Four New Chloro-Eremophilane Sesquiterpenes from an Antarctic Deep-Sea Derived Fungus, Penicillium sp. PR19N-1



Abstract
:
1. Introduction
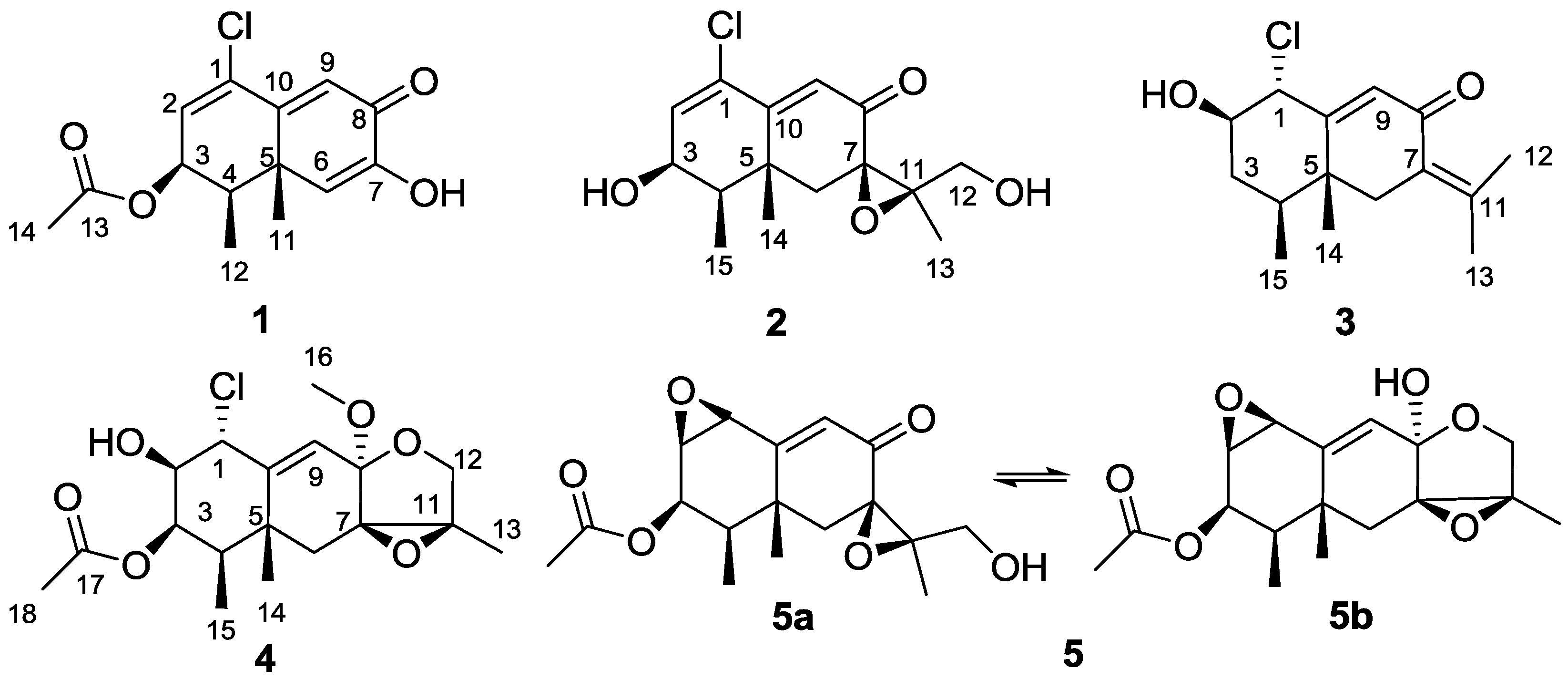
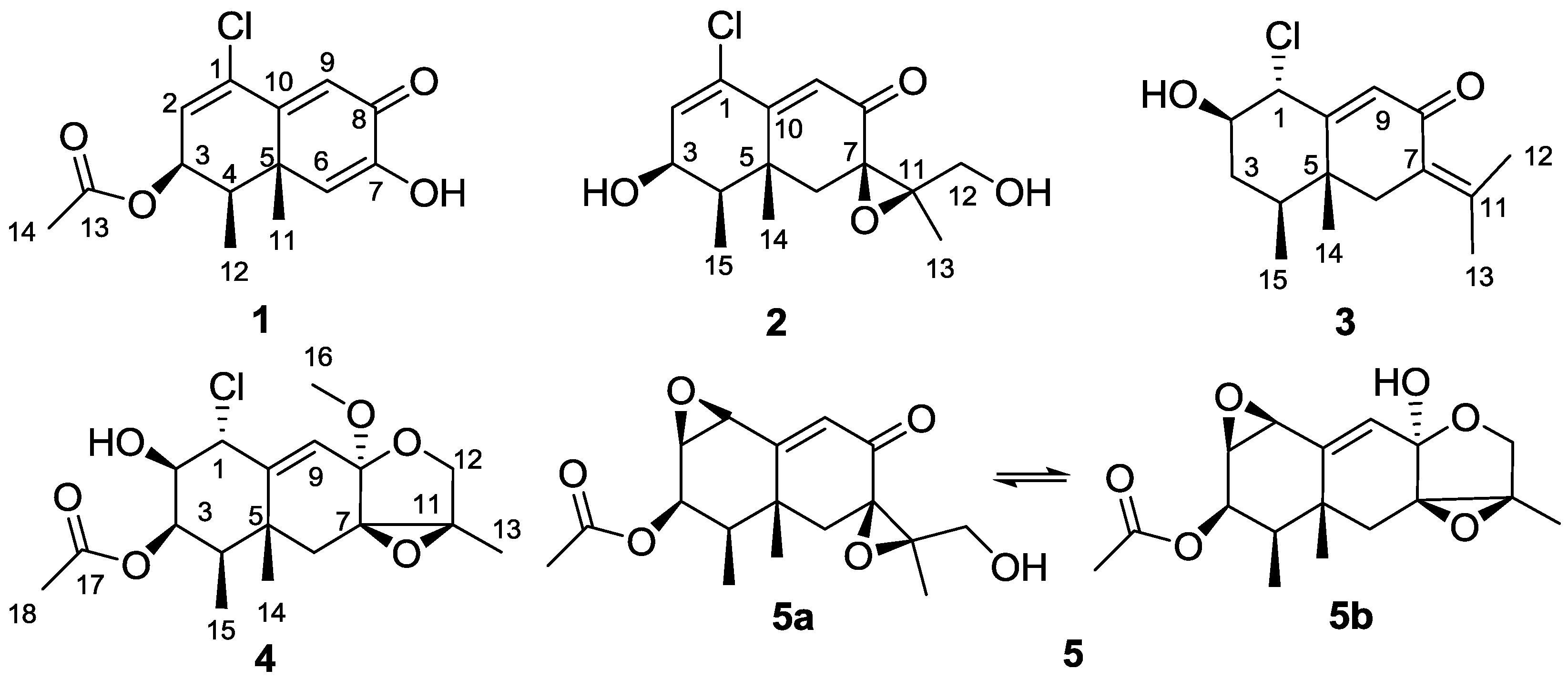
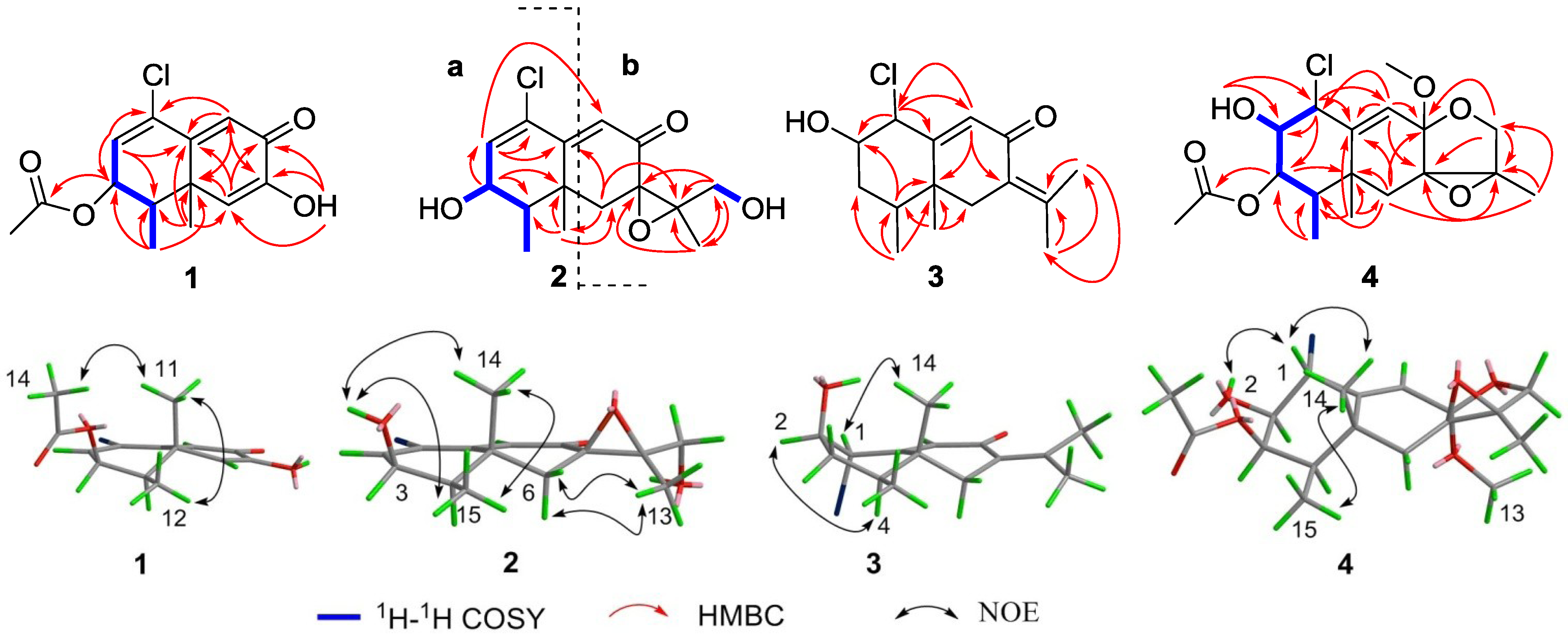
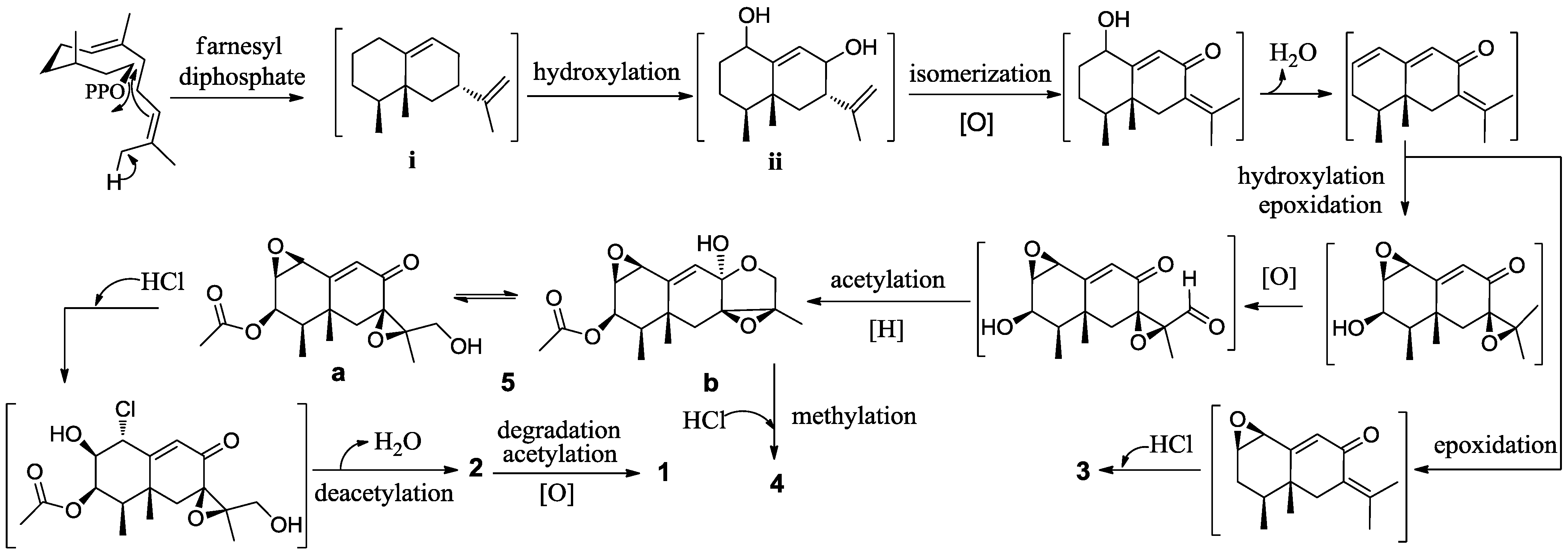
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
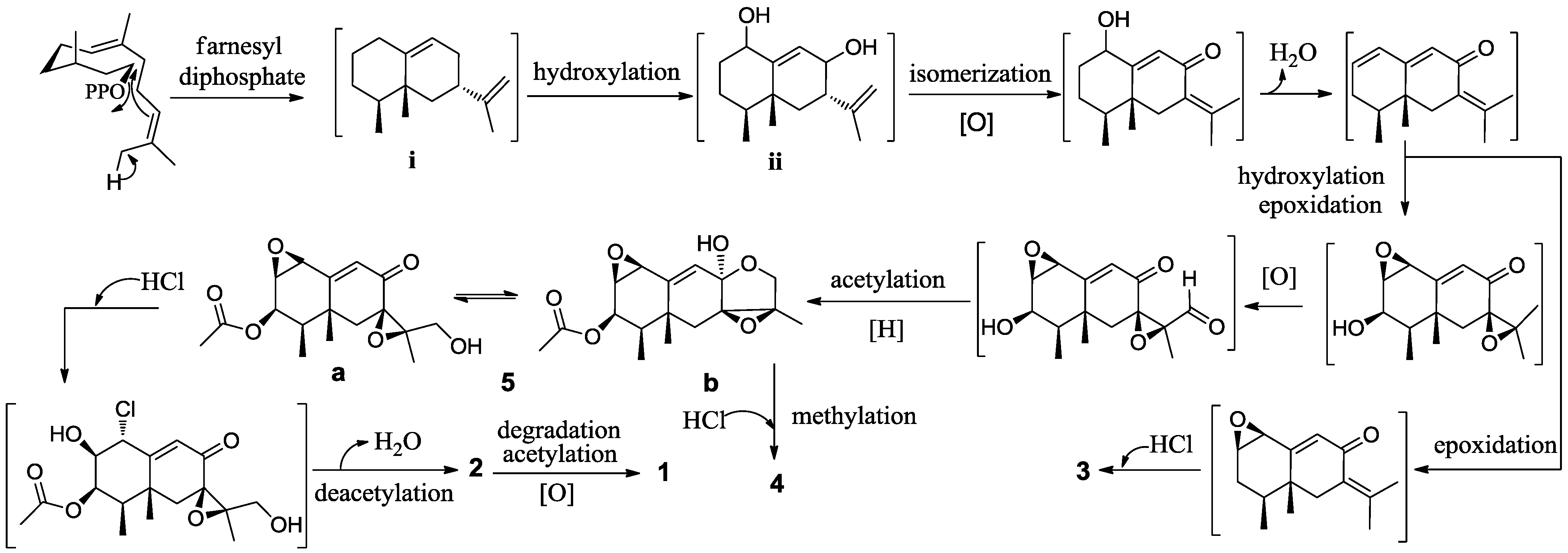{kind=link}
| No. | 1 a | 2 b | 3 a | 4 b |
|---|---|---|---|---|
| 1 | 133.5 s | 129.5 s | 67.6 d | 63.1 d |
| 2 | 129.1 d | 138.4 d | 74.9 d | 75.3 d |
| 3 | 70.3 d | 66.9 d | 37.1 t | 75.6 d |
| 4 | 37.7 d | 39.8 d | 38.5 d | 38.6 d |
| 5 | 42.7 s | 38.5 s | 42.8 s | 42.2 s |
| 6 | 121.3 d | 40.3 t | 41.1 t | 32.0 t |
| 7 | 146.3 s | 61.8 s | 126.6 s | 67.3 s |
| 8 | 181.1 s | 194.9 s | 191.1 s | 100.6 s |
| 9 | 123.2 d | 125.3 d | 127.9 d | 122.0 d |
| 10 | 159.7 s | 157.3 s | 159.9 s | 144.4 s |
| 11 | 23.7 q | 67.3 s | 144.3 s | 65.3 s |
| 12 | 11.5 q | 61.2 t | 22.6 q | 68.7 t |
| 13 | 170.4 s | 16.1 q | 22.2 q | 12.7 q |
| 14 | 20.9 q | 23.6 q | 16.9 q | 23.3 q |
| 15 | 11.0 q | 14.9 q | 11.2 q | |
| 16 | 50.9 q | |||
| 17 | 170.0 s | |||
| 18 | 20.8 q |
| No. | 1 a | 2 b | 3 a | 4 b |
|---|---|---|---|---|
| 1 | 4.48, dd (10.0, 2.0) | 4.72, dd (10.5, 1.3) | ||
| 2 | 6.36, d (5.0) | 6.63, d (6.4) | 3.63, m (10.0, 9.4, 5.0) | 3.36, ddd (10.5, 7.3, 3.7) |
| 3 | 5.50, dd (5.0, 5.0) | 4.14, ddd (6.4, 5.3, 4.2) | 1.61, m | 5.23, dd (3.7, 3.7) |
| 4 | 2.12, m | 1.80, qd (7.0, 4.2) | 2.04, m | 1.74, qd (6.9, 3.7) |
| 6 | 6.28, s | a: 2.33, d (14.6) | a: 2.88, d (13.7) | a: 1.89, d (14.4); |
| b: 1.89, d (15.1) | b: 2.26, d (13.7) | b: 1.83, d (14.4) | ||
| 9 | 6.76, s | 6.28, s | 6.41, d (2.0) | 5.94, d (1.3) |
| 11 | 1.42, s | |||
| 12 | 1.18, d (6.8) | 3.70, dd (6.0, 5.8) | 2.11, s | a: 3.89, d (10.4); |
| b: 3.53, d (10.4) | ||||
| 13 | 1.39, s | 1.87, s | 1.35, s | |
| 14 | 2.13, s | 1.20, brs | 1.04, s | 1.19, s |
| 15 | 1.04, d (7.0) | 1.02, d (6.4) | 0.84, d (6.9) | |
| 16 | 3.30, s | |||
| 18 | 2.08, s | |||
| 2-OH | 5.69, d (7.3) | |||
| 3-OH | 5.27, d (5.3) | |||
| 7-OH | 6.32, brs | |||
| 12-OH | 4.81, t (5.8) |

3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation and Extraction
3.4. Purification
3.5. Biological Assays
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Pettit, R.K. Culturability and secondary metabolite diversity of extreme microbes: Expanding contribution of deep sea and deep-sea vent microbes to natural product discovery. Mar. Biotechnol. 2011, 13, 1–11. [Google Scholar]
- Thornburg, C.C.; Zabriskie, T.M.; McPhail, K.L. Deep-sea hydrothermal vents: Potential hot spots for natural products discovery? J. Nat. Prod. 2010, 73, 489–499. [Google Scholar]
- Kellenberger, E.; Hofmann, A.; Quinn, J.R. Similar interactions of natural products with biosynthetic enzymes and therapeutic targets could explain why nature produces such a large proportion of existing drugs. Nat. Prod. Rep. 2011, 28, 1483–1492. [Google Scholar] [CrossRef]
- Fenical, W.; Jensen, P.R. Developing a new resource for drug discovery: Marine actinomycete bacteria. Nat. Chem. Biol. 2006, 2, 666–673. [Google Scholar]
- Li, D.H.; Wang, F.P.; Xiao, X.; Fang, Y.C.; Zhu, T.J.; Gu, Q.Q.; Zhu, W.M. Trisorbicillinone A, a novel sorbicillin trimer, from a deep sea fungus, Phialocephala sp. FL30r. Tetrahedron Lett. 2007, 48, 5235–5238. [Google Scholar]
- Du, L.; Li, D.H.; Zhu, T.J.; Cai, S.X.; Wang, F.P.; Xiao, X.; Gu, Q.Q. New alkaloids and diterpenes from a deep ocean sediment derived fungus Penicillium sp. Tetrahedron 2009, 65, 1033–1039. [Google Scholar]
- Zhou, L.N.; Zhu, T.J.; Cai, S.X.; Gu, Q.Q.; Li, D.H. Three new indole-containing diketopiperazine alkaloids from a deep-ocean sediment derived fungus Penicillium griseofulvum. Helv. Chim. Acta 2010, 93, 1758–1763. [Google Scholar]
- Skropeta, D. Deep-sea natural products. Nat. Prod. Rep. 2008, 25, 1131–1166. [Google Scholar] [CrossRef]
- Moreau, S.; Cacan, M. Eremofortin C, a new metabolite obtained from Penicillium roqueforti cultures and from biotransformation of PR toxin. J. Org. Chem. 1977, 42, 2632–2633. [Google Scholar]
- Moreau, S.; Biguet, J. Structures et stereochimie des sesquiterpenes de Penicillium roqueforti PR toxine et eremofortines A, B, C, D, E. Tetrahedron 1980, 36, 2989–2997. [Google Scholar]
- Watanabe, N.; Fujita, A.; Ban, N.; Yagi, A.; Etoh, H.; Ina, K. Bipolal: Anti-microalgal compound isolated as a candidate for marine antifouling produced by Bipolaris sp. F5206. J. Nat. Prod. 1995, 58, 463–466. [Google Scholar]
- Guerriero, A.; Cuomo, V.; Vanzanella, F.; Pietra, F. A novel glyceryl ester (glyceryl dendryphiellate A), a trinor-eremophilane (dendryphiellin A1), and eremophilanes (dendryphiellin E1 and E2) from the marine deutermycete Dendryphiella salina (Sutherland) Pugh et Nicot. Helv. Chim. Acta 1990, 73, 2090–2096. [Google Scholar]
- Nunes, F.M.; Oliveira, M.C.F.; Arriaga, Â.M.C.; Lemos, T.L.G.; Andrade-Neto, M.; Mattos, M.C.; Mafezoli, J.; Viana, F.M.P.; Ferreira, V.M.; Rodrigues-Filhod, E.; et al. A new eremophilane-type sesquiterpene from the phytopatogen fungus Lasiodiplodia theobromae (Sphaeropsidaceae). J. Braz. Chem. Soc. 2008, 19, 478–482. [Google Scholar]
- Guerriero, A.; D’Ambrosio, M.; Cuomo, V.; Vanzanella, F.; Pietra, F. Dendryphiellin A, the first fungal trinor-eremophilane. Isolation from the marine deuteromycete Dendryphiella salina (Sutherland) Pugh et Nicot. Helv. Chim. Acta 1989, 71, 57–61. [Google Scholar]
- McDonald, L.A.; Barbieri, L.R.; Bernan, V.S.; Janso, J.; Lassota, P.; Carter, G.T. 07H239-A, a new cytotoxic eremophilane sesquiterpene from the marine-dedived Xylariaceous fungus LL-07H239. J. Nat. Prod. 2004, 67, 1565–1567. [Google Scholar]
- Yamada, T.; Doi, M.; Miura, A.; Harada, W.; Hiramura, M.; Minoura, K.; Tanaka, R.; Numata, A. Absolute stereostructures of cell-adhesion inhibitors, peribysins A, E, F and G, produced by a sea hare-derived Periconia sp. J. Antibiot. 2005, 58, 185–191. [Google Scholar]
- Sørensen, D.; Raditsis, A.; Trimble, L.A.; Blackwell, B.A.; Sumarah, M.W.; Miller, J.D. Isolation and structure elucidation by LC-MS-SPE/NMR; PR toxin- and cuspidatol-related eremophilane sesquiterpenes from Penicillium roqueforti. J. Nat. Prod. 2007, 70, 121–123. [Google Scholar]
- Huang, Y.F.; Qiao, L.; Lv, A.L.; Pei, Y.H.; Tian, L. Eremophilane sesquiterpenes from the marine fungus Penicillium sp. BL-27-2. Chin. Chem. Lett. 2008, 19, 562–564. [Google Scholar]
- Shishova, E.T.; Costamzo, L.D.; Cane, D.E.; Christianson, D.W. X-ray crystal structure of aristolochene synthase from Aspergillus terreus and evolution of templates for the cyclization of farnesyl diphosphate. Biochemistry 2007, 46, 1941–1951. [Google Scholar]
- Agger, S.; Lopez-Gallego, F.; Schmidt-Dannert, C. Diversity of sesquiterpene synthases in the basidiomycete Coprinus cinereus. Mol. Microbiol. 2009, 72, 1181–1195. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Jonathan, T. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Nat. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Fraga, B.M. Natural sesquiterpenoids. Nat. Prod. Rep. 2012, 29, 1334–1366. [Google Scholar] [CrossRef]
- Fraga, B.M. Natural sesquiterpenoids. Nat. Prod. Rep. 2011, 28, 1580–1610. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wu, G.; Lin, A.; Gu, Q.; Zhu, T.; Li, D. Four New Chloro-Eremophilane Sesquiterpenes from an Antarctic Deep-Sea Derived Fungus, Penicillium sp. PR19N-1. Mar. Drugs 2013, 11, 1399-1408. https://doi.org/10.3390/md11041399
Wu G, Lin A, Gu Q, Zhu T, Li D. Four New Chloro-Eremophilane Sesquiterpenes from an Antarctic Deep-Sea Derived Fungus, Penicillium sp. PR19N-1. Marine Drugs. 2013; 11(4):1399-1408. https://doi.org/10.3390/md11041399
Chicago/Turabian StyleWu, Guangwei, Aiqun Lin, Qianqun Gu, Tianjiao Zhu, and Dehai Li. 2013. "Four New Chloro-Eremophilane Sesquiterpenes from an Antarctic Deep-Sea Derived Fungus, Penicillium sp. PR19N-1" Marine Drugs 11, no. 4: 1399-1408. https://doi.org/10.3390/md11041399
APA StyleWu, G., Lin, A., Gu, Q., Zhu, T., & Li, D. (2013). Four New Chloro-Eremophilane Sesquiterpenes from an Antarctic Deep-Sea Derived Fungus, Penicillium sp. PR19N-1. Marine Drugs, 11(4), 1399-1408. https://doi.org/10.3390/md11041399