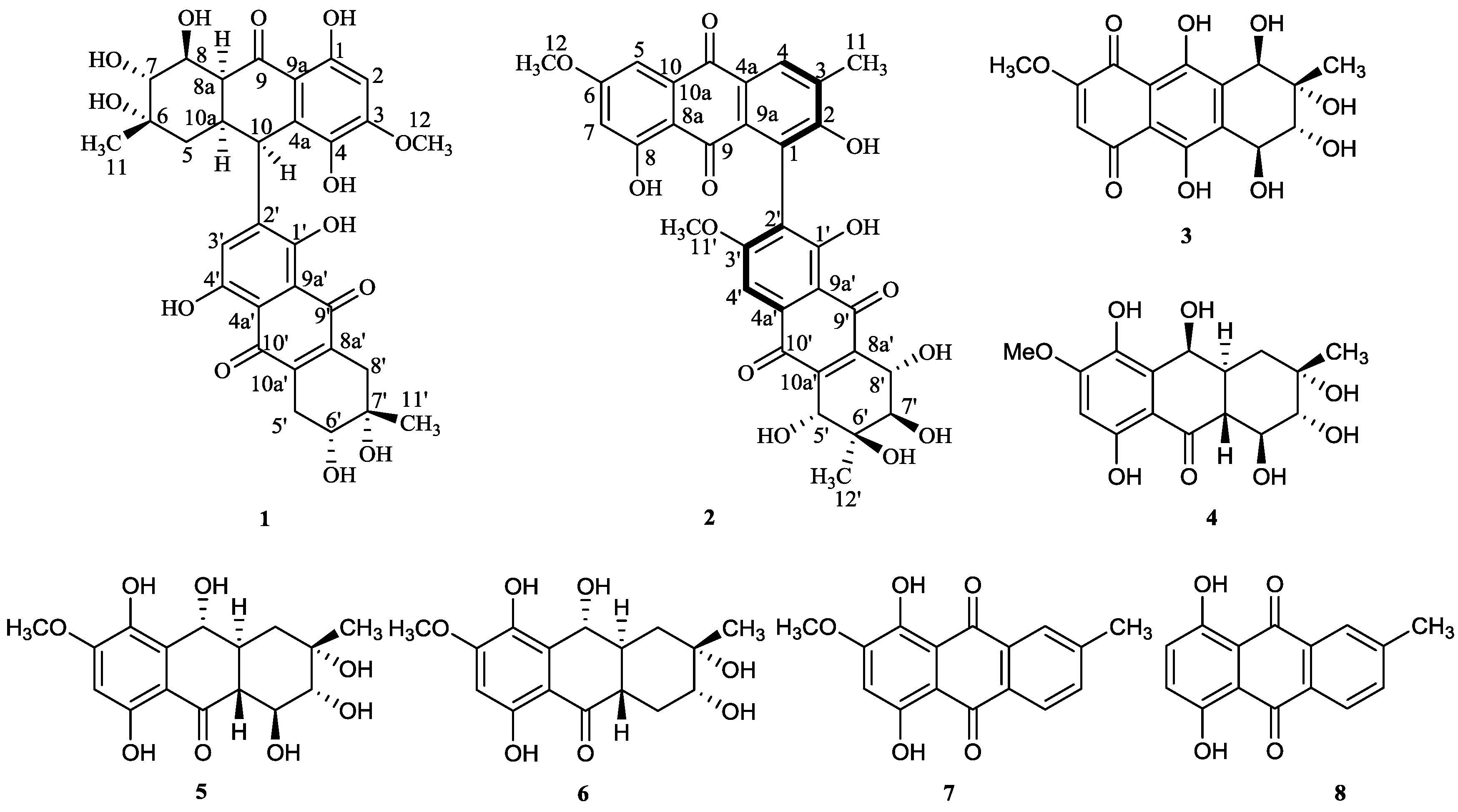
Alterporriol-Type Dimers from the Mangrove Endophytic Fungus, Alternaria sp. (SK11), and Their MptpB Inhibitions


 and
and
Abstract
:1. Introduction
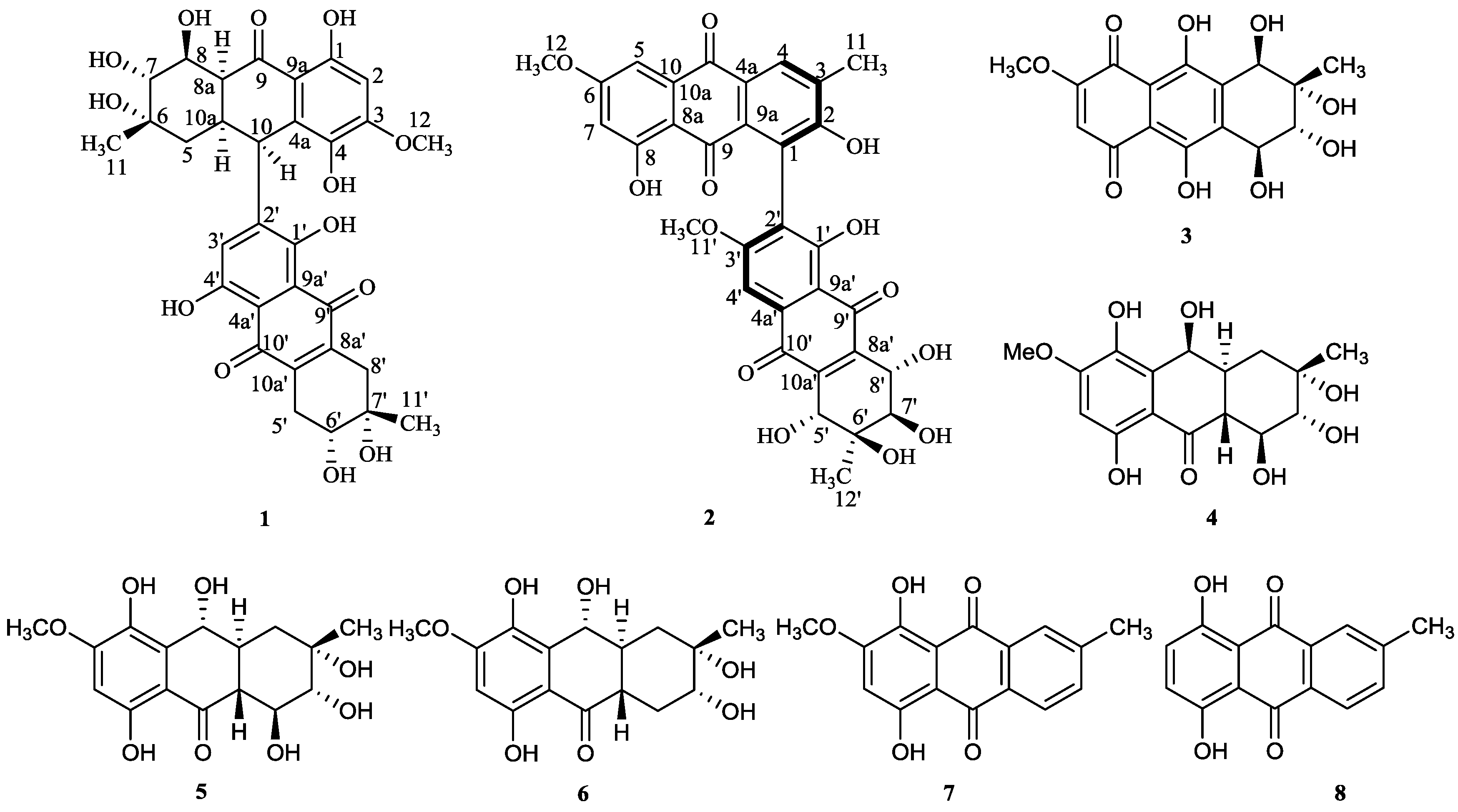
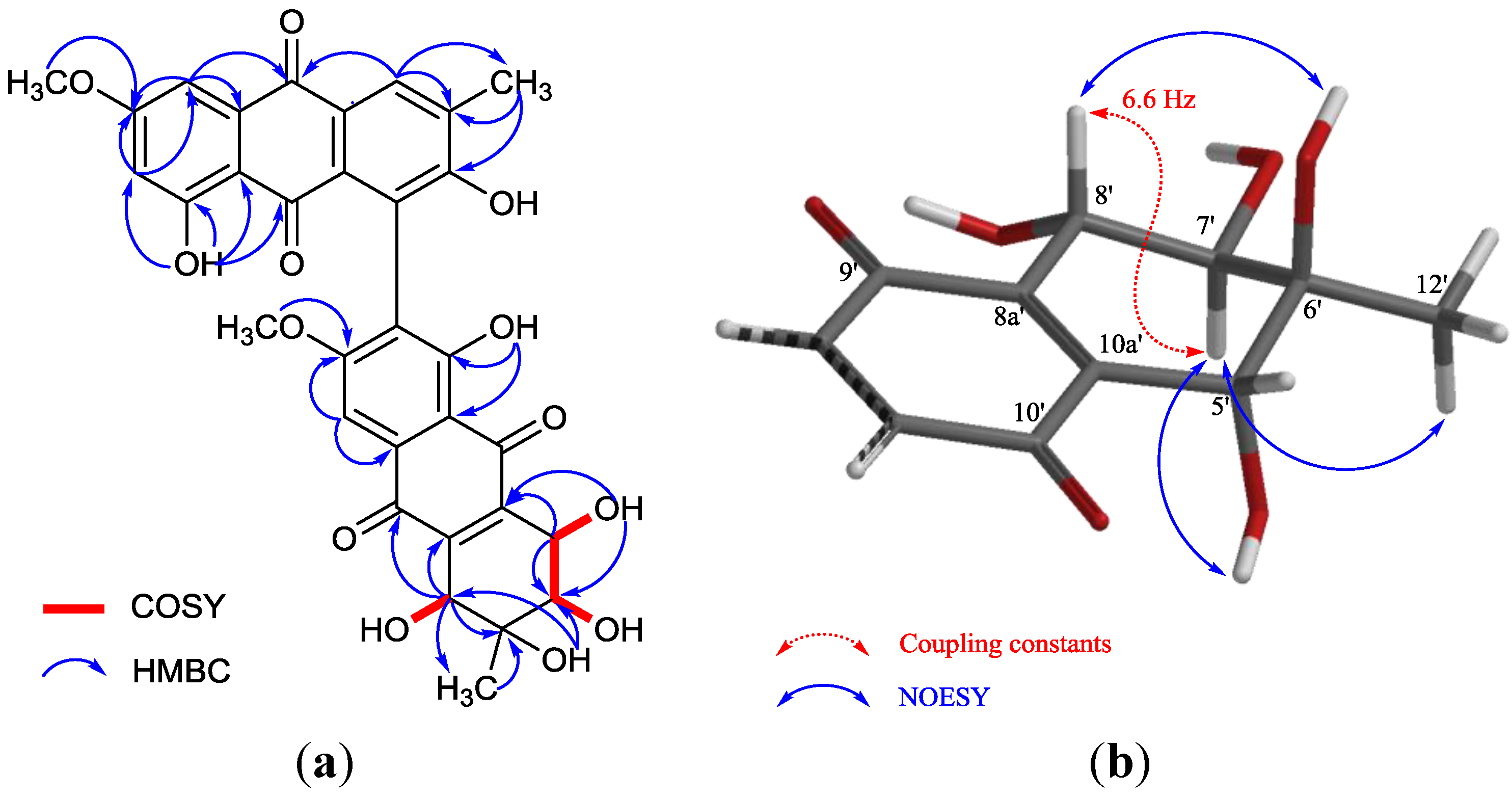
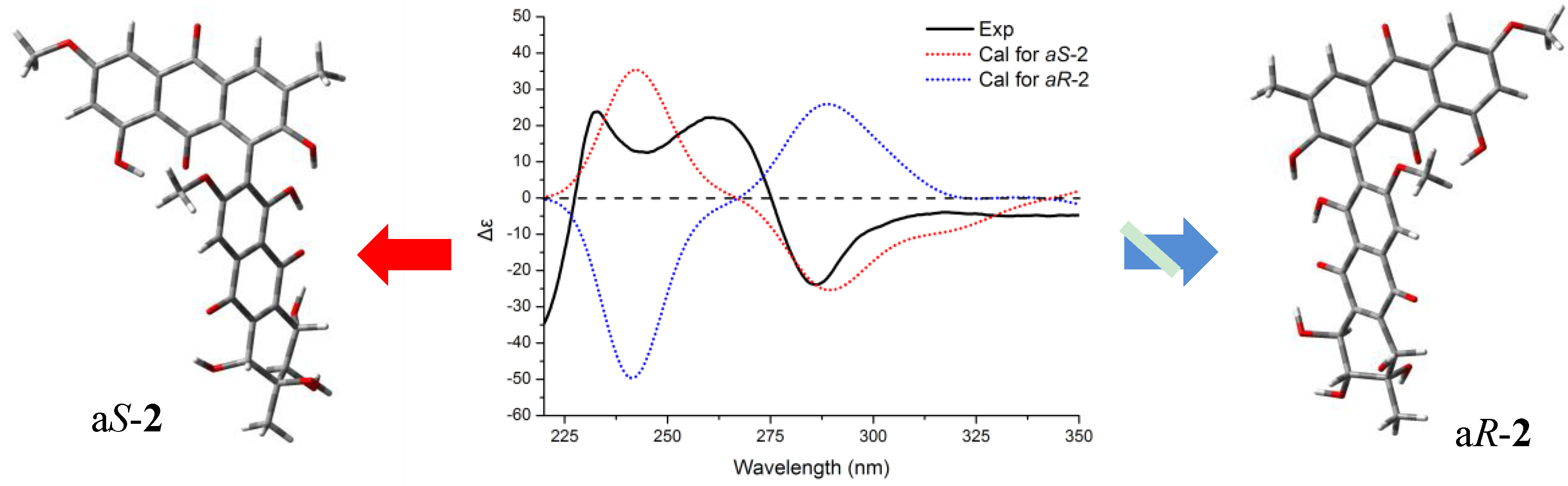
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δC | δH | HMBC b |
|---|---|---|---|
| 1 | 156.9, C | ||
| 2 | 99.5, CH | 6.73, s | 1, 3, 4, 9a |
| 3 | 157.0, CH | ||
| 4 | 136.5, C | ||
| 4a | 125.5,C | ||
| 5ax 5eq | 42.0, CH2 | 1.37, dd (12.8, 13.5); 1.65, dd (12.8, 3.8) | 10, 10a |
| 6 | 71.7, C | ||
| 7 | 75.6, CH | 3.20, dd (10.0, 4.9) | 8 |
| 8 | 73.1, CH | 3.45, ddd (10.0, 4.9, 4.6) | 7 |
| 8a | 46.0, CH | 2.99, ddd (4.6, 4.1, 1.2) | 5, 7, 8, 9, 10 |
| 9 | 206.4, C | ||
| 9a | 110.2, C | ||
| 10 | 37.1, C | 4.60, brs | 4, 4a, 5, 8a, 9a, 10a, 2′, 3′, 4′ |
| 10a | 35.1, C | 2.64, m | 11 |
| 11 | 26.7, CH3 | 1.08, s | 5, 6, 7 |
| 12 | 56.7, CH3 | 3.89, s | 3 |
| 1-OH | 12.46, s | 1, 2, 9a | |
| 4-OH | 8.57, s | 3, 4, 4a | |
| 6-OH | 4.06, s | ||
| 7-OH | 4.62, d (4.9) | ||
| 8-OH | 4.33, d (4.9) | ||
| 1′ | 161.8, C | ||
| 2′ | 146.3, C | ||
| 3′ | 127.6, CH | 6.34, s | 10, 1′, 4a′ |
| 4′ | 162.5, C | ||
| 4a′ | 110.5, C | ||
| 5′ax 5′eq | 30.2, CH2 | 2.65, m 2.80, dd (14.4, 5.0) | 6′, 10′ c, 10a′ |
| 6′ | 70.6, CH | 3.61, dt (11.8, 5.0) | 11′ c |
| 7′ | 69.4, C | ||
| 8′ | 35.9, CH2 | 2.74, d (14.1) 2.58, m | 7′, 8a′, 11′ |
| 8a′ | 142.3, C | ||
| 9′ | 180.9, C | ||
| 9a′ | 111.5, C | ||
| 10′ | 181.9, C | ||
| 10a′ | 142.8,C | ||
| 11′ | 25.7, CH3 | 1.18, s | 6′, 8′ |
| 1′-OH | 13.17, s | 1′, 2′, 9′ | |
| 4′-OH | 12.42, s | 3′, 4′, 4a′,10′ | |
| 6′-OH | 4.82, d (5.0) | ||
| 7′-OH | 4.49, s |
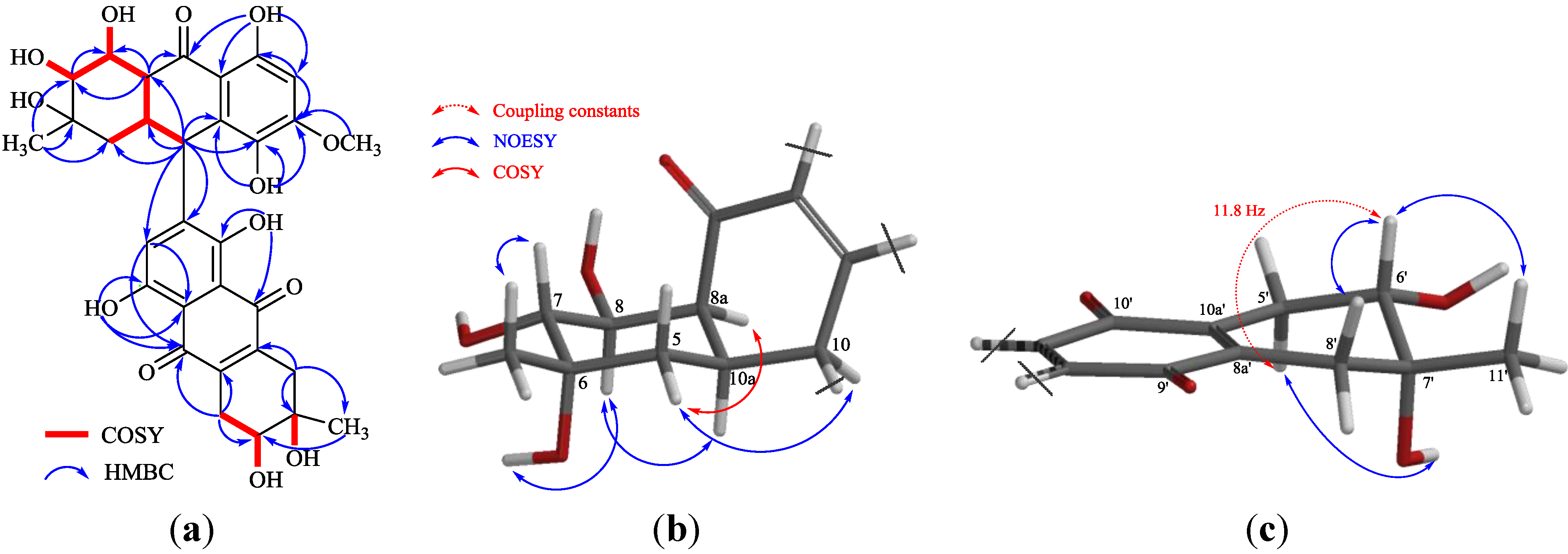
| Position | δC | δH | HMBC b |
|---|---|---|---|
| 1 | 125.5, C | ||
| 2 | 159.1, C | ||
| 3 | 131.0, C | ||
| 4 | 129.7, CH | 8.05, s | 2, 3, 10 c, 11 |
| 4a | 131.6, C | ||
| 5 | 106.6, CH | 7.16, d (2.3) | 6, 8a, 9, 10, 10a |
| 6 | 165.6, C | ||
| 7 | 106.9, C | 6.76, d (2.2) | 5, 6, 8a, 9 |
| 8 | 164.5, C | ||
| 8a | 110.3, C | ||
| 9 | 187.6, C | ||
| 9a | 106.1, C | ||
| 10 | 180.8, C | ||
| 10a | 134.5, C | ||
| 11 | 17.3, CH3 | 2.31, s | 2, 3, 9 |
| 12 | 56.8, CH3 | 3.89, s | 6 |
| 2-OH | 9.50, brs | ||
| 8-OH | 12.48, s | 7, 8, 8a, 9 | |
| 1′ | 163.9, C | ||
| 2′ | 126.0, C | ||
| 3′ | 164.2, C | ||
| 4′ | 104.1, CH | 6.96, s | 3′, 4a′, 9a′ |
| 4a′ | 123.0, C | ||
| 5′ | 68.4, CH | 4.05, s | 6′, 8a′, 10′,10a′, 12′ |
| 6′ | 73.0, C | ||
| 7′ | 68.2, CH | 3.57, dd (5.6, 6.6) | 8′ |
| 8′ | 73.8, CH | 4.48, m | 7′, 8a′, 10a′ |
| 8a′ | 142.3, C | ||
| 9′ | 189.1, C | ||
| 9a′ | 109.7, C | ||
| 10′ | 184.4, C | ||
| 10a′ | 143.6, C | ||
| 11′ | 56.3, CH3 | 3.71, s | 3′ |
| 12′ | 22.3, CH3 | 1.12, s | 5′, 6′ |
| 1′-OH | 13.09, s | 1′, 4′, 9a′ | |
| 5′-OH | 5.75, brs | ||
| 6′-OH | 4.38, s | 5′, 6′, 7′ | |
| 7′-OH | 4.86, d (5.6) | ||
| 8′-OH | 5.05, brs | 6′, 7′, 8a′ |
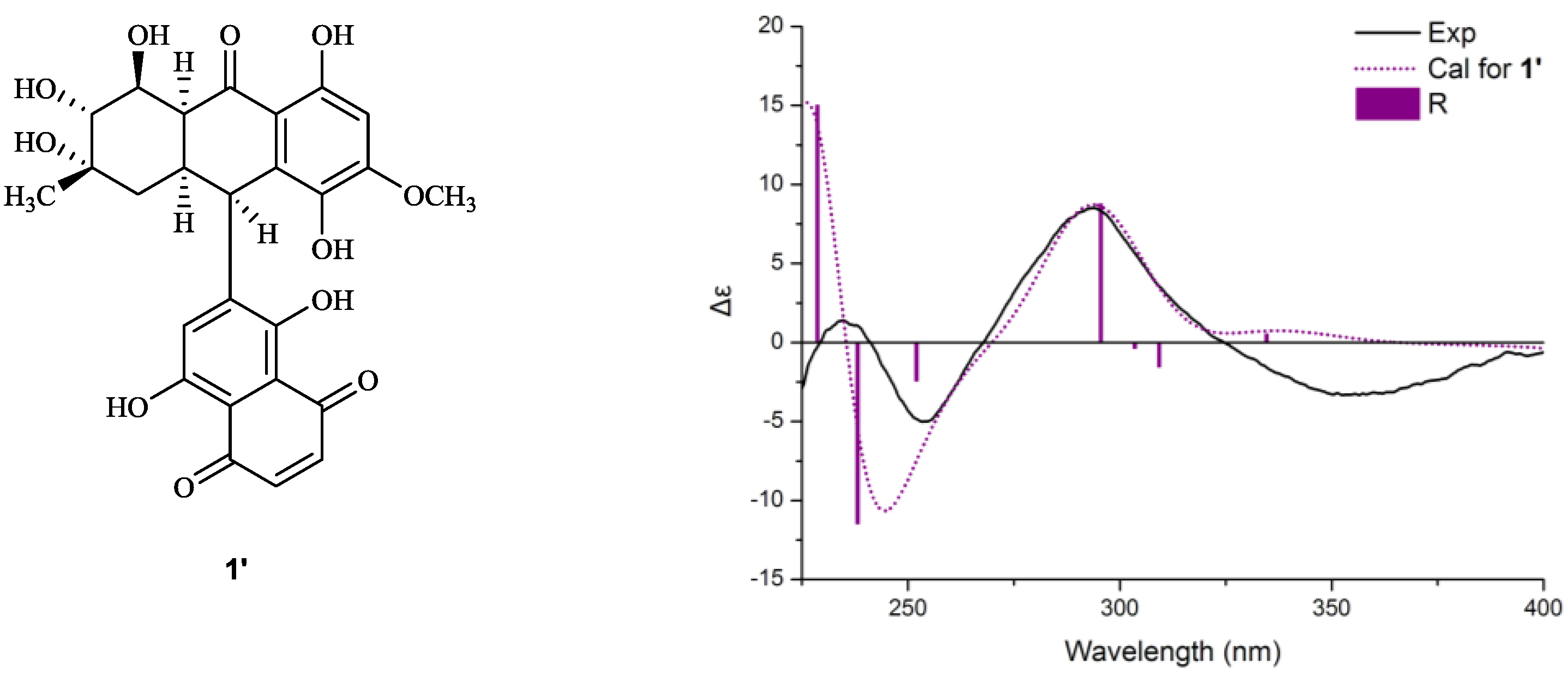
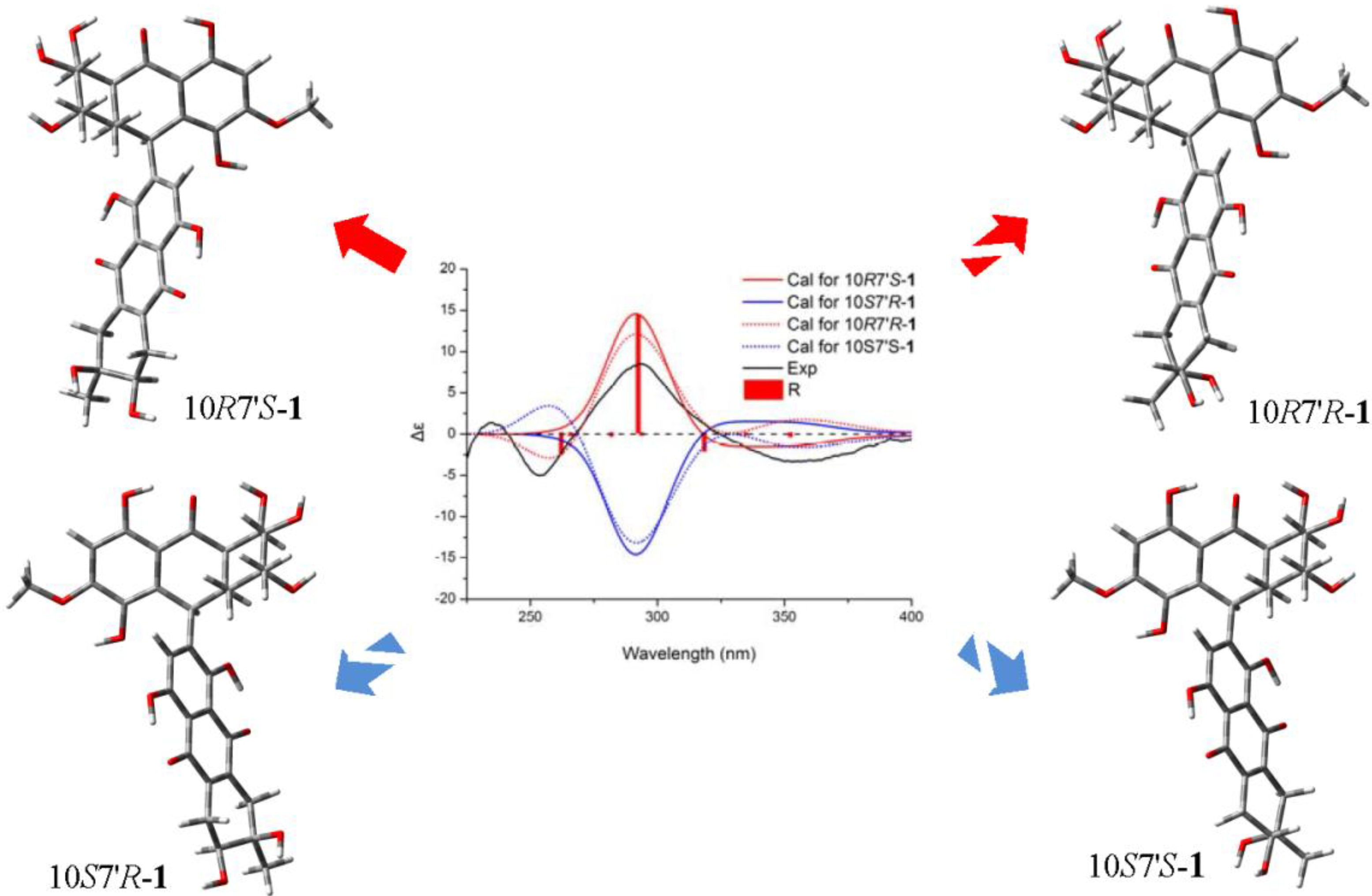
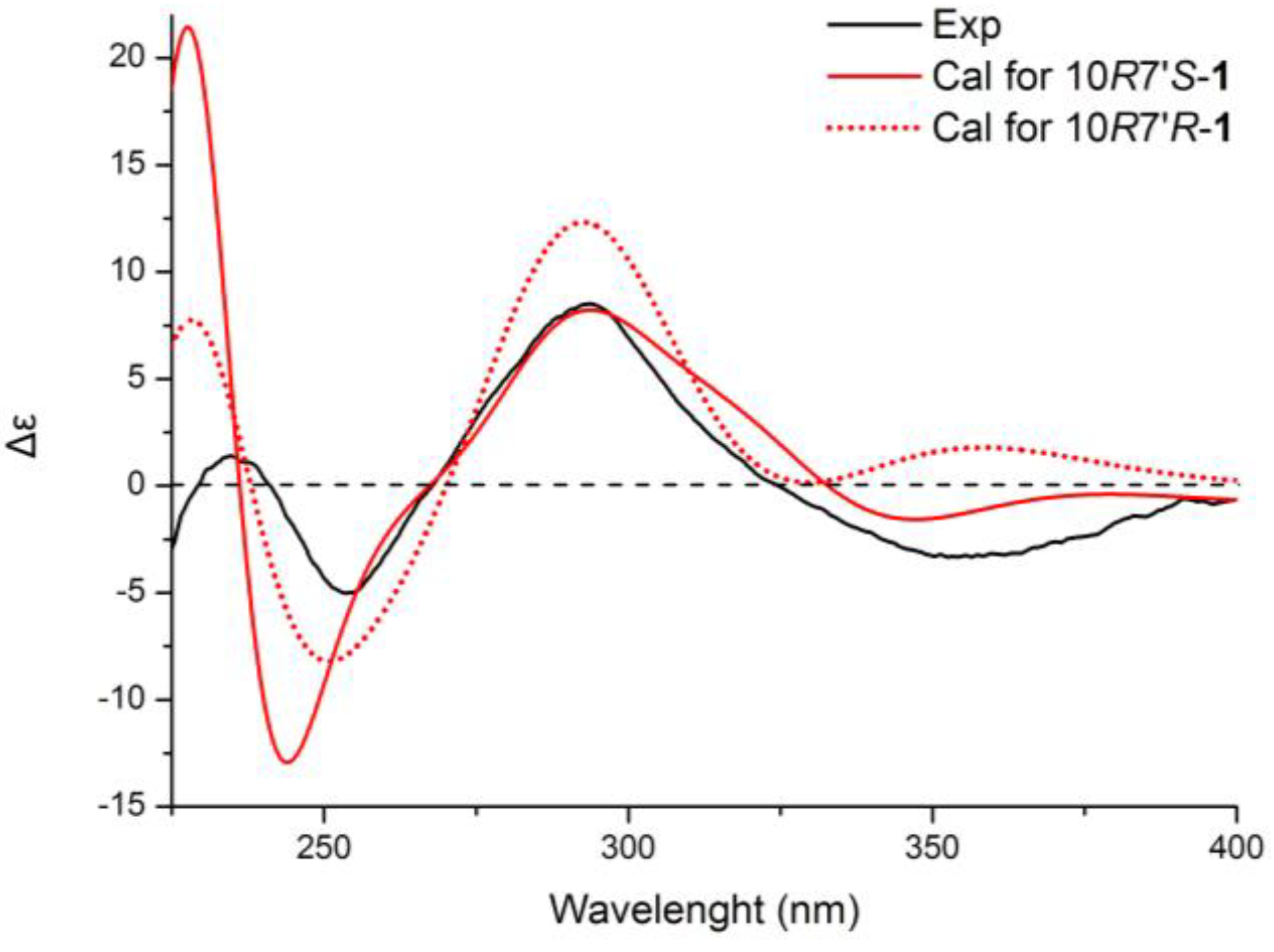
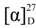 +75° (c 0.02, EtOH),
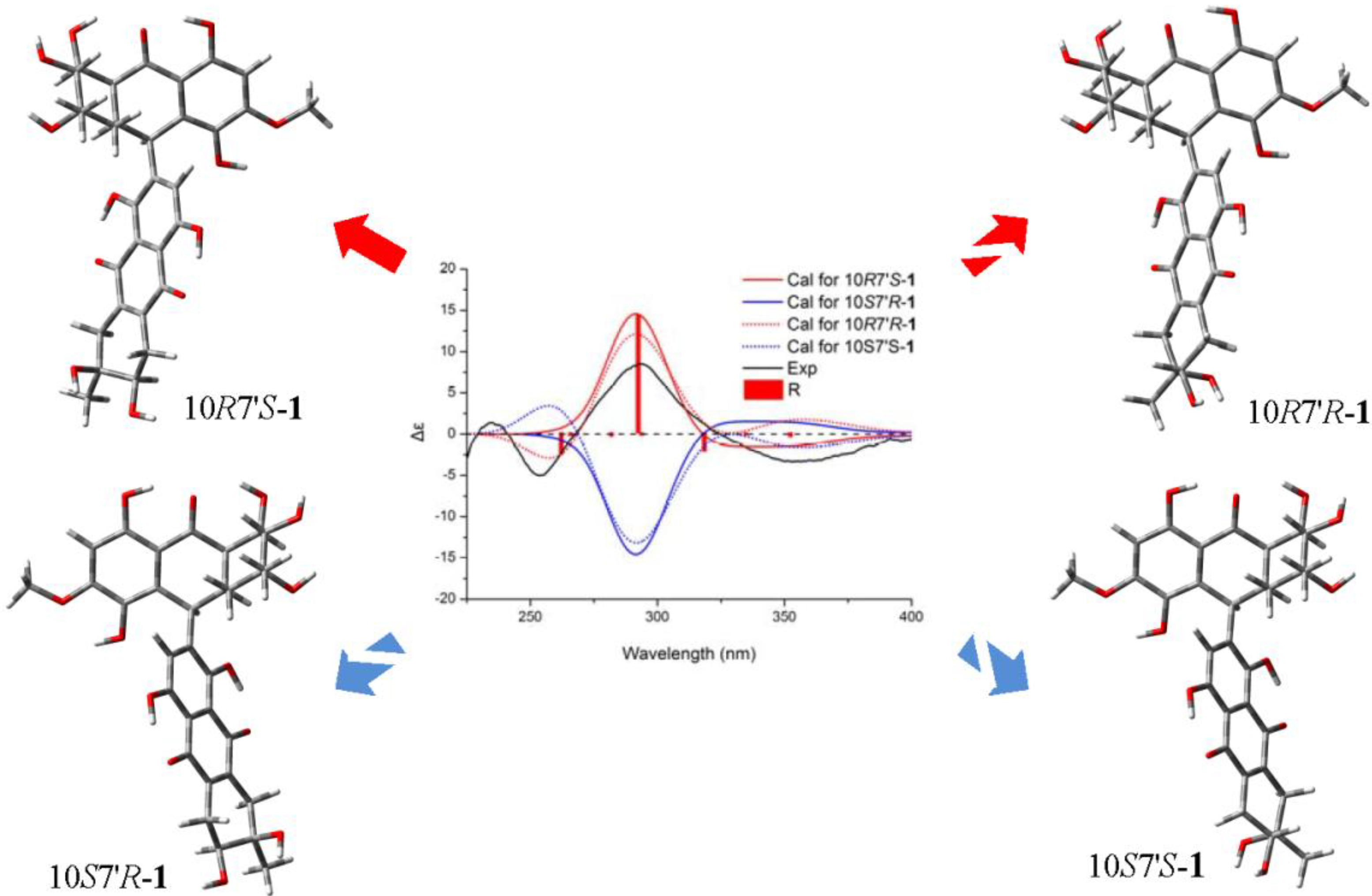
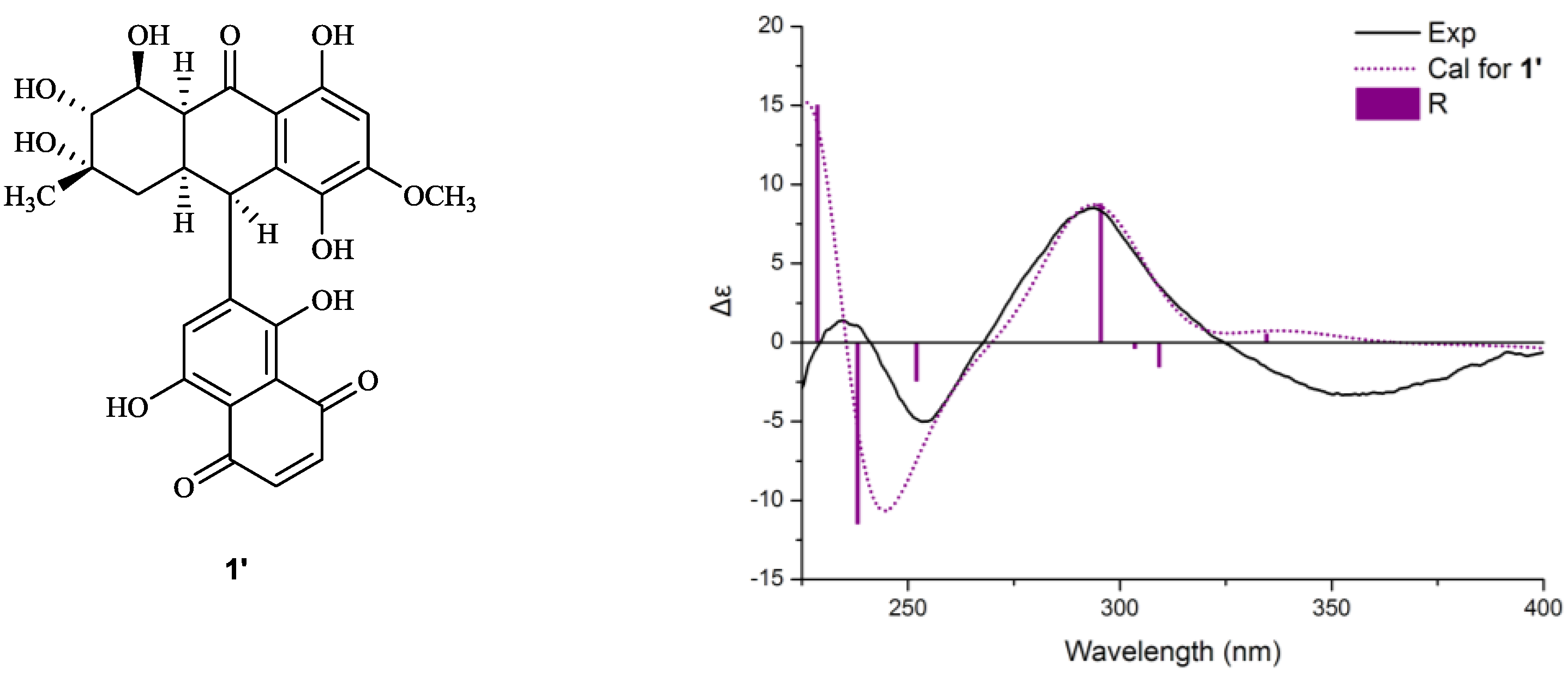
+75° (c 0.02, EtOH), 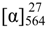 +208° (c 0.02, EtOH)) suggested that 2 was an atropisomer of alterporriol C [23]. Herein, we report the configuration of the chiral axis in the molecule by theoretical calculation.
+208° (c 0.02, EtOH)) suggested that 2 was an atropisomer of alterporriol C [23]. Herein, we report the configuration of the chiral axis in the molecule by theoretical calculation.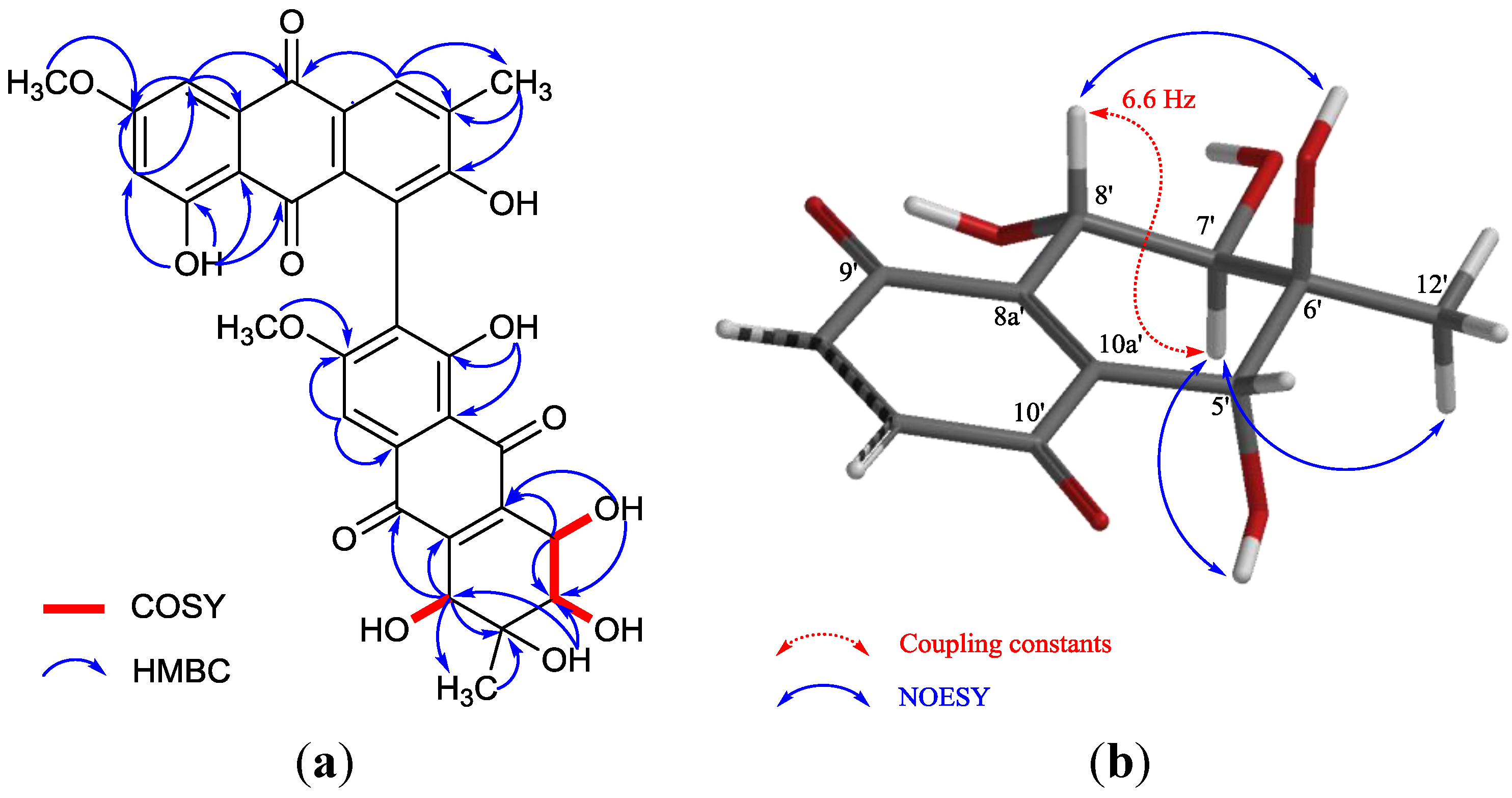
| Compound | IC50 (μM) |
|---|---|
| 1 | 64.70 |
| 2 | 8.70 |
| 3–8 | >100.00 |
| Control | 0.05 |
3. Experimental Section
3.1. General
3.2. Strain Isolation, Taxonomic Classification and Endophyte Fermentation
3.3. Extraction and Separation of Metabolites
+40° (c 0.02, EtOH); UV (MeOH) λmax 362.2, 288.8, 248.6, 220.4 nm; CD (MeOH) Δε355 (−3.3), Δε293 (+8.8), Δε254 (−5.1), Δε235 (+1.6), Δε210 (−18.6) cm2 mol−1; IR (KBr) νmax 3432, 2916, 2857, 1653, 1437, 1383, 1077, 480 cm−1; For 1H, 13C and 2D NMR spectroscopic data, see Table 1; HRESIMS m/z = 611.1793 (calcd. for C31H31O13, 611.1759). +75° (c 0.02, EtOH), +208° (c 0.02, EtOH); UV (MeOH) λmax 314.6, 280.4, 256.8, 226.4 nm; CD (MeOH) Δε285 (−23.9), Δε259 (+22.2), Δε245 (+12.2), Δε233 (+25.1), Δε218 (−39.5) cm2 mol−1; IR (KBr) νmax 3406, 2919, 2519, 1604, 1429, 1384, 1284, 1161, 1070, 878, 711, 613, 546, 483 cm−1; for 1H, 13C and 2D NMR spectroscopic data, see Table 2; HRESIMS m/z = 617.1335 (calcd. for C32H25O13, 617.1290).3.4. Calculation of ECD Spectra
3.5. Materials and Methods for mPTPB Assay
3.5.1. Cloning, Expression and Purification of MptpB
3.5.2. MptpB Inhibition Assay
4. Conclusions
Supplementary Files
Abbreviation
| B3LYP | Becke-3-Parameter-Lee-Yang-Parr |
| CAM–B3LYP | Coulomb Attenuating Method based on B3LYP |
| CCSD(T) | Coupled-Cluster Singles and Doubles (Triple) |
| Fr. | Fraction |
| IEF-PCM | Integral-Equation-Formalism Polarizable Continuum Model |
| ITS | Internal Transcribed Spacer |
| SVP | Split Valence plus Polarization |
| TMS | Tetramethylsilane |
| ZINDO/S-CI | Zerner’s Intermediate Neglect of Differential Overlap-Spectroscopic-Configuration Interaction |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2013. Available online: http://www.who.int/tb/publications/global_report/en/ (assessed on 21 December 2013).
- Singh, R.; Singh, A.; Tyagi, A.K. Deciphering the genes involved in pathogenesis of Mycobacterium tuberculosis. Tuberculosis (Edinb.) 2005, 85, 325–335. [Google Scholar] [CrossRef]
- Castandet, J.; Prost, J.F.; Peyron, P.; Astarie-Dequeker, C.; Anes, E.; Cozzone, A.J.; Griffiths, G.; Maridonneau-Parini, I. Tyrosine phosphatase MptpA of Mycobacterium tuberculosis inhibits phagocytosis and increases actin polymerization in macrophages. Res. Microbiol. 2005, 156, 1005–1013. [Google Scholar] [CrossRef]
- Ecco, G.; Vernal, J.; Razzera, G.; Martins, P.A.; Matiollo, C.; Terenzi, H. Mycobacterium tuberculosis tyrosine phosphatase A (PtpA) activity is modulated by S-nitrosylation. Chem. Commun. (Camb.) 2010, 46, 7501–7503. [Google Scholar] [CrossRef]
- Beresford, N.; Patel, S.; Armstrong, J.; Szöor, B.; Fordham-Skelton, A.P.; Tabernero, L. MptpB, a virulence factor from Mycobacterium tuberculosis, exhibits triple-specificity phosphatase activity. Biochem. J. 2007, 406, 13–18. [Google Scholar] [CrossRef]
- Silva, A.P.G.; Tabernero, L. New strategies in fighting TB: Targeting Mycobacterium tuberculosis-secreted phosphatases MptpA & MptpB. Future Med. Chem. 2010, 2, 1325–1337. [Google Scholar] [CrossRef]
- Zhou, B.; He, Y.; Zhang, X.; Xu, J.; Luo, Y.; Wang, Y.; Franzblau, S.G.; Yang, Z.; Chan, R.J.; Liu, Y.; et al. Targeting Mycobacterium protein tyrosine phosphatase B for antituberculosis agents. Proc. Natl. Acad. Sci. USA 2010, 107, 4573–4578. [Google Scholar] [CrossRef]
- Wang, C.; Wang, J.; Huang, Y.H.; Chen, H.; Li, Y.; Zhong, L.L.; Chen, Y.; Chen, S.P.; Wang, J.; Kang, J.L.; et al. Anti-mycobacterial activity of marine fungus-derived 4-deoxybostrycin and nigrosporin. Molecules 2013, 18, 1728–1740. [Google Scholar] [CrossRef]
- Huang, X.S.; Huang, H.B.; Li, H.X.; Sun, X.F.; Huang, H.R.; Lu, Y.J.; Lin, Y.C.; Long, Y.H.; She, Z.G. Asperterpenoid A, a new sesterterpenoid as an inhibitor of Mycobacterium tuberculosis protein tyrosine phosphatase B from the culture of Aspergillus sp. 16-5c. Org. Lett. 2013, 15, 721–723. [Google Scholar] [CrossRef]
- Li, H.X.; Jiang, J.Y.; Liu, Z.M.; Lin, S.E.; Xia, G.P.; Xia, X.K.; Ding, B.; He, L.; Lu, Y.J.; She, Z.G. Peniphenones A–D from the mangrove fungus Penicillium dipodomyicola HN4–3A as inhibitors of Mycobacterium tuberculosis Phosphatase MptpB. J. Nat. Prod. 2014, 77, 800–806. [Google Scholar] [CrossRef]
- Kimura, Y.; Shimada, A.; Nakajima, H.; Hamaski, T. Structures of naphthoquinones produced by the fungus, Fusarium sp., and their biological activity toward pollen germination. Agric. Biol. Chem. 1988, 52, 1253–1259. [Google Scholar] [CrossRef]
- Xia, X.K.; Huang, H.R.; She, Z.G.; Shao, C.L. 1H and 13C NMR assignments for five anthraquinones from the mangrove endophytic fungus Halorosellinia sp. (No. 1403). Magn. Reson. Chem. 2007, 45, 1006–1009. [Google Scholar]
- Sommart, U.; Rukachaisirikul, V.; Sukpondma, Y.; Phongpaichit, S.; Sakayaroj, J.; Kirtikara, K. Hydronaphthalenones and a dihydroramulosin from the endophytic fungus PSU-N24. Chem. Pharm. Bull. 2008, 56, 1687–1690. [Google Scholar] [CrossRef]
- Dreyer, D.L.; Arai, I.; Bachman, C.D.; Anderson, W.R.J.; Smith, R.G.; Daves, G.D.J. Toxins causing noninflammatory paralytic neuronopathy. Isolation and structure elucidation. J. Am. Chem. Soc. 1975, 97, 4985–4990. [Google Scholar] [CrossRef]
- Bringmann, G.; Mutanyatta-Comar, J.; Maksimenka, K.; Wanjohi, J.M.; Heydenreich, M.; Brun, R.; Müller, W.E.; Peter, M.G.; Midiwo, J.O.; Yenesew, A. Joziknipholones A and B: The first dimeric phenylanthraquinones, from the roots of Bulbine frutescens. Chem. Eur. J. 2008, 14, 1420–1429. [Google Scholar] [CrossRef]
- Noda, T.; Take, T.; Watanabe, T.; Abe, J. Structure of bostrycin. Tetrahedron 1970, 26, 1339–1346. [Google Scholar] [CrossRef]
- Bringmann, G.; Maksimenka, K.; Bruhn, T.; Reichert, M.; Harada, T.; Kuroda, R. Quantum chemical CD calculations of dioncophylline A in the solid state. Tetrahedron 2009, 65, 5720–5728. [Google Scholar] [CrossRef]
- Autschbach, J.; Nitsch-Velasquez, L.; Rudolph, M. Time-dependent density functional response theory for electronic chiroptical properties of chiral molecules. Top. Curr. Chem. 2011, 298, 1–98. [Google Scholar]
- Stephens, P.J.; Harada, N. ECD cotton effect approximated by the Gaussian curve and other methods. Chirality 2010, 22, 229–233. [Google Scholar]
- Suemitsu, R.; Ueshima, T.; Yamamoto, T.; Yanagawase, S. Alterporriol C: A modified bianthraquinone from Alternaria porri. Phytochemistry 1988, 27, 3251–3254. [Google Scholar] [CrossRef]
- Stoessl, A. Relative stereochemistry of altersolanol A. Can. J. Chem. 1969, 47, 777–784. [Google Scholar] [CrossRef]
- Debbab, A.; Aly, A.H.; Edrada-Ebel, R.; Wray, V.; Pretsch, A.; Pescitelli, G.; Kurtan, T.; Proksch, P. New anthracene derivatives—Structure elucidation and antimicrobial activity. Eur. J. Org. Chem. 2012, 2012, 1351–1359. [Google Scholar]
- Okamura, N.; Haraguchi, H.; Hashimoto, K.; Yagi, A. Altersolanol-related antimicrobial compounds from a strain of Alternaria solani. Phytochemistry 1993, 34, 1005–1009. [Google Scholar] [CrossRef]
- Hattori, T.; Sakurai, K.; Koike, N.; Miyano, S. Is the CD Exciton Chirality Method applicable to chiral 1,1′-biphenanthryl compounds? J. Am. Chem. Soc. 1998, 120, 9086–9087. [Google Scholar] [CrossRef]
- Tessier, A.M.; Delaveau, P.; Champion, B. New anthraquinones in Rubia cordifolia roots. Planta Med. 1981, 41, 337–343. [Google Scholar] [CrossRef]
- Archard, M.A.; Gill, M.; Strauch, R.J. Anthraquinones from the genus Cortinarius. Phytochemistry 1985, 24, 2755–2758. [Google Scholar] [CrossRef]
- Suemitsu, R.; Yamamoto, T.; Miyai, T.; Ueshima, T. Alterporriol A: A modified bianthraquinone from Alternaria porri. Phytochemistry 1987, 26, 3221–3224. [Google Scholar] [CrossRef]
- Suemitsu, R.; Sano, T.; Yamamoto, M.; Arimoto, Y.; Morimatsu, F.; Nabeshima, T. Structural elucidation of alterporriol B, a novel metabolic pigment produced by Alternaria porri (Ellis) ciferri. Agric. Biol. Chem. 1984, 48, 2611–2613. [Google Scholar] [CrossRef]
- Lazarovits, G.; Steele, R.W.; Stoessl, A. Dimers of altersolanol A from Alternaria solani. Z. Naturforsch. C 1988, 43, 813–817. [Google Scholar]
- Phuwapraisirisan, P.; Rangsan, J.; Siripong, P.; Tip-Pyang, S. New antitumour fungal metabolites from Alternaria porri. Nat. Prod. Res. 2009, 23, 1063–1071. [Google Scholar] [CrossRef]
- Debbab, A.; Aly, A.H.; Edrada-Ebel, R.; Wray, V.; Müller, W.E.; Totzke, F.; Zirrgiebel, U.; Schächtele, C.; Kubbutat, M.H.; Lin, W.H.; et al. Bioactive metabolites from the endophytic fungus Stemphylium globuliferum isolated from Mentha pulegium. J. Nat. Prod. 2009, 72, 626–631. [Google Scholar] [CrossRef]
- Huang, C.H.; Pan, J.H.; Chen, B.; Yu, M.; Huang, H.B.; Zhu, X.; Lu, Y.J.; She, Z.G.; Lin, Y.C. Three bianthraquinone derivatives from the mangrove endophytic fungus Alternaria sp. ZJ9-6B from the South China Sea. Mar. Drug 2011, 9, 832–843. [Google Scholar] [CrossRef]
- Zheng, C.J.; Shao, C.L.; Guo, Z.Y.; Chen, J.F.; Deng, D.S.; Yang, K.L.; Chen, Y.Y.; Fu, X.M; She, Z.G.; Lin, Y.C.; et al. Bioactive hydroanthraquinones and anthraquinone dimers from a soft coral-derived Alternaria sp. fungus. J. Nat. Prod. 2012, 75, 189–197. [Google Scholar] [CrossRef]
- Qhotsokoane-Lusunzi, M.A.; Karuso, P. Secondary metabolites from Basotho medicinal plants. I. Bulbine narcissifolia. J. Nat. Prod. 2001, 64, 1368–1372. [Google Scholar] [CrossRef]
- Hou, Y.; Cao, S.; Brodie, P.J.; Callmander, M.W.; Ratovoson, F.; Rakotobe, E.A.; Rasamison, V.E.; Ratsimbason, M.; Alumasa, J.N.; Roepe, P.D.; et al. Antiproliferative and antimalarial anthraquinones of Scutia myrtina from the Madagascar forest. Bioorg. Med. Chem. 2009, 17, 2871–2876. [Google Scholar] [CrossRef]
- Alemayehu, G.; Hailu, A.; Abegaz, B.M. Bianthraquinones from Senna didymobotrya. Phytochemistry 1996, 42, 1423–1425. [Google Scholar] [CrossRef]
- Lanzetta, R.; Parrilli, M.; Adinolfi, M.; Aquila, T.; Corsaro, M.M. Bianthrone C-glycosides. 2. Three new compounds from Asphodelus ramosus tubers. Tetrahedron 1990, 46, 1287–1294. [Google Scholar] [CrossRef]
- Dagne, E.; Berhanu, E.; Steglich, W. New bianthraquinone pigments from Kniphofia species. Bull. Chem. Soc. Ethiop. 1987, 1, 32–35. [Google Scholar]
- Yagi, A.; Makino, K.; Nishioka, I. Studies on the constituents of Aloe saponaria Haw. IV. The structures of bianthraquinoid pigments. Chem. Pharm. Bull. 1978, 26, 1111–1116. [Google Scholar] [CrossRef]
- Yang, Q.Y.; Yao, C.S.; Fang, W.S. A new triglucosylated naphthalene glycoside from Aloe vera L. Fitoterapia 2010, 81, 59–62. [Google Scholar] [CrossRef]
- Shin, K.H.; Woo, W.S.; Lim, S.S.; Shim, C.S.; Chung, H.S.; Kennelly, E.J.; Kinghorn, A.D. Elgonica-dimers A and B, two potent alcohol metabolism inhibitory constituents of Aloe arborescens. J. Nat. Prod. 1997, 60, 1180–1182. [Google Scholar] [CrossRef]
- Choi, J.S.; Lee, S.K.; Sung, C.K.; Jung, J.H. Phytochemical study on Aloe vera. Arch. Pharm. Res. 1996, 19, 163–167. [Google Scholar] [CrossRef]
- Conner, J.M.; Gray, A.I.; Waterman, P.G.; Reynolds, T. Novel anthrone-anthraquinone dimers from Aloe elgonica. J. Nat. Prod. 1990, 53, 1362–1364. [Google Scholar] [CrossRef]
- Carroll, A.R.; Nash, B.D.; Duffy, S.; Avery, V.M. Albopunctatone, an antiplasmodial anthrone-anthraquinone from the Australian ascidian Didemnum albopunctatum. J. Nat. Prod. 2012, 75, 1206–1209. [Google Scholar] [CrossRef]
- LaPlante, S.R.; Fader, L.D.; Fandrick, K.R.; Fandrick, D.R.; Hucke, O.; Kemper, R.; Miller, S.P.F.; Edwards, P.J. Assessing atropisomer axial chirality in drug discovery and development. J. Med. Chem. 2011, 54, 7005–7022. [Google Scholar] [CrossRef]
- Polavarapu, P.L.; Jeirath, N.; Kurtán, T.; Pescitelli, G.; Krohn, K. Determination of the absolute configurations at stereogenic centers in the presence of axial chirality. Chirality 2009, 21, E202–E207. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xia, G.; Li, J.; Li, H.; Long, Y.; Lin, S.; Lu, Y.; He, L.; Lin, Y.; Liu, L.; She, Z. Alterporriol-Type Dimers from the Mangrove Endophytic Fungus, Alternaria sp. (SK11), and Their MptpB Inhibitions. Mar. Drugs 2014, 12, 2953-2969. https://doi.org/10.3390/md12052953
Xia G, Li J, Li H, Long Y, Lin S, Lu Y, He L, Lin Y, Liu L, She Z. Alterporriol-Type Dimers from the Mangrove Endophytic Fungus, Alternaria sp. (SK11), and Their MptpB Inhibitions. Marine Drugs. 2014; 12(5):2953-2969. https://doi.org/10.3390/md12052953
Chicago/Turabian StyleXia, Guoping, Jia Li, Hanxiang Li, Yuhua Long, Shao'e Lin, Yongjun Lu, Lei He, Yongcheng Lin, Lan Liu, and Zhigang She. 2014. "Alterporriol-Type Dimers from the Mangrove Endophytic Fungus, Alternaria sp. (SK11), and Their MptpB Inhibitions" Marine Drugs 12, no. 5: 2953-2969. https://doi.org/10.3390/md12052953
APA StyleXia, G., Li, J., Li, H., Long, Y., Lin, S., Lu, Y., He, L., Lin, Y., Liu, L., & She, Z. (2014). Alterporriol-Type Dimers from the Mangrove Endophytic Fungus, Alternaria sp. (SK11), and Their MptpB Inhibitions. Marine Drugs, 12(5), 2953-2969. https://doi.org/10.3390/md12052953