investig. 2001, 108, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Scherzinger, E.; Sittler, A.; Schweiger, K.; Heiser, V.; Lurz, R.; Hasenbank, R.; Bates, G.P.; Lehrach, H.; Wanker, E.E. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: Implications for Huntington’s disease pathology. Proc. Natl. Acad. Sci. USA 1999, 96, 4604–4609. [Google Scholar] [CrossRef] [PubMed]
- Rakhit, R.; Cunningham, P.; Furtos-Matei, A.; Dahan, S.; Qi, X.F.; Crow, J.P.; Cashman, N.R.; Kondejewski, L.H.; Chakrabartty, A. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J. Biol. Chem. 2002, 277, 47551–47556. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Nakajo, S.; Tu, P.-H.; Tomita, T.; Nakaya, K.; Lee, V.M.-Y.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar] [PubMed]
- Swietnicki, W.; Morillas, M.; Chen, S.G.; Gambetti, P.; Surewicz, W.K. Aggregation and fibrillization of the recombinant human prion protein huPrP90–231. Biochemistry 2000, 39, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ(1–42) are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Creighton, T.E. Protein Folding, 1st ed.; W.H. Freeman and Company: New York, NY, USA, 1992. [Google Scholar]
- Fink, A.L. Protein aggregation: Folding aggregates, inclusion bodies and amyloid. Fold. Des. 1998, 3, R9–R23. [Google Scholar] [CrossRef]
- Roberts, C.J. Protein aggregation and its impact on product quality. Curr. Opin. Biotechnol. 2014, 30, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C.; Janin, J. Principles of protein-protein recognition. Nature 1975, 256, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Brange, J.; Andersen, L.; Laursen, E.D.; Meyn, G.; Rasmussen, E. Toward understanding insulin fibrillation. J. Pharm. Sci. 1997, 86, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Brummitt, R.K.; Andrews, J.M.; Jordan, J.L.; Fernandez, E.J.; Roberts, C.J. Thermodynamics of amyloid dissociation provide insights into aggregate stability regimes. Biophys. Chem. 2012, 168, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Maurer, R.W.; Hunter, A.K.; Robinson, A.S.; Roberts, C.J. Aggregates of α-chymotrypsinogen anneal to access more stable states. Biotechnol. Bioeng. 2014, 111, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.D.; Pirttilä, T.; Mehta, S.P.; Sersen, E.A.; Aisen, P.S.; Wisniewski, H.M. Plasma and cerebrospinal fluid levels of amyloid β proteins 1–40 and 1–42 in Alzheimer disease. Arch. Neurol. 2000, 57, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Terzi, E.; Hölzemann, G.; Seelig, J. Self-association of β-amyloid peptide (1–40) in solution and binding to lipid-membranes. J. Mol. Biol. 1995, 252, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Tseng, B.P.; Rydel, R.E.; Podlisny, M.B.; Selkoe, D.J. The oligomerization of amyloid β-protein begins intracellularly in cells derived from human brain. Biochemistry 2000, 39, 10831–10839. [Google Scholar] [CrossRef] [PubMed]
- Koppaka, V.; Paul, C.; Murray, I.V.; Axelsen, P.H. Early synergy between Abeta42 and oxidatively damaged membranes in promoting amyloid fibril formation by Abeta40. J. Biol. Chem. 2003, 278, 36277–36284. [Google Scholar] [CrossRef] [PubMed]
- Lindström, F.; Bokvist, M.; Sparrman, T.; Gröbner, G. Association of amyloid-β peptide with membrane surfaces monitored by solid state NMR. Phys. Chem. Chem. Phys. 2002, 4, 5524–5530. [Google Scholar] [CrossRef]
- Klein, W.L.; Stine, W.B.; Teplow, D.B. Small assemblies of unmodified amyloid β-protein are the proximate neurotoxin in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, S.; Hambsch, B.; Hesse, L.; Stumm, J.; Schmidt, C.; Beher, D.; Bayer, T.A.; Beyreuther, K.; Multhaup, G. Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer’s disease. J. Biol. Chem. 2011, 276, 33923–33929. [Google Scholar] [CrossRef] [PubMed]
- Devanathan, S.; Salamon, Z.; Lindblom, G.; Gröbner, G.; Tollin, G. Effects of sphingomyelin, cholesterol and zinc ions on the binding, insertion and aggregation of the amyloid Aβ(1–40) peptide in solid-supported lipid bilayers. FEBS J. 2006, 273, 1389–1402. [Google Scholar] [CrossRef] [PubMed]
- Qing, G.Y.; Zhao, S.L.; Xiong, Y.T.; Lv, Z.Y.; Jiang, F.L.; Liu, Y.; Chen, H.; Zhang, M.X.; Sun, T.L. Chiral effect at protein/graphene interface: A bioinspired perspective to understand amyloid formation. J. Am. Chem. Soc. 2014, 136, 10736–10742. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, S.; Goodwin, H.; Cumings, J.N. The distribution of phosphorus-containing lipid compounds in the human brain. J. Neurochem. 1961, 8, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Kuchinka, E.; Seelig, J. Interaction of melittin with phosphatidylcholine membranes. Binding isotherm and lipid head-group conformation. Biochemistry 1989, 28, 4216–4221. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.; Berry, R.S. Proteins with H-bond packing defects are highly interactive with lipid bilayers: Implications for amyloidogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2391–2396. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Goto, Y.; Takai, M.; Matsuno, R.; Inoue, Y.; Konno, T. Novel polymer biomaterials and interfaces inspired from cell membrane functions. Biochim. Biophys. Acta 2011, 1810, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Liu, X.Y.; Ma, L.; Cheng, C.; Shi, W.B.; Nie, C.X.; Zhao, C.S. Heparin-mimicking multilayer coating on polymeric membrane via LbL assembly of cyclodextrin-based supramolecules. ACS Appl. Mater. Interfaces 2014, 6, 21603–21614. [Google Scholar] [CrossRef] [PubMed]
- Jean, L.; Lee, C.F.; Vaux, D.J. Enrichment of amyloidogenesis at an air-water interface. Biophys. J. 2012, 102, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Gidalevitz, D.; Huang, Z.; Rice, S.A. Protein folding at the air-water interface studied with X-ray reflectivity. Proc. Natl. Acad. Sci. USA 1999, 96, 2608–2611. [Google Scholar] [CrossRef] [PubMed]
- Treuheit, M.J.; Kosky, A.A.; Brems, D.N. Inverse relationship of protein concentration and aggregation. Pharm. Res. 2002, 19, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, T.L.; Boonstra, E.F.; Hammond, J.M. Kinetics of protein denaturation at gas-liquid interfaces. J. Colloid Interface Sci. 1980, 74, 441–450. [Google Scholar] [CrossRef]
- Maa, Y.F.; Hsu, C.C. Protein denaturation by combined effect of shear and air-liquid interface. Biotechnol. Bioeng. 1997, 54, 503–512. [Google Scholar] [CrossRef]
- Morinaga, A.; Hasegawa, K.; Nomura, R.; Ookoshi, T.; Ozawa, D.; Goto, Y.; Yamada, M.; Naiki, H. Critical role of interfaces and agitation on the nucleation of Aβ amyloid fibrils at low concentrations of Aβ monomers. Biochim. Biophys. Acta 2010, 1804, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.D.; Lansbury, P.T., Jr. Models of amyloid seeding in Alzheimer’s disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 1997, 66, 385–407. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Nagaraj, R. Self-assembly of short amyloidogenic peptides at the air–water interface. J. Colloid Interface Sci. 2011, 360, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Kowalewski, T.; Holtzman, D.M. In situ atomic force microscopy study of Alzheimer’s β-amyloid peptide on different substrates: New insights into mechanism of β-sheet formation. Proc. Natl. Acad. Sci. USA 1999, 96, 3688–3693. [Google Scholar] [CrossRef] [PubMed]
- Bam, N.B.; Cleland, J.L.; Yang, J.; Manning, M.C.; Carpenter, J.F.; Kelley, R.F.; Randolph, T.W. Tween protects recombinant human growth hormone against agitation-induced damage via hydrophobic interactions. J. Pharm. Sci. 1998, 87, 1554–1559. [Google Scholar] [CrossRef] [PubMed]
- Chou, D.K.; Krishnamurthy, R.; Randolph, T.W.; Carpenter, J.F.; Manning, M.C. Effects of Tween 20® and Tween 80® on the stability of albutropin during agitation. J. Pharm. Sci. 2005, 94, 1368–1381. [Google Scholar] [CrossRef] [PubMed]
- Serno, T.; Carpenter, J.F.; Randolph, T.W.; Winter, G. Inhibition of agitation-induced aggregation of an IgG-antibody by hydroxypropyl-β-cyclodetxrin. J. Pharm. Sci. 2010, 99, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Wiesbauer, J.; Prassl, R.; Nidetzky, B. Renewal of the air-water interface as a critical system parameter of protein stability: Aggregation of the human growth hormone and its prevention by surface-active compounds. Langmuir 2013, 29, 15240–15250. [Google Scholar] [CrossRef] [PubMed]
- Kabanov, A.V.; Batrakova, E.V.; Alakhov, V.Y. Pluronic® block copolymers as novel polymer therapeutics for drug and gene delivery. J. Control. Release 2002, 82, 189–212. [Google Scholar] [CrossRef]
- Giacomelli, C.E.; Norde, W. Influence of hydrophobic Teflon particles on the structure of amyloid β-peptide. Biomacromolecules 2003, 4, 1719–1726. [Google Scholar] [CrossRef] [PubMed]
- Schladitz, C.; Vieira, E.P.; Hermel, H.; Möhwald, H. Amyloid-β-sheet formation at the air-water interface. Biophys. J. 1999, 77, 3305–3310. [Google Scholar] [CrossRef]
- Jorgensen, L.; Bennedsen, P.; Hoffmann, S.V.; Krogh, R.L.; Pinholt, C.; Groenning, M.; Hostrup, S.; Bukrinsky, J.T. Adsorption of insulin with varying self-association profiles to a solid Teflon surface-Influence on protein structure, fibrillation tendency and thermal stability. Eur. J. Pharm. Sci. 2011, 42, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Marcinowski, K.J.; Shao, H.; Clancy, E.L.; Zagorski, M.G. Solution structure model of residues 1–28 of the amyloid β-peptide when bound to micelles. J. Am. Chem. Soc. 1998, 120, 11082–11091. [Google Scholar] [CrossRef]
- Blackley, H.K.L.; Sanders, G.H.W.; Davies, M.C.; Roberts, C.J.; Tendler, S.J.B.; Wilkinson, M.J. In-situ atomic force microscopy study of β-amyloid fibrillization. J. Mol. Biol. 2000, 298, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Adachi, T.; Bout, D.V.; Zhu, X.-Y. A mobile precursor determines amyloid-β peptide fibril formation at interfaces. J. Am. Chem. Soc. 2012, 134, 14172–14178. [Google Scholar] [CrossRef] [PubMed]
- Anthony, S.; Zhang, L.; Granick, S. Methods to track single-molecule trajectories. Langmuir 2006, 22, 5266–5272. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.D.; Hebda, J.A.; Miranker, A.D. Conserved and cooperative assembly of membrane-bound α-helical states of islet amyloid polypeptide. Biochemistry 2006, 45, 9496–9508. [Google Scholar] [CrossRef] [PubMed]
- Sigel, A.; Sigel, H.; Sigel, R.K.O. Neurodegenerative Diseases and Metal Ions, 1st ed.; John Wiley and Sons: Chichester, UK, 2006. [Google Scholar]
- Faller, P.; Hureau, C.; Penna, G.L. Metal ions and intrinsically disordered proteins and peptides: From Cu/Zn amyloid-β to general principles. Acc. Chem. Res. 2014, 47, 2252–2259. [Google Scholar] [CrossRef] [PubMed]
- Zatta, P.; Drago, D.; Bolognin, S.; Sensi, S.L. Alzheimer’s disease, metal ions and metal homeostatic therapy. Trends Pharmacol. Sci. 2009, 30, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Gaeta, A.; Hider, R.C. The crucial role of metal ions in neurodegeneration: The basis for a promising therapeutic strategy. Br. J. Pharmacol. 2005, 146, 1041–1059. [Google Scholar] [CrossRef] [PubMed]
- Miller, Y.; Ma, B.; Nussinov, R. Zinc ions promote Alzheimer Aβ aggregation via population shift of polymorphic states. Proc. Natl. Acad. Sci. USA 2010, 107, 9490–9495. [Google Scholar] [CrossRef] [PubMed]
- Pagel, K.; Seri, T.; von Berlepsch, H.; Griebel, J.; Kirmse, R.; Böttcher, C.; Koksch, B. How metal ions affect amyloid formation: Cu2+- and Zn2+-sensitive peptides. ChemBioChem 2008, 9, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Hoernke, M.; Koksch, B.; Brezesinski, G. Amyloidogenic peptides at hydrophobic-hydrophilic interfaces: Coordination affinities and the chelate effect dictate the competitive binding of Cu2+ and Zn2+. Chem. Phys. Chem. 2011, 12, 2225–2229. [Google Scholar] [CrossRef] [PubMed]
- Hoernke, M.; Koksch, B.; Brezesinski, G. Influence of the hydrophobic interface and transition metal ions on the conformation of amyloidogenic model peptides. Biophys. Chem. 2010, 150, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.P.; Perrin, D.D. Copper(II) and zinc(II) complexes of glycylglycyl-l-histidine and derivatives. J. Chem. Soc. Dalton Trans. 1977, 18, 53–57. [Google Scholar] [CrossRef]
- Hoernke, M.; Falenski, J.A.; Schwieger, C.; Koksch, B.; Brezesinski, G. Triggers for β-sheet formation at the hydrophobic-hydrophilic interface: High concentration, in-plane orientational order, and metal ion complexation. Langmuir 2011, 27, 14218–14231. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Ray, I.; Chauhan, V.P.S. Interaction of amyloid beta-protein with anionic phospholipids: Possible involvement of Lys28 and C-terminus aliphatic amino acids. Neurochem. Res. 2000, 25, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.X.; Tuominen, E.K.J.; Kinnunen, P.K.J. Formation of amyloid fibers triggered by phosphatidylserine-containing membranes. Biochemistry 2004, 43, 10302–10307. [Google Scholar] [CrossRef] [PubMed]
- Terzi, E.; Holzemann, G.; Seelig, J. Interaction of Alzheimer β-amyloid peptide(1–40) with lipid membranes. Biochemistry 1997, 36, 14845–14852. [Google Scholar] [CrossRef] [PubMed]
- Ege, C.; Lee, K.Y.C. Insertion of Alzheimer’s Aβ40 peptide into lipid monolayers. Biophys. J. 2004, 87, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Terzi, E.; Hölzemann, G.; Seelig, J. Alzheimer β-amyloid peptide 25–35: Electrostatic interactions with phospholipid membranes. Biochemistry 1994, 33, 7434–7441. [Google Scholar] [CrossRef] [PubMed]
- Chi, E.Y.; Ege, C.; Winans, A.; Majewski, J.; Wu, G.H.; Kjaer, K.; Lee, K.Y.C. Lipid membrane templates the ordering and induces the fibrillogenesis of Alzheimer’s disease amyloid-β peptide. Proteins Struct. Funct. Bioinf. 2008, 72, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.M.; Dubey, M.; Camp, P.J.; Vernon, B.C.; Biernat, J.; Mandelkow, E.; Majewski, J.; Chi, E.Y. Interaction of tau protein with model lipid membranes induces tau structural compaction and membrane disruption. Biochemsitry 2012, 51, 2539–2550. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Spillantini, M.G.; Hasegawa, M.; Smith, M.J.; Crowther, R.A. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383, 550–553. [Google Scholar] [CrossRef] [PubMed]
- King, M.E.; Gamblin, T.C.; Kuret, J.; Binder, L.I. Differential assembly of human tau isoforms in the presence of arachidonic acid. J. Neurochem. 2000, 74, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Chirita, C.; Necula, M.; Kuret, J. Anionic micelles and vesicles induce tau fibrillization in vitro. J. Biol. Chem. 2003, 278, 25644–25650. [Google Scholar] [CrossRef] [PubMed]
- Moores, B.; Drolle, E.; Attwood, S.J.; Simons, J.; Leonenko, Z. Effect of surfaces on amyloid fibril formation. PLoS ONE 2011, 6, e25954. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.M.; Zhao, J.; Yu, X.; Zhao, C.; Li, L.Y.; Zheng, J. Alzheimer Aβ(1–42) monomer adsorbed on the self-assembled monolayers. Langmuir 2010, 26, 12722–12732. [Google Scholar] [CrossRef] [PubMed]
- Giacomelli, C.E.; Norde, W. Conformational changes of the amyloid β-peptide (1–40) adsorbed on solid surfaces. Macromol. Biosci. 2005, 5, 401–407. [Google Scholar] [CrossRef] [PubMed]
- McMasters, M.J.; Hammer, R.P.; McCarley, R.L. Surface-induced aggregation of beta amyloid peptide by ω-substituted alkanethiol monolayers supported on gold. Langmuir 2005, 21, 4464–4470. [Google Scholar] [CrossRef] [PubMed]
- McUmber, A.C.; Randolph, T.W.; Schwartz, D.K. Electrostatic interactions influence protein adsorption (but not desorption) at the silica–aqueous interface. J. Phys. Chem. Lett. 2015, 6, 2583–2587. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gan, H.; Sun, T.L. Chiral design for polymeric biointerface: The influence of surface chirality on protein adsorption. Adv. Funct. Mater. 2011, 21, 3276–3281. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Prashar, D.; Luk, Y.Y. Anti-fouling chemistry of chiral mono layers: Enhancing biofilm resistance on racemic surface. Langmuir 2011, 27, 6124–6131. [Google Scholar] [CrossRef] [PubMed]
- Geva, M.; Frolow, F.; Eisenstein, M.; Addadi, L. Antibody recognition of chiral surfaces. Enantiomorphous crystals of leucine-leucine-tyrosine. J. Am. Chem. Soc. 2003, 125, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.L.; Han, D.; Rhemann, K.; Chi, L.F.; Fuchs, H. Stereospecific interaction between immune cells and chiral surfaces. J. Am. Chem. Soc. 2007, 129, 1496–1497. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Köwitsch, A.; Zhou, G.Y.; Groth, T.; Fuhrmann, B.; Niepel, M.; Amado, E.; Kressler, J. Enantiopure chiral poly(glycerol methacrylate) self-assembled monolayers knock down protein adsorption and cell adhesion. Adv. Healthcare Mater. 2013, 2, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Hellstrand, E.; Boland, B.; Walsh, D.M.; Linse, S. Amyloid β-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process. ACS Chem. Neurosci. 2010, 1, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.B.; Zhang, M.X.; Lu, P.; Guo, G.L.; Wang, D.; Sun, T.L. Chirality-assisted ring-like aggregation of Aβ(1–40) at liquid-solid interfaces: A stereoselective two-step assembly process. Angew. Chem. Int. Ed. 2015, 54, 2245–2250. [Google Scholar] [CrossRef] [PubMed]
- Bailo, E.; Deckert, V. Tip-enhanced raman scattering. Chem. Soc. Rev. 2008, 37, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.L.; Xu, C.; Gao, N.; Ren, J.; Qu, X.G. Opposing enantiomers of tartaric acid anchored on a surface generate different insulin assemblies and hence contrasting cellular responses. Chem. Sci. 2014, 5, 4367–4374. [Google Scholar] [CrossRef]
- Krysmann, M.J.; Castelletto, V.; Kelarakis, A.; Hamley, I.W.; Hule, R.A.; Pochan, D.J. Self-assembly and hydrogelation of an amyloid peptide fragment. Biochemistry 2008, 47, 4597–4605. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Leapman, R.D.; Guo, Z.H.; Yau, W.M.; Mattson, M.P.; Tycko, R. Self-propagating, molecular-level polymorphism in Alzheimer’s β-amyloid fibrils. Science 2005, 307, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Lee, R.T. Carbohydrate-protein interactions: Basis of glycobiology. Acc. Chem. Res. 1995, 28, 321–327. [Google Scholar] [CrossRef]
- Yanagisawa, K. Role of gangliosides in Alzheimer’s disease. Biochim. Biophys. Acta 2007, 1768, 1943–1951. [Google Scholar] [CrossRef] [PubMed]
- McLaurin, J.; Franklin, T.; Zhang, X.Q.; Deng, J.P.; Fraser, P.E. Interactions of Alzheimer amyloid-β peptides with glycosaminoglycans: Effects on fibril nucleation and growth. Eur. J. Biochem. 1999, 266, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Cohlberg, J.A.; Li, J.; Uversky, V.N.; Fink, A.L. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from α-synuclein in vitro. Biochemistry 2002, 41, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Matsumoto, E.; Onogi, S.; Miura, Y. Aggregation of Alzheimer amyloid peptide (1–42) on the multivalent sulfonated sugar interface. Bioconjug. Chem. 2010, 21, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Ban, T.; Morigaki, K.; Yagi, H.; Kawasaki, T.; Kobayashi, A.; Yuba, S.; Naiki, H.; Goto, Y. Real-time and single fibril observation of the formation of amyloid β spherulitic structures. J. Biol. Chem. 2006, 281, 33677–33683. [Google Scholar] [CrossRef] [PubMed]
- Giulian, D.; Haverkamp, L.J.; Yu, J.H.; Karshin, W.; Tom, D.; Li, J.; Kazanskaia, A.; Kirkpatrick, J.; Roher, A.E. The HHQK domain of β-amyloid provides a structural basis for the immunopathology of Alzheimer’s disease. J. Biol. Chem. 1998, 273, 29719–29726. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Mihara, H. Peptide and protein mimetics inhibiting amyloid β-peptide aggregation. Acc. Chem. Res. 2008, 41, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.P.; Kelly, J.W. Templates that induce α-helical, β-sheet, and loop conformations. Chem. Rev. 1995, 95, 2169–2187. [Google Scholar] [CrossRef]
- Eisele, Y.S. From soluble Aβ to progressive Aβ aggregation: Could prion-like templated misfolding play a role? Brain Pathol. 2013, 23, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Dorgeret, B.; Khemtémourian, L.; Correia, I.; Soulier, J.-L.; Lequin, O.; Ongeri, S. Sugar-based peptidomimetics inhibit amyloid β-peptide aggregation. Eur. J. Med. Chem. 2011, 46, 5959–5969. [Google Scholar] [CrossRef] [PubMed]
- Norlin, N.; Hellberg, M.; Filippov, A.; Sousa, A.A.; Gröbner, G.; Leapman, R.D.; Almqvist, N.; Antzutkin, O.N. Aggregation and fibril morphology of the Arctic mutation of Alzheimer’s Aβ peptide by CD, TEM, STEM and in situ AFM. J. Struct. Biol. 2012, 180, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Han, S.Y.; Xu, W.W.; Cao, M.W.; Wang, J.Q.; Xia, D.H.; Xu, H.; Zhao, X.B.; Lu, J.R. Interfacial adsorption of cationic peptide amphiphiles: A combined study of in situ spectroscopic ellipsometry and liquid AFM. Soft Matter 2012, 8, 645–652. [Google Scholar] [CrossRef]
- Ye, S.J.; Nguyen, K.T.; le Clair, S.V.; Chen, Z. In situ molecular level studies on membrane related peptides and proteinsins in real time using sum frequency generation vibrational spectroscopy. J. Struct. Biol. 2009, 168, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Scheuring, S.; Lévy, D.; Rigaud, J.-L. Watching the components of photosynthetic bacterial membranes and their in situ organisation by atomic force microscopy. Biochim. Biophys. Acta 2005, 1712, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.E.; Epand, R.F.; Hsu, J.C.Y.; Mo, G.C.H.; Epand, R.M.; Yip, C.M. Cationic peptide-induced remodelling of model membranes: Direct visualization by in situ atomic force microscopy. J. Struct. Biol. 2008, 162, 121–138. [Google Scholar] [CrossRef] [PubMed]
- Qing, G.Y.; Sun, T.L. Chirality-triggered wettability switching on a smart polymer surface. Adv. Mater. 2011, 23, 1615–1620. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.L.; Qing, G.Y.; Su, B.L.; Jiang, L. Functional biointerface materials inspired from nature. Chem. Soc. Rev. 2011, 40, 2909–2921. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.S.; Zhang, M.X.; Qing, G.Y.; Sun, T.L. Dynamic biointerfaces: From recognition to function. Small 2015, 11, 1097–1112. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.D.; Tian, Y.M.; Cai, M.J.; Wang, F.; Wu, J.Z.; Gao, J.; Liu, S.H.; Jiang, J.G.; Jiang, S.B.; Wang, H.D. Studying the nucleated mammalian cell membrane by single molecule approaches. PLoS ONE 2014, 9, e91595. [Google Scholar] [CrossRef] [PubMed]
- Denzer, A.J.; Nabholz, C.E.; Spiess, M. Transmembrane orientation of signal-anchor proteins is affected by the folding state but not the size of the N-terminal domain. EMBO J. 1995, 14, 6311–6317. [Google Scholar] [PubMed]
- Bretscher, M.S. Membrane structure: Some general principles. Science 1973, 181, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Pierres, A.; Monnet-Corti, V.; Benoliel, A.-M.; Bongrand, P. Do membrane undulations help cells probe the world? Trends Cell Biol. 2009, 19, 428–433. [Google Scholar] [CrossRef] [PubMed]
- González-Domínguez, J.M.; Gutiérrez, F.A.; Hernández-Ferrer, J.; Ansón-Casaos, A.; Rubianes, M.D.; Rivas, G.; Martínez, M.T. Peptide-based biomaterials. Linking l-tyrosine and poly l-tyrosine to graphene oxide nanoribbons. J. Mater. Chem. B 2015, 3, 3870–3884. [Google Scholar] [CrossRef]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Q.; Tang, Q.; Xiong, Y.; Qing, G.; Sun, T. Protein/Peptide Aggregation and Amyloidosis on Biointerfaces. Materials 2016, 9, 740. https://doi.org/10.3390/ma9090740
Lu Q, Tang Q, Xiong Y, Qing G, Sun T. Protein/Peptide Aggregation and Amyloidosis on Biointerfaces. Materials. 2016; 9(9):740. https://doi.org/10.3390/ma9090740
Chicago/Turabian StyleLu, Qi, Qiuhan Tang, Yuting Xiong, Guangyan Qing, and Taolei Sun. 2016. "Protein/Peptide Aggregation and Amyloidosis on Biointerfaces" Materials 9, no. 9: 740. https://doi.org/10.3390/ma9090740