All commercial reagents and anhydrous solvents were obtained from commercial sources and were distilled from standard drying agents unless otherwise specified. 1H NMR spectra were recorded on a Bruker AVANCE III 400 spectrometer (Bruker Corp., Billerica, MA, USA) and referenced to tetramethylsilane in appropriate organic solutions. Chemical shifts were expressed as δ units using tetramethylsilane as internal standard (in NMR description: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; bs, broad signal; J, joules; Hz, hertz). Mass spectra were measured with an Agilent 6120 spectrometer (Agilent Tech., Santa Clara, CA, USA) using an ESI+APCI source coupled to an Agilent 1200 HPLC system (Agilent Tech.) operating in reverse mode. High-resolution mass spectra were recorded on an ESI-ion trap mass spectrometer (Shimadzu LCMS-IT-TOF, Shimadzu, Kyoto, Japan), and the results agreed with the theoretical values to within 5 ppm. Purity of all biologically evaluated compounds was greater than 95%.
3.1. Chemistry
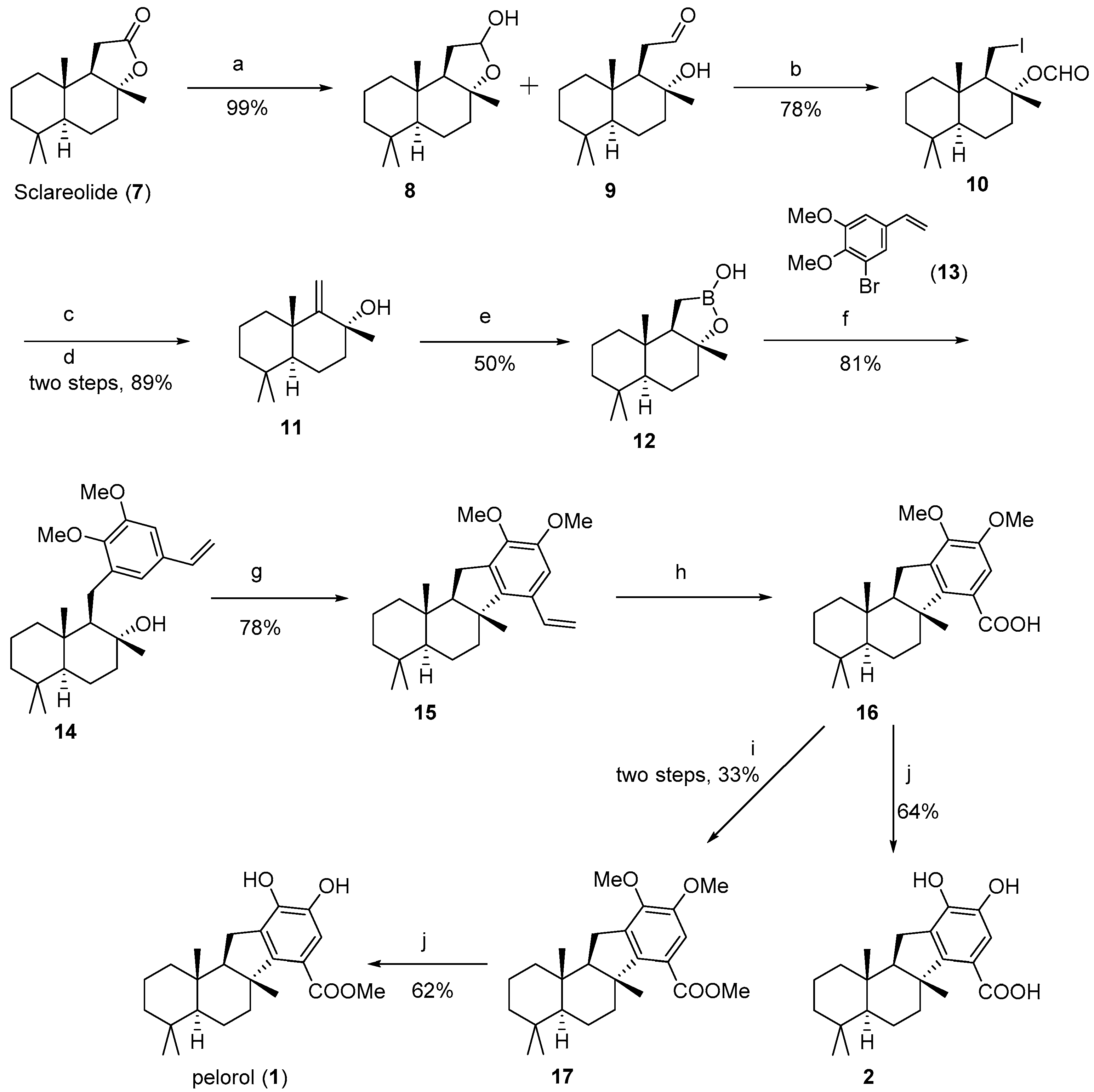
(
1R,
2R,
4aS,
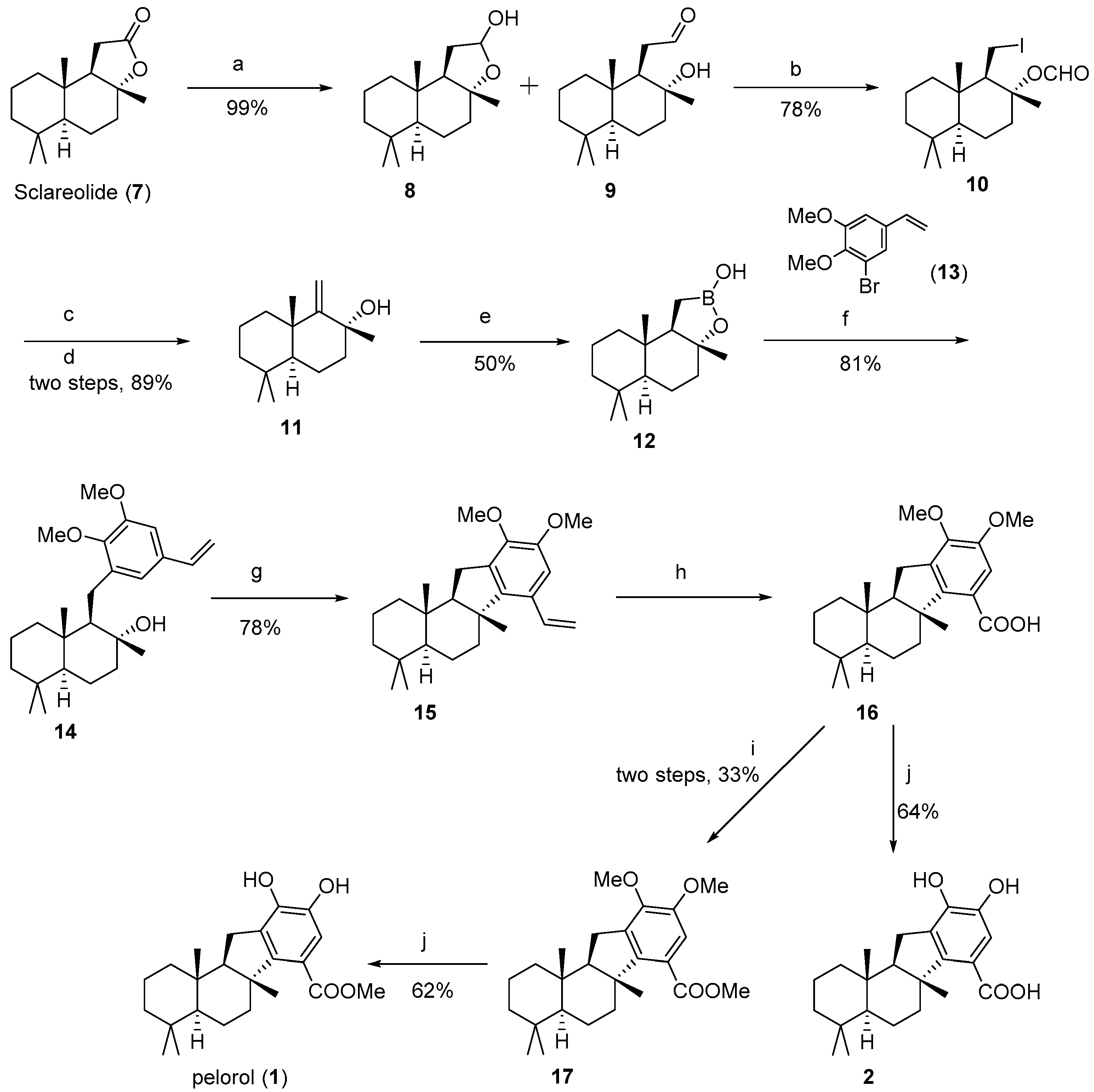
8aS)-1-(Iodoethyl)-2,5,5,8a-tetramethyldecahydronaphthalen-2-yl formate (
10) [
14]: To a solution of (+)-sclareolide (10.00 g, 40.0 mmol, 1.0 equiv.) in 200 mL CH
2Cl
2 was added 1.0 M solution of DIBAL in heptane (48.0 mL, 48.0 mmol, 1.2 equiv.) dropwise at −78 °C. The reaction mixture was then stirred for another 60 min. Water was slowly added, the reaction mixture was warmed to room temperature and stirred overnight, extracted with CH
2Cl
2 (4 × 50 mL), washed with saturated brine (50 mL), dried over Na
2SO
4. Evaporation of the solvent afforded a white solid, which was used immediately in the next step.
The white solid in 90 mL anhydrous toluene was treated with PIDA (18.04 g, 56 mmol, 1.4 equiv.) and I
2 (12.18 g, 48 mmol, 1.2 equiv.). The purple reaction mixture was vigorously stirred at 74 °C for 60 min under the irradiation of flood lamp (150 W). The reaction mixture was cooled, concentrated under reduced pressure, diluted with saturated brine, and extracted with EtOAc. The organic layers were combined, washed with saturated Na
2S
2O
3, dried over Na
2SO
4. The solution was filtered and concentrated under reduced pressure. The residue was quickly diluted with anhydrous MeOH, stirred and cooled at −78 °C. The resulting white solid
10 (7.87 g, 78% yield) was collected via filtration.
1H NMR (400 MHz, CDCl
3) δ: 8.02 (s, 1H), 3.36 (dd,
J = 10.7, 2.9 Hz, 1H), 3.13 (dd,
J = 10.7, 4.6 Hz, 1H), 2.55 (d,
J = 12.7 Hz, 1H), 2.46 (d,
J = 2.0 Hz, 1H), 1.93–1.85 (m, 2H), 1.69 (d,
J = 3.5 Hz, 1H), 1.57 (s, 3H), 1.47 (d,
J = 1.0 Hz, 3H), 1.38 (s, 1H), 1.25 (s, 2H), 1.07 (d,
J = 2.3 Hz, 1H), 0.88 (s, 3H), 0.85 (d,
J = 0.8 Hz, 3H), 0.79 (s, 3H).
13C NMR (101 MHz, CDCl
3) δ: 160.3, 88.3, 62.1, 55.3, 41.6, 40.8, 39.6, 38.9, 33.2, 33.1, 21.4, 20.4, 19.7, 18.4, 14.8, −3.2. (
Figure S1).
(+)-Drim-9(11)-en-8-ol (
11) [
14]: A solution of compound
10 (2.32 g, 6.13 mmol, 1.0 equiv.) in anhydrous pyridine (20 mL) was treated with silver (I) fluoride (1.16 g, 9.2 mmol, 1.5 equiv.) at room temperature. The reaction flask was covered with aluminum foil to prevent light. The solution was stirred at room temperature for 12 h, then concentrated
in vacuo, diluted with Et
2O (20 mL), and filtered through celite pad. The dark grey solid (AgF) can be recycled. The filter cake was washed with Et
2O (20 mL). The filtrate was combined, washed with saturated NaHCO
3 and saturated Na
2S
2O
3 (
, 45 mL), then washed with brine (50 mL), dried over Na
2SO
4. The solution was filtered and concentrated under reduced pressure to afford unstable pale yellow syrup which was used immediately in the next step.
A solution of yellow syrup in 90 mL MeOH was treated with powdered, oven-dried K
2CO
3 (1.01 g, 7.35 mmol, 1.2 equiv.) at 0 °C. The reaction was warmed to room temperature and vigorously stirred for 2 h. The mixture was concentrated under reduced pressure, diluted with Et
2O (20 mL), washed with H
2O (60 mL), and extracted with Et
2O (2 × 20 mL). The combined Et
2O extracts were dried over Na
2SO
4, filtered and concentrated under reduced pressure to afford compound
11 as oyster white solid (1.18 g, 89% yield).
1H NMR (400 MHz, CDCl
3) δ: 5.21 (s, 1H), 4.83 (s, 1H), 2.01 (d,
J = 9.5 Hz, 1H), 1.79 (d,
J = 12.6 Hz, 1H), 1.69 (t,
J = 13.5 Hz, 2H), 1.53 (d,
J = 14.8 Hz, 1H), 1.47 (s, 1H), 1.40 (s, 3H), 1.38–1.30 (m, 2H), 1.25 (s, 1H), 1.15 (d,
J = 14.2 Hz, 1H), 1.08 (s, 3H), 0.98 (d,
J = 11.0 Hz, 1H), 0.87 (s, 3H), 0.84 (s, 3H).
13C NMR (101 MHz, CDCl
3) δ: 166.6, 103.7, 73.3, 53.5, 44.2, 41.8, 40.0, 39.0, 33.8, 33.2, 30.6, 29.68, 22.4, 21.6, 20.2, 19.1. (
Figure S2).
Borono-sclareolide (
12) [
14]: Yield 50%, white solid.
1H NMR (400 MHz, CDCl
3) δ: 6.62 (s, 1H), 1.95 (d,
J = 11.6 Hz, 1H), 1.79–1.74 (m, 1H), 1.63 (d,
J = 8.0 Hz, 2H), 1.56–1.50 (m, 1H), 1.42 (s, 3H), 1.38–1.34 (m, 1H), 1.33-1.23 (m, 1H), 1.17 (s, 4H), 0.98–0.90 (m, 2H), 0.85 (s, 3H), 0.82 (s, 3H), 0.80 (s, 3H), 0.75 (d,
J = 7.6 Hz, 1H).
13C NMR (101 MHz, CDCl
3) δ: 83.9, 61.6, 56.9, 42.5, 40.4, 39.5, 37.0, 33.3, 33.2, 23.1, 21.0, 20.9, 18.3, 14.6. (
Figure S3).
(
1R,
2R,
4aS,
8aS)-1-(2,3-diMethoxy-5-vinylbenzyl)-2,5,5,8a-tetramethyldecahydronaphthalen-2-ol (
14): To the borono-sclareolide
12 (1.00 g, 4.0 mmol, 1.0 equiv.) was added bromide
13 (1.94 g, 8 mmol, 2.0 equiv.),
S-Phos (0.25 g, 0.6 mmol, 0.15 equiv.), CsF (1.82 g, 12 mmol, 3.0 equiv.) and 50 mL anhydrous dioxane. The solution was degassed for 20 min by bubbling N
2 under sonication. Pd(OAc)
2 (0.09 g, 0.4 mmol, 0.1 equiv.) was immediately added under N
2, then the reaction mixture was heated at 50 °C for 18 h. The solution was cooled to room temperature and filtered through a celite pad, concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (petroleum ether/EtOAc = 30:1) to afford
14 (1.22 g, 80.5% yield) as colorless oil.
1H NMR (400 MHz, CDCl
3) δ: 6.87 (d,
J = 1.6 Hz, 1H), 6.81 (d,
J = 1.7 Hz, 1H), 6.63 (dd,
J = 17.5, 10.8 Hz, 1H), 5.63 (d,
J = 17.5 Hz, 1H), 5.18 (d,
J = 10.9 Hz, 1H), 3.86 (s, 3H), 3.85 (s, 3H), 2.84 (dd,
J = 14.5, 5.1 Hz, 1H), 2.66 (s, 1H), 2.60 (d,
J = 4.9 Hz, 1H), 1.86 (d,
J = 12.5 Hz, 1H), 1.75 (d,
J = 12.8 Hz, 1H), 1.62 (s, 3H), 1.41 (d,
J = 14.0 Hz, 2H), 1.33 (d,
J = 16.6 Hz, 2H), 1.28 (s, 3H), 1.10 (dd,
J = 13.2, 3.6 Hz, 1H), 0.91 (s,3H), 0.85 (s, 3H), 0.83-0.81 (m, 1H), 0.80 (s, 3H).
13C NMR (101 MHz, CDCl
3) δ: 152.6, 146.3, 138.1, 136.8, 133.5, 121.5, 112.9, 107.0, 73.7, 62.9, 60.7, 56.1, 55.7, 43.8, 41.8, 40.3, 39.5, 33.5, 33.3, 25.1, 24.2, 21.6, 20.3, 18.6, 15.5. (
Figure S4).
(
4aS,
6aR,
11aR,
11bS)-9,10-diMethoxy-4,4,6a,11b-tetramethyl-7-vinyl-2,3,4,4a,5,6,6a,11,11a,11b-decahydro-1
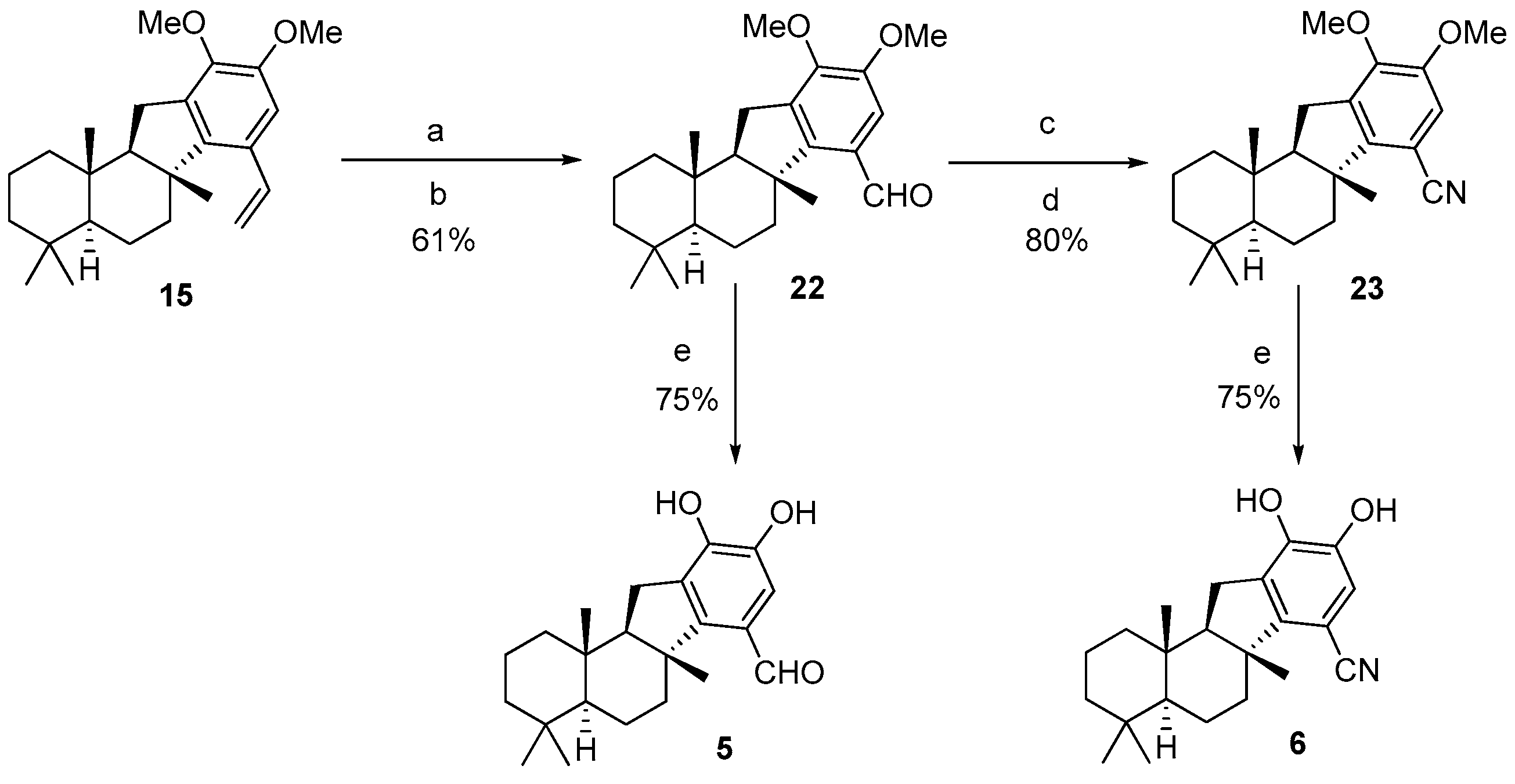
H-benzo[a]fluorene (
15): To a stirred solution of
14 (232 mg, 0.6 mmol, 1.0 equiv.) in CH
2Cl
2 (60 mL), SnCl
4 (0.6 mL) was added dropwise at −78 °C under N
2. The resulting mixture was stirred for an additional 1 h. Upon the completion of the reaction (monitored by TLC), the solution was diluted with CH
2Cl
2 (90 mL) and poured into ice. The aqueous phase was extracted with CH
2Cl
2 (2 × 20 mL), the combined CH
2Cl
2 extracts were dried over Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (petroleum ether/EtOAc = 15:1) to give
15 as yellow-green crystal (173 mg, 78.3% yield).
1H NMR (400 MHz, CDCl
3) δ: 7.06 (dd,
J = 17.4, 10.9 Hz, 1H), 6.81 (s, 1H), 5.49 (dd,
J = 17.3, 1.3 Hz, 1H), 5.17 (dd,
J = 10.9, 1.3 Hz, 1H), 3.86 (s, 3H), 3.84 (s, 3H), 2.72 (dd,
J = 14.7, 6.2 Hz, 1H), 2.56–2.47 (m, 1H), 2.42–2.35 (m, 1H), 1.73 (s, 3H), 1.61 (s, 2H), 1.43 (s, 2H), 1.26 (s, 1H), 1.18 (d,
J = 4.3 Hz, 1H), 1.11 (s, 3H), 1.04 (s, 3H), 1.01–0.95 (m, 2H), 0.87 (s, 6H).
13C NMR (101 MHz, CDCl
3) δ: 150.6, 145.5, 145.3, 135.8, 134.3, 128.3, 113.6, 107.7, 64.2, 60.4, 57.0, 56.0, 47.9, 42.5, 40.1, 39.5, 37.1, 33.4, 33.1, 25.3, 21.6, 21.1, 19.7, 18.4, 16.1. (
Figure S5).
(
4aS,
6aR,
11aR,
11bS)-Methyl-9,10-dimethoxy-4,4,6a,11b-tetramethyl-2,3,4,4a,5,6,6a,11,11a,11b-decahydro-1
H-benzo[a]fluorene-7-carboxylate (
17): To a mixture of
15 (100 mg, 0.27 mmol, 1.0 equiv.) and ferric chloride hexahydrate (40 mg) was added TBHP (1.2 mL, 70% aqueous solution), CTAB (2 g) and 8 mL water. NaOH (40 mg, 1.0 mmol) was added after 3 h, and the mixture was heated at 90 °C for 48 h. After the completion of the reaction, dilute HCl and crushed ice were added. The mixture was extracted with EtOAc (2 × 50 mL), washed with saturated brine, dried over Na
2SO
4, then concentrated to dryness. The residue was dissolved in 2 mL DMF, then potassium carbonate (350 mg) and MeI (0.4 mL) were added. The solution was stirred overnight, quenched with 10 mL brine. The mixture was extracted with Et
2O (2 × 20 mL), washed with brine, dried over MgSO
4, and evaporated to dryness. The residue was purified by column chromatography on silica gel (petroleum ether/EtOAc = 8:1) to give
17 as colorless oil (34 mg, 33.3% yield).
1H NMR (400 MHz, CDCl
3) δ: 7.03 (s, 1H), 3.88 (s, 3H), 3.86 (s, 3H), 3.84 (s, 3H), 2.71 (dd,
J = 14.7, 6.2 Hz, 1H), 2.60–2.52 (m, 1H), 2.46 (dd,
J = 9.1, 3.2 Hz, 1H), 1.66 (s, 2H), 1.40 (d,
J = 6.2 Hz, 2H), 1.32 (s, 3H), 1.24 (s, 3H), 1.06 (s, 3H), 0.99 (d,
J = 6.7 Hz, 1H), 0.94 (s, 1H), 0.90 (d,
J = 2.8 Hz, 2H), 0.86 (s, 3H), 0.85 (s, 3H).
13C NMR (101 MHz, CDCl
3) δ: 168.3, 149.8, 148.8, 148.3, 137.1, 121.2, 111.5, 64.8, 60.4, 57.1, 56.0, 51.8, 48.1, 42.5, 40.2, 37.1, 36.4, 33.4, 33.1, 25.3, 21.1, 20.0, 19.5, 18.35, 16.3. (
Figure S6).
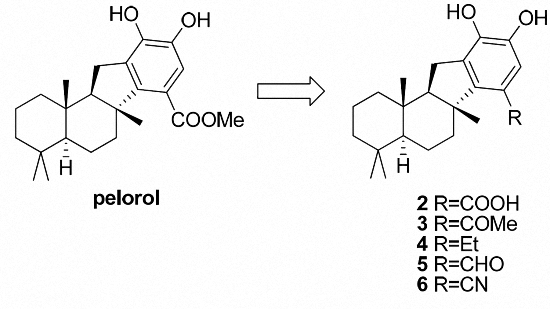
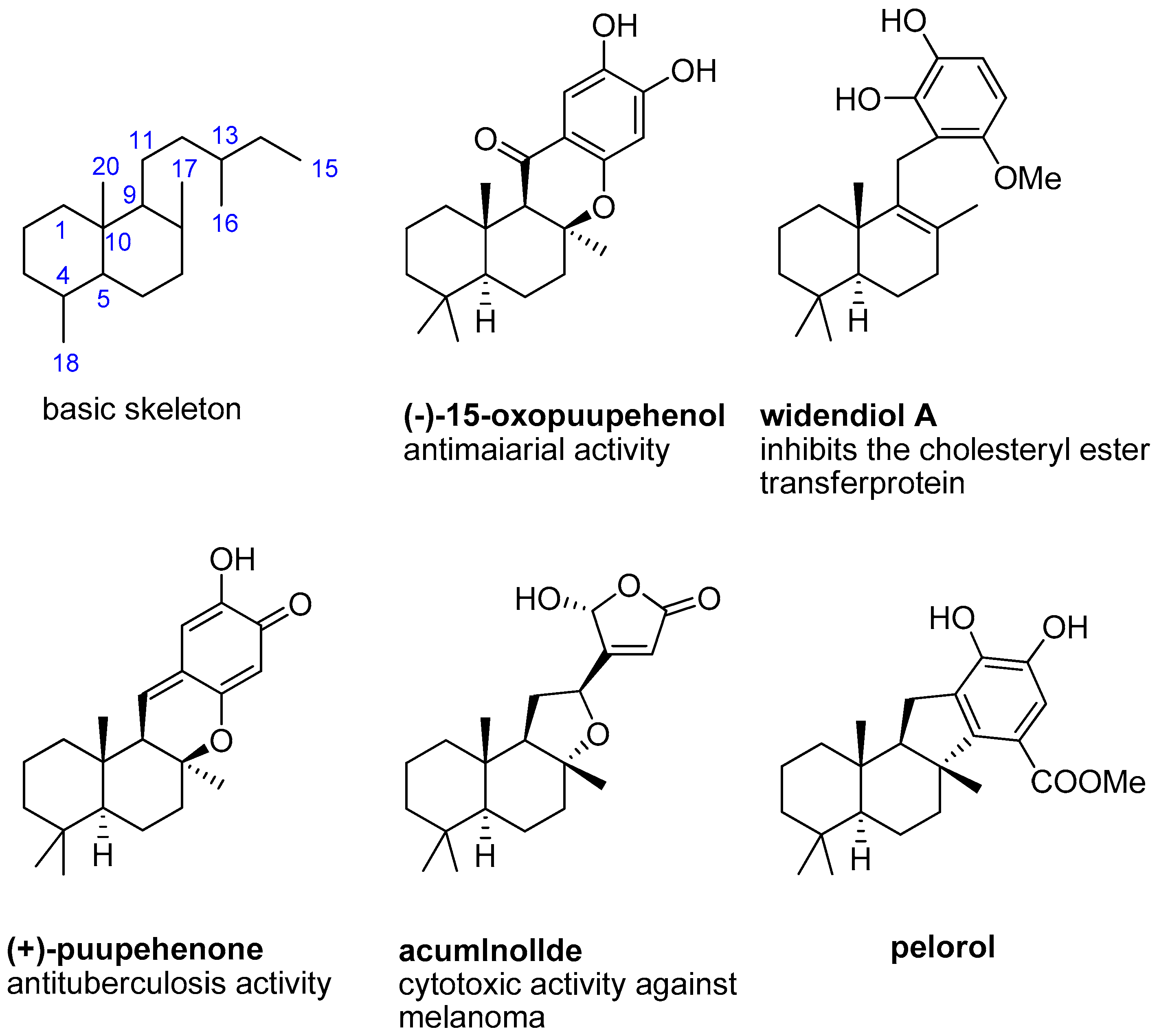
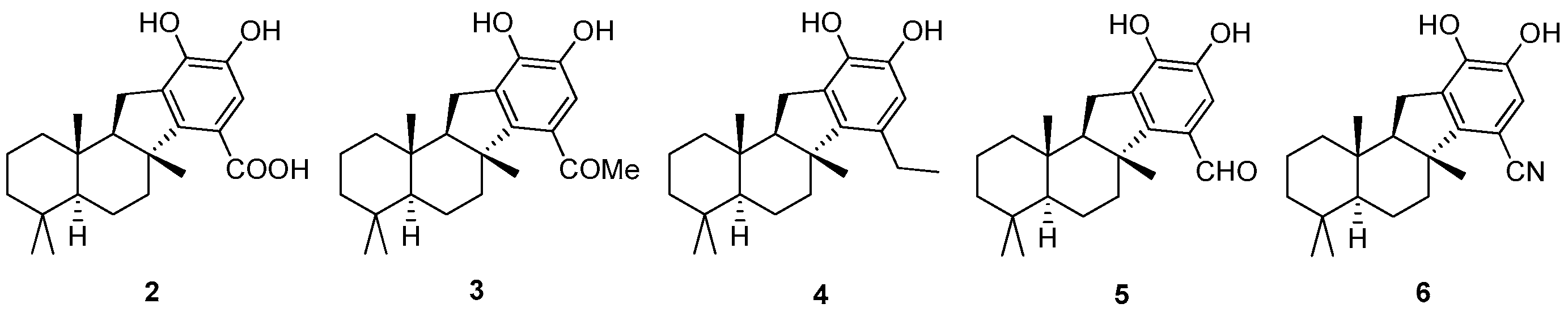
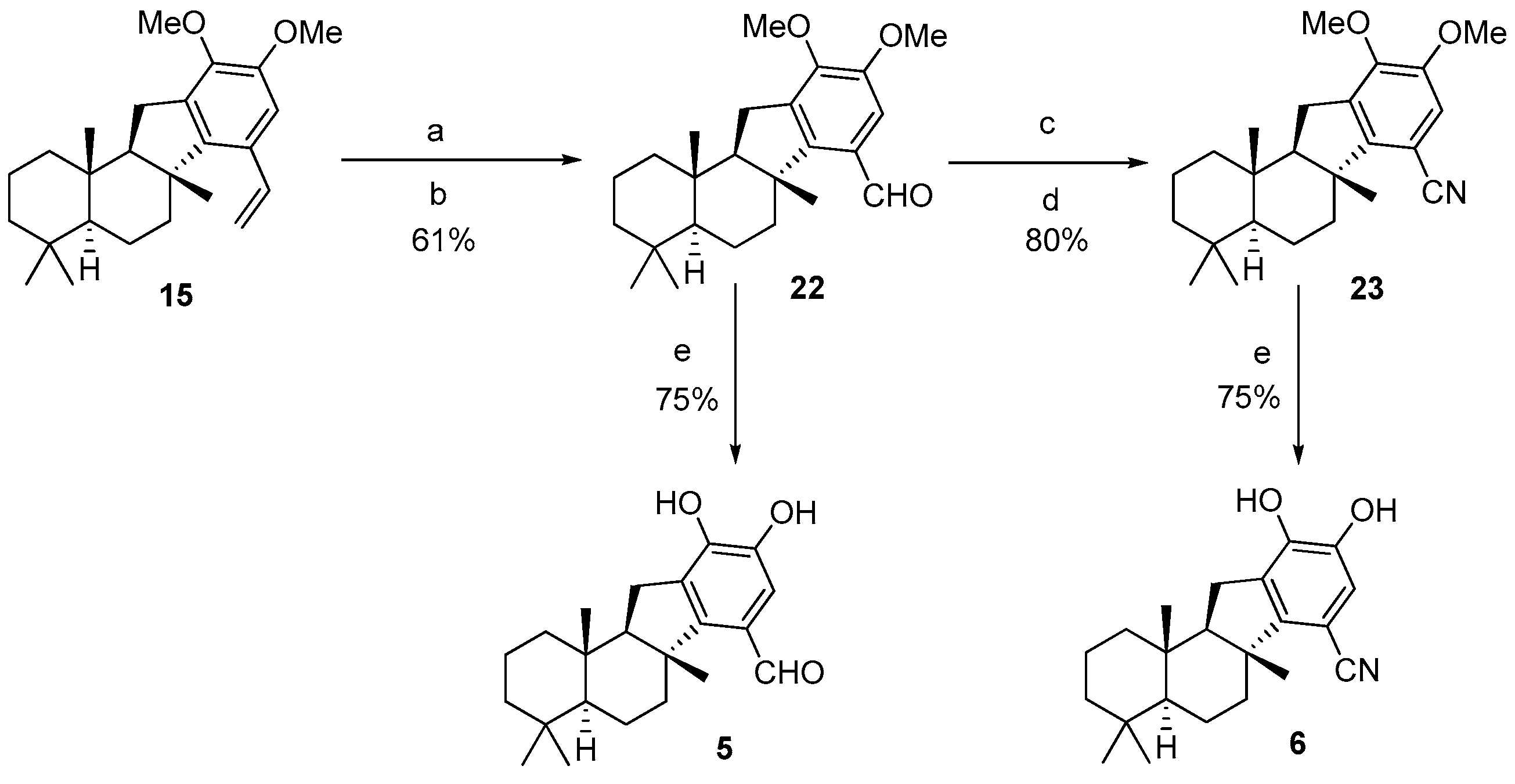
Pelorol (
1): To a solution of methyl ester
17 (11 mg, 0.028 mmol, 1.0 equiv.) in 4 mL CH
2Cl
2 was added BI
3 (129 mg, 0.336 mmol, 12 equiv.) in 2 mL CH
2Cl
2 dropwise at −78 °C under N
2. The mixture was stirred for an additional 0.5 h, then poured into H
2O and extracted with CH
2Cl
2 (2 × 50 mL). The combined CH
2Cl
2 extracts were dried over Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (petroleum ether/EtOAc = 3:1) to provide
1 as colorless oil (6.5 mg, 61.6% yield).
1H NMR (400 MHz, CDCl
3) δ: 7.09 (s, 1H), 5.59 (s, 2H), 3.83 (s, 3H), 2.61 (d,
J = 6.2 Hz, 1H), 2.54–2.47 (m, 2H), 1.66 (d,
J = 6.2 Hz, 2H), 1.58–1.53 (m, 2H), 1.41 (dd,
J = 8.8, 3.4 Hz, 2H), 1.25 (s, 3H), 1.23 (s, 3H), 1.05 (s, 3H), 0.98 (d,
J = 4.5 Hz, 1H), 0.96–0.93 (m, 1H), 0.85 (d,
J = 5.6 Hz, 6H).
13C NMR (101 MHz, CDCl
3) δ: 168.3, 149.4, 143.7, 140.6, 130.0, 118.0, 114.8, 65.2, 57.1, 51.7, 48.5, 42.5, 40.2, 37.2, 36.5, 33.3, 33.1, 24.3, 21.1, 19.9, 19.5, 18.4, 16.3. (
Figure S12). HRMS (ESI):
m/z [M-H]
− calcd. for [C
23H
32O
4]
−: 371.2228, found: 371.2220.
= −64.5 (
c = 0.41, MeOH).
(
4aS,
6aR,
11aR,
11bS)-9,10-diHydroxy-4,4,6a,11b-tetramethyl-2,3,4,4a,5,6,6a,11,11a,11b-decahydro-1
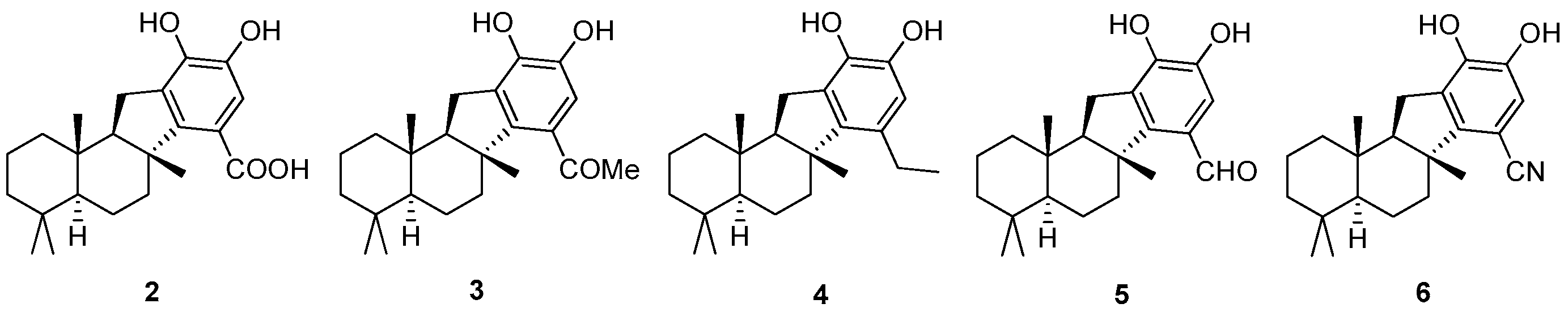
H-benzo[a]fluorene-7-carboxylic acid (
2): To a solution of
16 (13 mg, 0.033 mmol, 1.0 equiv.) in 8 mL CH
2Cl
2 was slowly added BI
3 (158 mg, 0.396 mmol, 12 equiv.) in 2 mL CH
2Cl
2 at −78 °C under N
2. The mixture was stirred for an additional 0.5 h, then poured into H
2O and extracted with CH
2Cl
2 (2 × 50 mL). The combined CH
2Cl
2 extracts were dried over Na
2SO
4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (petroleum ether/EtOAc = 1:1) to provide
2 as colorless oil (7.7 mg, 64.1% yield).
1H NMR (400 MHz, DMSO-
d6) δ: 12.15 (s, 1H), 8.98 (d,
J = 113.4 Hz, 2H), 6.91 (s, 1H), 2.63 (d,
J = 12.4 Hz, 1H), 2.34 (d,
J = 13.2 Hz, 1H), 1.97 (s, 1H), 1.54 (s, 1H), 1.49–1.45 (m, 2H), 1.39–1.33 (m, 2H), 1.21 (s, 3H), 1.16 (d,
J = 7.1 Hz, 2H), 1.11 (s, 3H), 0.99 (s, 3H), 0.90 (s, 1H), 0.81 (s, 6H).
13C NMR (101 MHz, DMSO-
d6) δ: 169.1, 147.2, 145.2, 142.9, 130.3, 117.8, 115.1, 65.1, 60.2, 57.1, 48.1, 42.5, 37.1, 36.7, 33.6, 33.2, 24.9, 21.5, 20.1, 19.5, 18.4, 16.5. (
Figure S13). HRMS (ESI):
m/z [M-H]
− calcd. for [C
22H
30O
4]
−: 357.2071, found: 357.2059.
= −11.61 (
c = 0.37, MeOH).
(
1R,
2R,
4aS,
8aS)-1-(5-Ethyl-2,3-diMethoxybenzyl)-2,5,5,8a-tetramethyldecahydronaphthalen-2-ol (
19) [
14]: Yield 73%, colorless oil.
1H NMR (400 MHz, CDCl
3) δ: 6.66 (s, 1H), 6.56 (s, 1H), 3.83 (s, 6H), 2.80 (s, 2H), 2.61–2.53 (m, 3H), 1.82 (s, 2H), 1.67 (s, 1H), 1.61 (s, 3H), 1.41 (d,
J = 6.6 Hz, 2H), 1.33 (d,
J = 15.8 Hz, 2H), 1.28 (s, 3H), 1.20 (t,
J = 7.5 Hz, 3H), 1.09 (d,
J = 3.4 Hz, 1H), 0.90 (s, 3H), 0.85 (s, 3H), 0.80 (s, 3H).
13C NMR (101 MHz, CDCl
3) δ: 152.3, 144.3, 140.0, 137.6, 122.0, 109.6, 73.7, 62.8, 60.56, 56.1, 55.6, 43.7, 41.8, 40.3, 39.4, 33.5, 33.3, 28.8, 25.3, 24.3, 21.6, 20.3, 18.6, 15.7, 15.4. (
Figure S7).
(
4aS,
6aR,
11aR,
11bS)-7-Ethyl-9,10-diMethoxy-4,4,6a,11b-tetramethyl-2,3,4,4a,5,6,6a,11,11a,11b-decahydro-1
H-benzo[a]fluorene (
20) [
14]: Yield 92%, colorless oil.
1H NMR (400 MHz, CDCl
3) δ: 6.53 (s, 1H), 3.85 (s, 3H), 3.84 (s, 3H), 2.76–2.68 (m, 2H), 2.59 (dd,
J = 30.3, 10.4 Hz, 2H), 2.41 (d,
J = 11.8 Hz, 1H), 1.81 (s, 1H), 1.79–1.73 (m, 3H), 1.61 (d,
J = 13.1 Hz, 3H), 1.44 (d,
J = 10.6 Hz, 3H), 1.29–1.25 (m, 3H), 1.13 (s, 3H), 1.07 (s, 3H), 1.00 (s, 1H), 0.90 (s, 6H).
13C NMR (101 MHz, CDCl
3) δ: 150.4, 144.8, 143.6, 135.8, 133.7, 111.1, 64.3, 60.3, 57.1, 56.0, 47.9, 42.6, 40.2, 39.3, 37.1, 33.4, 33.1, 25.3, 24.7, 21.4, 21.2, 19.7, 18.4, 16.2, 16.1. (
Figure S8).
1-((
4aS,
6aR,
11aR,
11bS)-9,10-diMethoxy-4,4,6a,11b-tetramet-hyl-2,3,4,4a,5,6,6a,11,11a,11b-decahydro-1
H-benzo[a]fluoren-7-yl)ethanone (
21) [
14]: Yield 27%, white solid.
1H NMR (400 MHz, CDCl
3) δ: 6.81 (s, 1H), 3.88 (s, 3H), 3.86 (s, 3H), 3.64 (d,
J = 4.3 Hz, 1H), 2.71 (dd,
J = 14.7, 6.2 Hz, 1H), 2.58 (d,
J = 13.0 Hz, 1H), 2.52 (s, 3H), 2.32 (d,
J = 12.5 Hz, 1H), 1.62 (d,
J = 5.9 Hz, 6H), 1.40 (d,
J = 9.1 Hz, 2H), 1.25 (s, 3H), 1.14 (s, 1H), 1.05 (s, 3H), 0.92 (s, 1H), 0.85 (d,
J = 5.6 Hz, 6H).
13C NMR (101 MHz, CDCl
3) δ: 201.9, 149.6, 147.9, 147.6, 137.4, 130.5, 110.0, 65.0, 60.5, 57.2, 56.2, 48.0, 42.5, 40.2, 37.1, 36.4, 33.4, 33.1, 30.3, 25.2, 21.1, 20.6, 19.4, 18.4, 16.3. (
Figure S9).
1-((
4aS,
6aR,
11aR,
11bS)-9,10-diHydroxy-4,4,6a,11b-tetramethyl-2,3,4,4a,5,6,6a,11,11a,11b-decahydro-1
H-benzo[a]fluoren-7-yl)ethanone (
3): To a solution of
21 (34 mg, 0.088 mmol, 1.0 equiv.) in 5 mL CH
2Cl
2 was slowly added BI
3 (413 mg, 1.05 mmol, 12 equiv.) in 2 mL CH
2Cl
2 at −78 °C under N
2. The mixture was stirred for an additional 0.5 h, then poured into H
2O and extracted with CH
2Cl
2 (2 × 30 mL). The combined CH
2Cl
2 extracts were dried over Na
2SO
4, filtered and concentrated. The residue was purified by column chromatography on silica gel (petroleum ether/EtOAc = 4:1) to provide
3 as colorless oil (23.5 mg, 75% yield).
1H NMR (400 MHz, CDCl
3) δ: 6.94 (s, 1H), 5.80 (d,
J = 157.3 Hz, 2H), 2.61 (dd,
J = 14.2, 6.3 Hz, 1H), 2.51 (d,
J = 12.9 Hz, 1H), 2.46 (s, 3H), 2.41 (d,
J = 12.6 Hz, 1H), 1.64 (d,
J = 5.9 Hz, 2H), 1.53 (dd,
J = 9.7, 3.0 Hz, 2H), 1.64 (d,
J = 5.9 Hz, 2H), 1.53 (dd,
J = 9.7, 3.0 Hz, 2H), 1.42 (d,
J = 4.8 Hz, 2H), 1.26 (s, 6H), 1.23 (s, 2H), 1.04 (s, 3H), 0.84 (d,
J = 6.3 Hz, 6H).
13C NMR (101 MHz, CDCl
3) δ: 201.5, 148.3, 143.3, 140.8, 130.3, 127.3, 113.7, 65.3, 57.2, 48.5, 42.5, 40.3, 37.1, 36.3, 33.4, 33.1, 29.7, 24.3, 21.1, 20.3, 19.5, 18.4, 16.3. (
Figure S14). HRMS (ESI):
m/z [M-H]
− calcd. for [C
23H
32O
3]
−: 355.2279, found: 355.2270.
= −18.0 (
c = 0.36, MeOH).
(
4aS,
6aR,
11aR,
11bS)-7-Ethyl-4,4,6a,11b-tetramethyl-2,3,4,4a,5,6,6a,11,11a,11b-decahydro-1
H-benzo [a]fluorene-9,10-diol (
4): To a solution of
20 (34 mg, 0.092 mmol, 1.0 equiv.) in 5 mL CH
2Cl
2 was slowly added BI
3 (431 mg, 1.10 mmol, 12 equiv.) in 2 mL CH
2Cl
2 at −78 °C under N
2. The mixture was stirred for an additional 0.5 h, then poured into H
2O and extracted with CH
2Cl
2 (2 × 30 mL). The combined CH
2Cl
2 extracts were dried over Na
2SO
4, filtered and concentrated. The residue was purified by column chromatography on silica gel (petroleum ether/EtOAc = 4:1) to provide
4 as colorless oil (29.9 mg, 95% yield).
1H NMR (400 MHz, CDCl
3) δ: 6.49 (s, 1H), 2.61 (s, 2H), 2.48 (s, 2H), 2.35 (s, 1H), 1.77 (s, 2H), 1.69 (s, 1H), 1.60–1.53 (m, 3H), 1.43 (d,
J = 4.1 Hz, 2H), 1.25 (s, 3H), 1.18 (s, 3H), 1.08 (s, 3H), 1.03 (s, 3H), 0.87 (s, 6H).
13C NMR (101 MHz, CDCl
3) δ: 144.8, 141.4, 137.9, 130.9, 128.7, 113.7, 64.6, 57.1, 48.1, 42.5, 40.1, 39.3, 37.1, 33.4, 33.1, 24.5, 24.2, 21.4, 21.1, 19.7, 18.4, 16.2, 15.9. (
Figure S15). HRMS (ESI):
m/z [M-H]
− calcd. for [C
23H
34O
2]
−: 341.2486, found: 341.2481.
= −161.3 (
c = 0.54, MeOH).
(
4aS,
6aR,
11aR,
11bS)-9,10-diMethoxy-4,4,6a,11b-tetramethyl-2,3,4,4a,5,6,6a,11,11a,11b-decahydro-1
H-benzo[a]fluorene-7-carbaldehyde (
22): To compound
15 (60 mg, 0.16 mmol, 1.0 equiv.) in 4 mL solvent (THF:acetone:H
2O = 2:1:1) at room temperature was added NMO (0.7 mL) and H
4K
2O
6Os (1 mg). The reaction mixture was stirred at room temperature for 12 h. Then the mixture was quenched with 5 mL saturated NaHS
2O
3, extracted with CH
2Cl
2 (2 × 30 mL). The combined CH
2Cl
2 extracts were dried over Na
2SO
4, filtered and concentrated. The residue was dissolved in 10 mL CH
2Cl
2, then NaIO
4 (68.4 mg in 2 mL H
2O) was added at room temperature for 3 h. The mixture was extracted with EtOAc (2 × 30 mL), washed with saturated brine (50 mL), dried over Na
2SO
4, filtered and concentrated. The residue was purified by column chromatography on silica gel (petroleum ether/EtOAc = 8:1) to provide
22 as colorless oil (36.8 mg, 61% yield).
1H NMR (400 MHz, CDCl
3) δ: 10.32 (s, 1H), 7.21 (s, 1H), 3.88 (s, 3H), 3.82 (s, 3H), 2.70 (d,
J = 6.2 Hz, 1H), 2.53 (s, 1H), 2.42 (d,
J = 12.1 Hz, 1H), 1.87 (s, 1H), 1.73 (d,
J = 12.8 Hz, 2H), 1.63 (d,
J = 3.3 Hz, 1H), 1.56 (s, 1H), 1.52 (s, 1H), 1.46–1.40 (m, 1H), 1.37 (d,
J = 13.3 Hz, 2H), 1.19 (s, 2H), 1.14 (s, 1H), 1.02 (s, 3H), 0.96 (s, 2H), 0.83 (s, 6H).
13C NMR (101 MHz, CDCl
3) δ: 189.9, 153.2, 150.7, 136.1, 127.0, 108.1, 63.9, 60.4, 56.7, 55.9, 48.7, 42.4, 40.8, 40.1, 37.1, 33.3, 33.0, 25.5, 23.7, 21.1, 19.8, 18.3, 16.1. (
Figure S10).
(
4aS,
6aR,
11aR,
11bS)-9,10-diMethoxy-4,4,6a,11b-tetramethyl-2,3,4,4a,5,6,6a,11,11a,11b-decahydro-1
H-benzo[a]fluorene-7-carbonitrile (
23): To a solution of
22 (49 mg, 0.13 mmol, 1.0 equiv.) and Et
3N (25.6 mg, 0.14 mmol, 1.1 equiv.) in 2 mL CH
2Cl
2 was slowly added NH
2OH·HCl (10 mg, 1.4 mmol, 10 equiv.) at 0 °C. The mixture was stirred for an additional 4 h at room temperature. Then Cl
3COCOOCCl
3 (43 mg, 0.15 mmol, 1.2 equiv.) was added, releasing a large number of bubbles. The reaction mixture was stirred at room temperature for 12 h, then extracted with CH
2Cl
2 (2 × 50 mL). The combined CH
2Cl
2 extracts were dried over Na
2SO
4, filtered and concentrated. The residue was purified by column chromatography on silica gel (petroleum ether/EtOAc = 8:1) to provide
23 as colorless oil (38.7 mg, 79.6% yield).
1H NMR (400 MHz, CDCl
3) δ: 6.86 (s, 1H), 3.91 (s, 3H), 3.83 (s, 3H), 2.71 (d,
J = 6.2 Hz, 1H), 2.62 (s, 1H), 2.53 (s, 1H), 1.82–1.76 (m, 1H), 1.72 (d,
J = 6.5 Hz, 2H), 1.61 (s, 2H), 1.47 (s, 2H), 1.25 (d,
J = 5.4 Hz, 2H), 1.14 (s, 3H), 1.04 (s, 3H), 0.99 (s, 1H), 0.95 (d,
J = 10.3 Hz, 1H), 0.88 (s, 6H).
13C NMR (101 MHz, CDCl
3) δ: 151.8, 149.5, 148.8, 135.5, 117.0, 112.9, 98.2, 63.1, 59.5, 56.2, 55.2, 46.3, 41.5, 39.1, 36.1, 35.8, 32.4, 32.1, 24.4, 20.8, 20.1, 18.3, 17.2, 15.1. (
Figure S11).
(
4aS,
6aR,
11aR,
11bS)-9,10-diHydroxy-4,4,6a,11b-tetramethyl-2,3,4,4a,5,6,6a,11,11a,11b-decahydro-1
H-benzo[a]fluorene-7-carbaldehyde (
5): To a solution of
22 (34 mg, 0.092 mmol, 1.0 equiv.) in 5 mL CH
2Cl
2 was slowly added BI
3 ( 431 mg, 1.10 mmol, 12 equiv.) in 2 mL CH
2Cl
2 at −78 °C under N
2. The mixture was stirred for an additional 0.5 h, then poured into H
2O and extracted with CH
2Cl
2 (2 × 30 mL). The combined CH
2Cl
2 extracts were dried over Na
2SO
4, filtered and concentrated. The residue was purified by column chromatography on silica gel (petroleum ether/EtOAc = 4:1) to provide
5 as colorless oil (23.5 mg, 75% yield).
1H NMR (400 MHz, CDCl
3) δ: 10.26 (s, 1H), 7.37 (s, 1H), 6.80 (s, 1H), 6.14 (s, 1H), 2.72 (dd,
J = 14.7, 5.9 Hz, 1H), 2.54 (d,
J = 13.4 Hz, 2H), 1.92 (s, 1H), 1.76 (s, 3H), 1.58 (d,
J = 12.1 Hz, 1H), 1.43 (d,
J = 11.4 Hz, 2H), 1.26 (s, 2H), 1.22 (s, 3H), 1.07 (s, 3H), 1.00 (d,
J = 13.8 Hz, 2H), 0.88 (s, 6H).
13C NMR (101 MHz, CDCl
3) δ: 190.5, 154.5, 147.3, 142.0, 129.2, 124.3, 111.2, 64.2, 56.8, 49.0, 42.4, 40.7, 40.1, 37.1, 33.3, 33.1, 24.6, 23.8, 21.1, 19.8, 18.3, 16.1. (
Figure S16). HRMS (ESI):
m/z [M-H]
− calcd. for [C
22H
30O
3]
−: 341.2122, found: 341.2107.
= 48.6 (
c = 0.23, MeOH).
(
4aS,
6aR,
11aR,
11bS)-9,10-diHydroxy-4,4,6a,11b-tetramethyl-2,3,4,4a,5,6,6a,11,11a,11b-decahydro-1
H-benzo[a]fluorene-7-carbonitrile (
6): To a solution of
23 (11 mg, 0.03 mmol, 1.0 equiv.) in 5 mL CH
2Cl
2 was slowly added BI
3 (140 mg, 0.36 mmol, 12 equiv.) in 1 mL CH
2Cl
2 at −78 °C under N
2. The mixture was stirred for an additional 0.5 h, then poured into H
2O. The solution was extracted with CH
2Cl
2 (2 × 30 mL). The combined CH
2Cl
2 extracts were dried over Na
2SO
4, filtered and concentrated. The residue was purified by column chromatography on silica gel (petroleum ether/EtOAc = 3:1) to provide
6 as colorless oil (7.6 mg, 75% yield).
1H NMR (400 MHz, CDCl
3) δ: 6.89 (s, 1H), 4.13 (d,
J = 7.2 Hz, 2H), 2.60 (dd,
J = 20.5, 8.7 Hz, 2H), 2.45 (s, 1H), 2.06 (s, 1H), 1.73 (d,
J = 12.9 Hz, 3H), 1.40 (s, 1H), 1.25 (s, 3H), 1.10 (s, 2H), 1.02 (s, 2H), 0.86 (s, 9H).
13C NMR (101 MHz, CDCl
3) δ: 152.9, 145.6, 141.8, 129.8, 118.3, 116.7, 95.3, 64.4, 57.2, 47.6, 42.5, 40.1, 37.0, 36.8, 33.4, 33.1, 24.5, 21.9, 21.1, 19.4, 18.3, 16.2. (
Figure S17). HRMS (ESI):
m/z [M-H]
− calcd. for [C
22H
29NO
2]
−: 338.2126, found: 338.2119.
= 10.2 (
c = 0.53, MeOH).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}