Bivalve Omics: State of the Art and Potential Applications for the Biomonitoring of Harmful Marine Compounds



 ,
, 
Abstract
:
1. Introduction
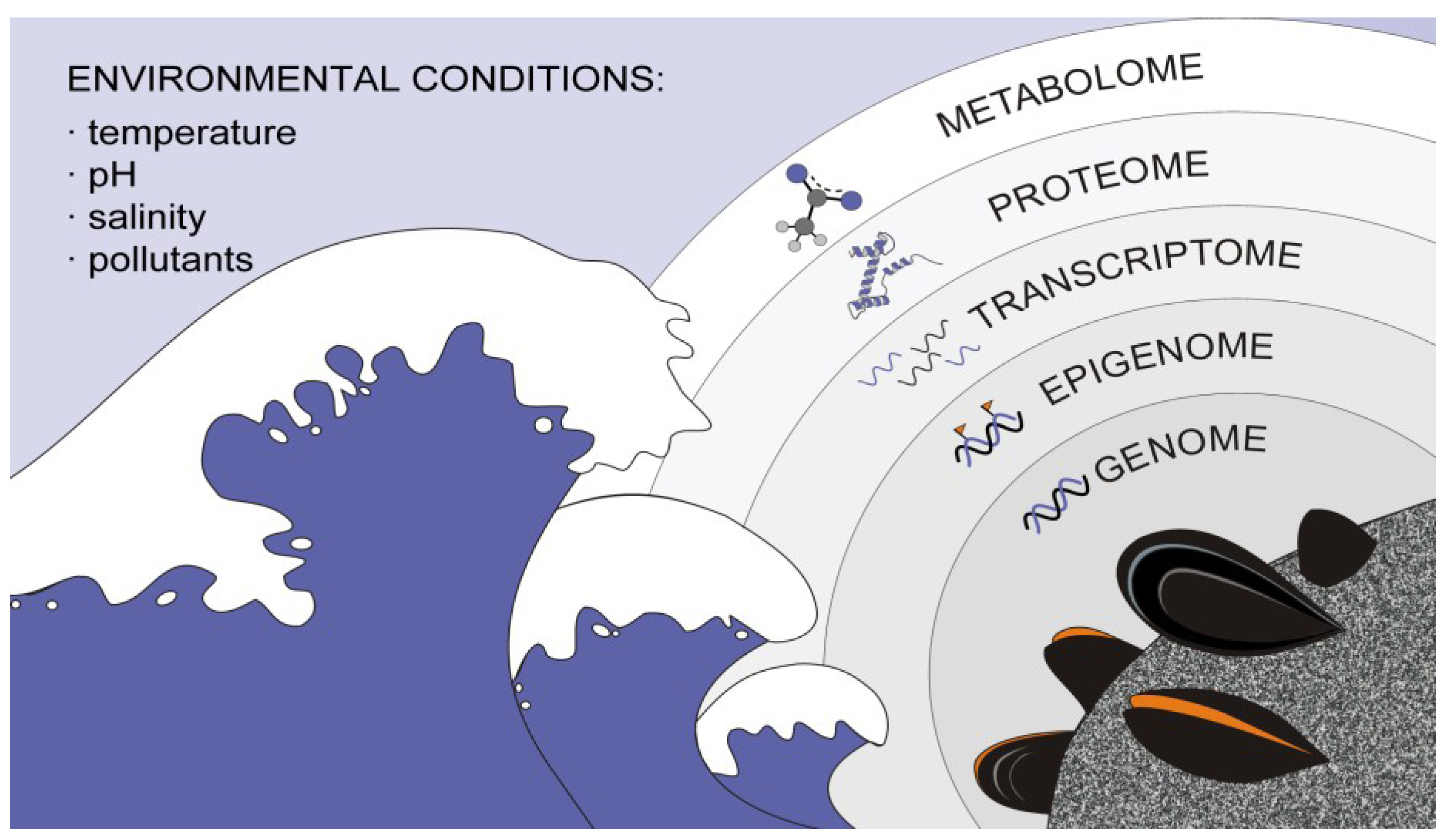
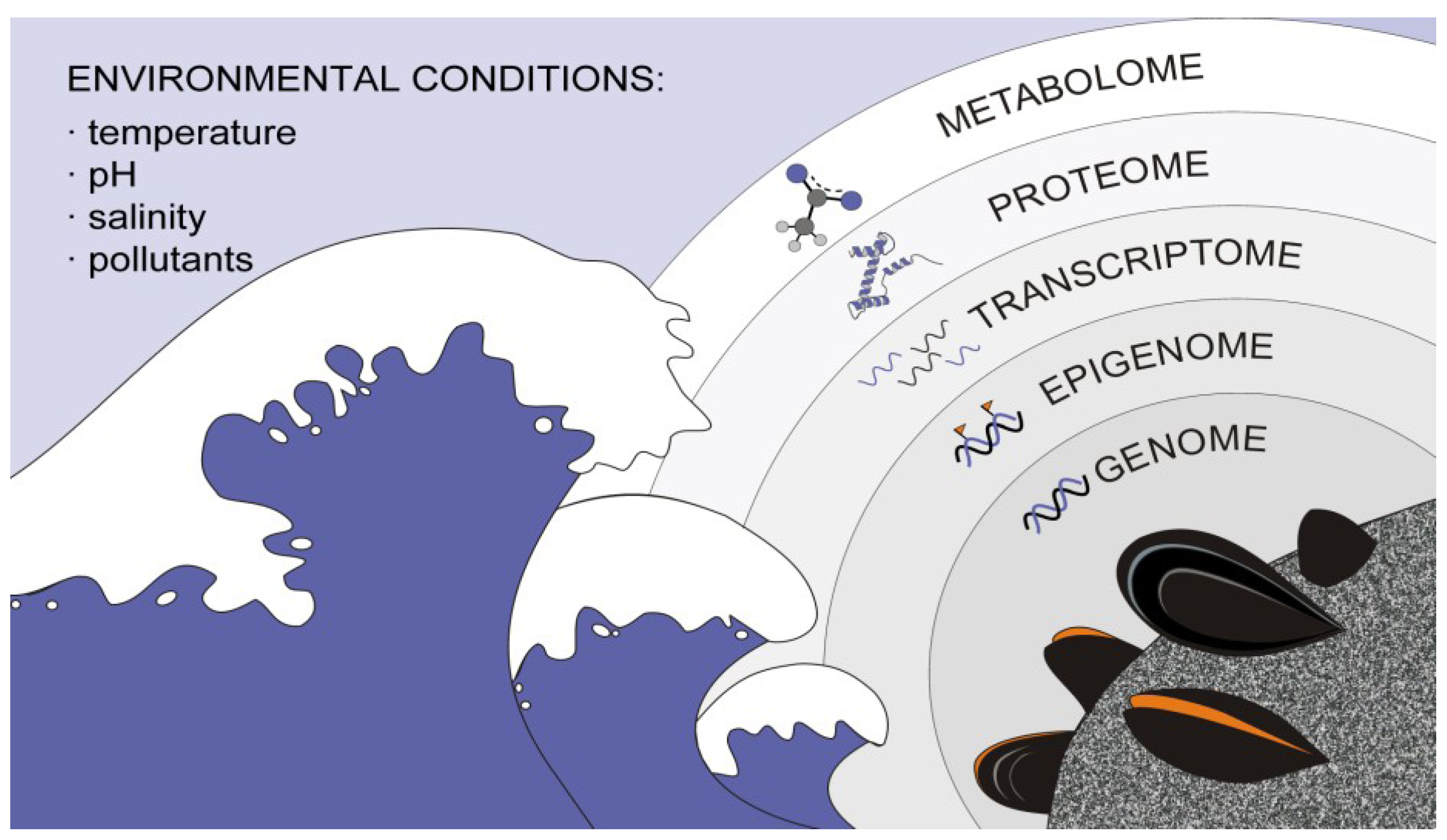
2. Leading Edge on Bivalve High-Throughput and NGS Data Analysis
2.1. Bivalve Genomes
2.2. Bivalve Transcriptomes
2.3. Bivalve Proteomes
2.4. Bivalve Metabolomics
2.5. Bivalve Epigenomics
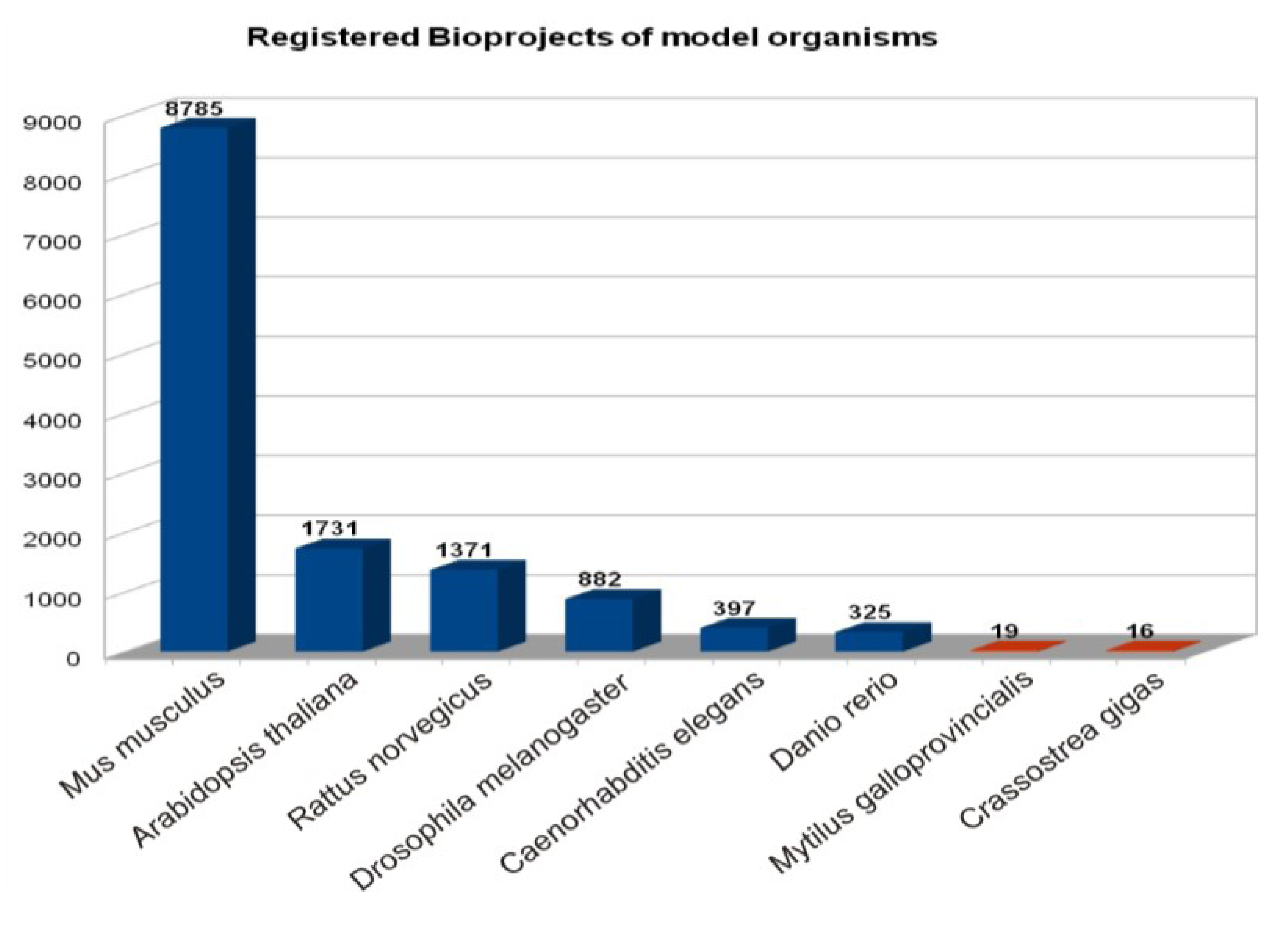
3. Resources for the Study of Bivalve Omic Data
3.1. General Resources

{kind=link}
{kind=link}
{kind=link}
{kind=link}
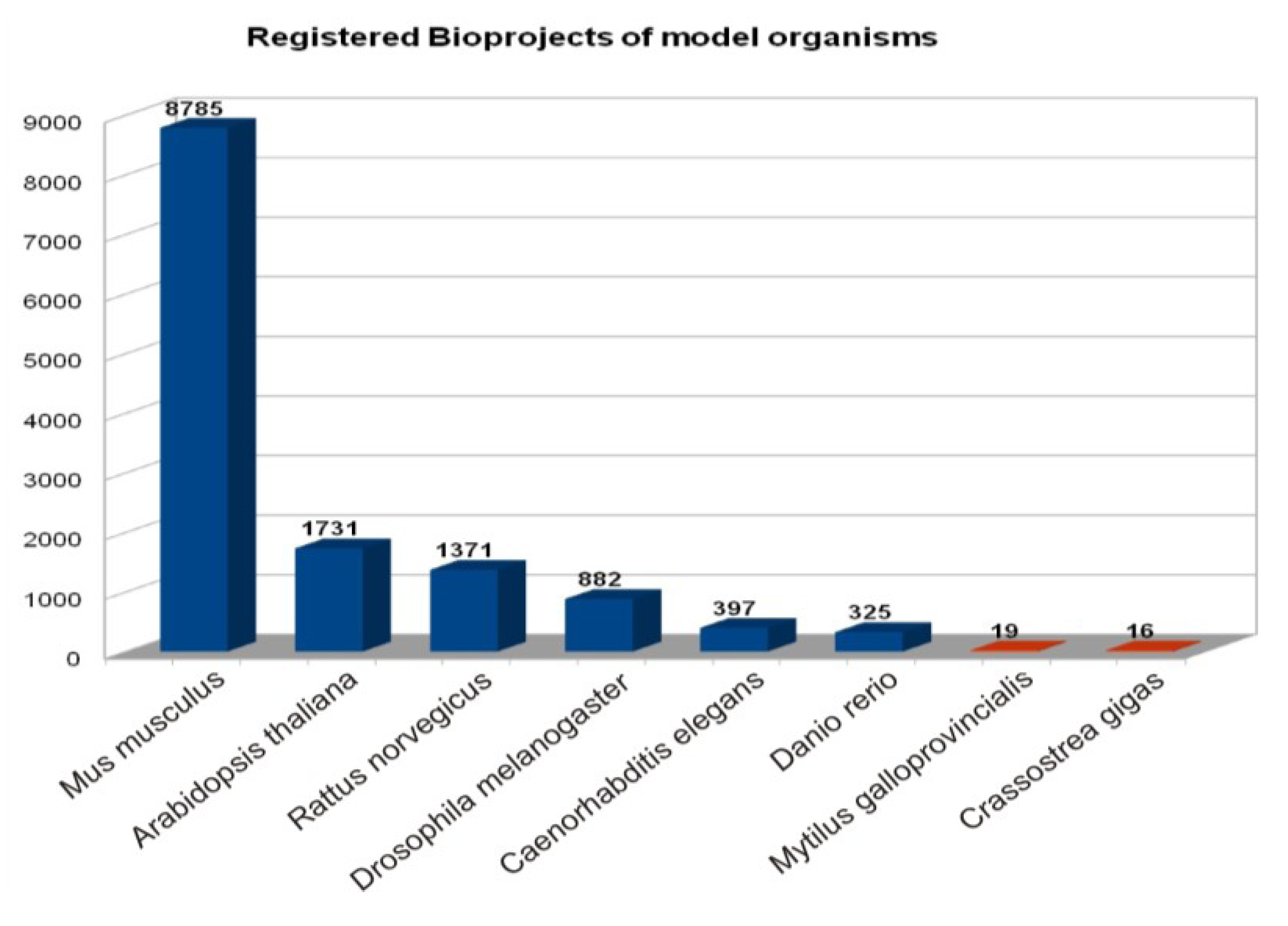
| Species | Bioproject | Genome | SRA Datasets | ||||
|---|---|---|---|---|---|---|---|
| Number | Type | Total | 454 | Illumina | AB SOLiD | ||
| Alasmidonta varicosa | 1 | transcriptome/gene expression | YES (no data) | 1 | 1 | - | - |
| Arctica islandica | 1 | genome | YES (no data) | 12 | 12 | - | - |
| Argopecten irradians | 3 | transcriptome/gene expression | - | 1 | 1 | - | - |
| Bankia setacea | 1 | genome | YES (no data) | 1 | 1 | - | - |
| Bathymodiolus azoricus | 1 | transcriptome/gene expression | YES (no data) | 1 | 1 | - | - |
| Chlamys farreri | 1 | transcriptome/gene expression | - | - | - | - | - |
| Chamelea gallina | - | - | - | 1 | 1 | - | - |
| Crassostrea angulata | 1 | transcriptome/gene expression | - | - | - | - | - |
| Crassostrea gigas | 16 | 1 genome, 15 transcriptome/gene expression | YES (scaffold or contigs status) | 159 | 2 | 155 | 2 |
| Crassostrea hongkongensis | 1 | proteome | - | - | - | - | - |
| Crassostrea virginica | 3 | transcriptome/gene expression | - | - | - | - | - |
| Ennucula tenuis | - | - | - | 1 | 1 | - | - |
| Glossus humanus | 1 | exome | YES (no data) | - | - | - | - |
| Hyriopsis cumingii | 1 | transcriptome/gene expression | - | 1 | 1 | - | - |
| Laternula elliptica | 1 | transcriptome/gene expression | - | 3 | 3 | - | - |
| Macoma balthica | - | - | - | 3 | 3 | - | - |
| Mercenaria mercenaria | 1 | transcriptome/gene expression | YES (no data) | 2 | - | - | 2 |
| Meretrix meretrix | 1 | transcriptome/gene expression | - | 1 | 1 | - | - |
| Mizuhopecten (Patinopecten) yessoensis | 1 | transcriptome/gene expression | - | 2 | 2 | - | - |
| Mya arenaria | 2 | transcriptome/gene expression | YES (no data) | - | - | - | - |
| Mytilus californianus | 4 | transcriptome/gene expression | - | - | - | - | - |
| Mytilus edulis | - | - | - | 44 | 44 | - | - |
| Mytilus galloprovincialis | 19 | transcriptome/gene expression | - | 12 | 6 | 6 | - |
| Nodipecten subnodosus | - | - | - | 2 | 2 | - | - |
| Nucula nitidosa | - | - | - | 1 | 1 | - | - |
| Ostrea lurida | 2 | transcriptome/gene expression | YES (no data) | 1 | - | 1 | - |
| Pinctada fucata | 1 | transcriptome/gene expression | YES (no data) | 10 | 7 | 3 | - |
| Pinctada margaritifera | 2 | transcriptome/gene expression | YES (no data) | 1 | 1 | - | - |
| Pinctada maxima | 7 | transcriptome/gene expression | - | 1 | 1 | - | - |
| Pteria penguin | 1 | transcriptome/gene expression | YES (no data) | - | - | - | - |
| Ruditapes decussatus | 3 | transcriptome/gene expression | - | - | - | - | - |
| Ruditapes philippinarum | 6 | transcriptome/gene expression | - | 28 | 2 | 26 | - |
| Solemya velum | - | - | - | 2 | 1 | 1 | - |
| Spisula solidissima | 1 | genome | YES (no data) | - | - | - | - |
| Tegillarca granosa | 1 | transcriptome/gene expression | - | 1 | 1 | - | - |
| Yoldia limatula | - | - | - | 1 | 1 | - | - |
3.2. Specialized Resources
3.2.1. Databases and Knowledge Repositories
| Database | Organism/# Sequences | Tissues/URL | |
|---|---|---|---|
| Species-centered | |||
| Mytibase | M. galloprovincialis 7112 | Digestive gland, gills, hemocytes [95] | |
| GigasDatabase | C. gigas 29745 | Digestive gland, gills, gonad, hemocytes, mantle-edge, muscle [96] | |
| RuphiBase | R. philippinarum 32606 | Mixed tissues [97] | |
| ChameleaBase | C. gallina 36422 | Muscle [98] | |
| DeepSeaVent | B. azoricus 35903 | Gills [99] | |
| Functionally-centered | |||
| Chromevaloa | M.galloprovincialis 14408 | Digestive gland [100] | |
3.2.2. Array Technology
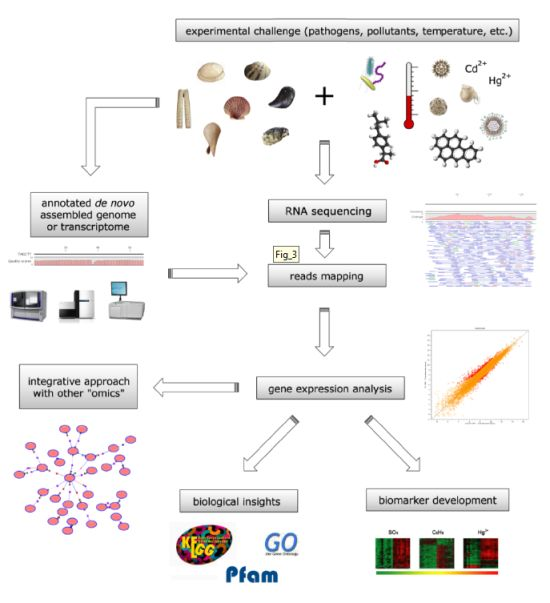
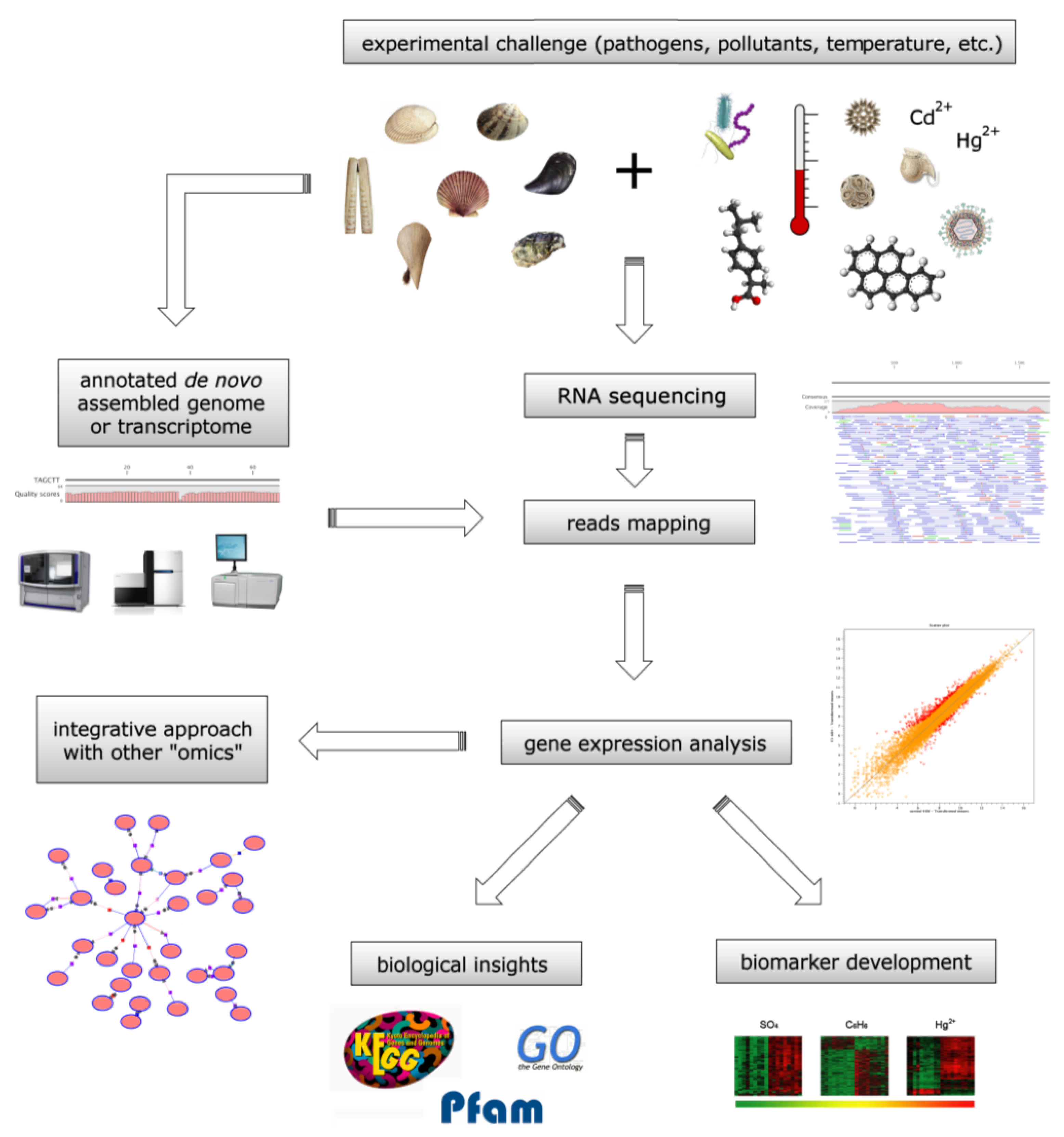
4. Bivalve Omic Approaches for the Biomonitoring of Marine Compounds
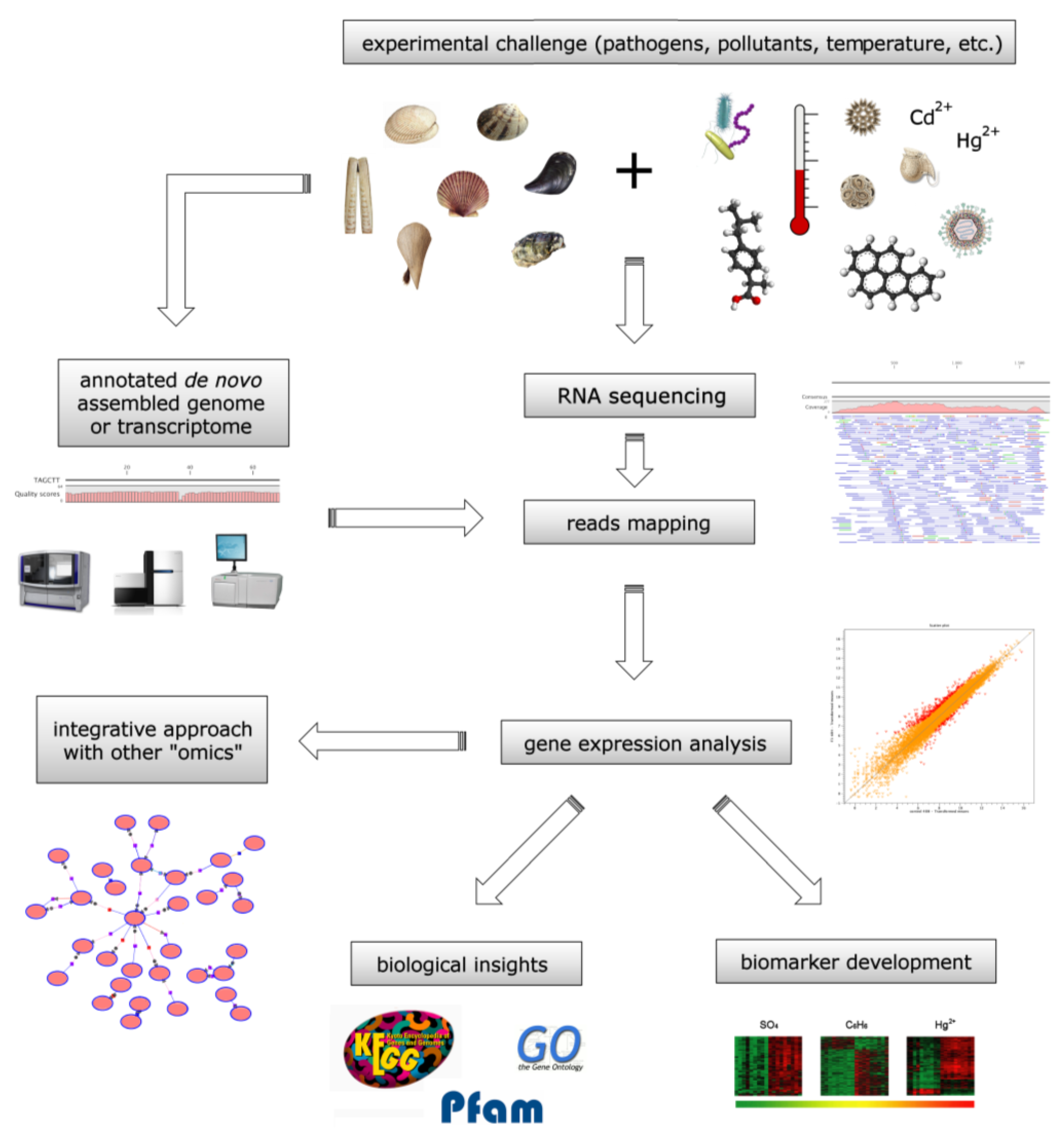
4.1. Bivalve Transcriptomes as Biomonitoring Tools
4.2. Toxin Biomonitoring during Harmful Algae Blooms
4.3. Evaluation of the Harmful Effects of Anthropic Pollutants
4.4. Proteomic Biomonitoring of Harmful Marine Compounds
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ruppert, E.E.; Fox, R.S.; Barnes, R.D. Invertebrate Zoology: A Functional Evolutionary Approach, 7th ed.; Cengage Learning: Stamford, CT, USA, 2004. [Google Scholar]
- Gosling, E.M. Bivalve Molluscs: Biology, Ecology and Culture; Oxford Fishing New Books, Blackwell Science: Oxford, UK, 2003. [Google Scholar]
- Newell, R. Ecosystem influences of natural and cultivated populations of suspension-feeding bivalve molluscs: A review. J. Shellfish Res. 2004, 23, 51–61. [Google Scholar]
- Campos, A.; Tedesco, S.; Vasconcelos, V.; Cristobal, S. Proteomic research in bivalves: Towards the identification of molecular markers of aquatic pollution. J. Proteomics 2012, 75, 4346–4359. [Google Scholar] [CrossRef]
- Suarez-Ulloa, V.; Fernandez-Tajes, J.; Aguiar-Pulido, V.; Rivera-Casas, C.; Gonzalez-Romero, R.; Ausio, J.; Mendez, J.; Dorado, J.; Eirin-Lopez, J.M. The CHROMEVALOA database: A resource for the evaluation of okadaic acid contamination in the marine environment based on the chromatin-associated transcriptome of the mussel Mytilus galloprovincialis. Mar. Drugs 2013, 11, 830–841. [Google Scholar] [CrossRef]
- Milan, M.; Pauletto, M.; Patarnello, T.; Bargelloni, L.; Marin, M.G.; Matozzo, V. Gene transcription and biomarker responses in the clam Ruditapes philippinarum after exposure to ibuprofen. Aquat. Toxicol. 2013, 126, 17–29. [Google Scholar] [CrossRef]
- Zhang, G.; Fang, X.; Guo, X.; Li, L.; Luo, R.; Xu, F.; Yang, P.; Zhang, L.; Wang, X.; Qi, H.; et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 2012, 490, 49–54. [Google Scholar] [CrossRef]
- Collin, H.; Meistertzheim, A.L.; David, E.; Moraga, D.; Boutet, I. Response of the Pacific oyster Crassostrea gigas, Thunberg 1793, to pesticide exposure under experimental conditions. J. Exp. Biol. 2010, 213, 4010–4017. [Google Scholar] [CrossRef] [Green Version]
- Luchmann, K.H.; Mattos, J.J.; Siebert, M.N.; Dorrington, T.S.; Toledo-Silva, G.; Stoco, P.H.; Grisard, E.C.; Bainy, A.C. Suppressive subtractive hybridization libraries prepared from the digestive gland of the oyster Crassostrea brasiliana exposed to a diesel fuel water-accommodated fraction. Environ. Toxicol. Chem. 2012, 31, 1249–1253. [Google Scholar] [CrossRef]
- Fernandez-Tajes, J.; Arias-Perez, A.; Fernandez-Moreno, M.; Mendez, J. Sharp decrease of genetic variation in two Spanish localities of razor clam Ensis siliqua: Natural fluctuation or Prestige oil spill effects? Ecotoxicology 2012, 21, 225–233. [Google Scholar] [CrossRef]
- Florez-Barros, F.; Prado-Alvarez, M.; Mendez, J.; Fernandez-Tajes, J. Evaluation of genotoxicity in gills and hemolymph of clam Ruditapes decussatus fed with the toxic dinoflagellate Prorocentrum lima. J. Toxicol. Environ. Health A 2011, 74, 971–979. [Google Scholar] [CrossRef]
- Wells, P.G.; Depledge, M.H.; Butler, J.N.; Manock, J.J.; Knap, A.H. Rapid toxicity assessment and biomonitoring of marine contaminants—exploiting the potential of rapid biomarker assays and microscale toxicity tests. Mar. Pollut. Bull. 2001, 42, 799–804. [Google Scholar] [CrossRef]
- Manfrin, C.; Dreos, R.; Battistella, S.; Beran, A.; Gerdol, M.; Varotto, L.; Lanfranchi, G.; Venier, P.; Pallavicini, A. Mediterranean mussel gene expression profile induced by okadaic acid exposure. Environ. Sci. Technol. 2010, 44, 8276–8283. [Google Scholar] [CrossRef]
- Takeuchi, T.; Kawashima, T.; Koyanagi, R.; Gyoja, F.; Tanaka, M.; Ikuta, T.; Shoguchi, E.; Fujiwara, M.; Shinzato, C.; Hisata, K.; et al. Draft genome of the pearl oyster Pinctada fucata: A platform for understanding bivalve biology. DNA Res. 2012, 19, 117–130. [Google Scholar] [CrossRef]
- Craft, J.A.; Gilbert, J.A.; Temperton, B.; Dempsey, K.E.; Ashelford, K.; Tiwari, B.; Hutchinson, T.H.; Chipman, J.K. Pyrosequencing of Mytilus galloprovincialis cDNAs: Tissue-specific expression patterns. PLoS One 2010, 5, e8875. [Google Scholar] [CrossRef]
- Moreira, R.; Balseiro, P.; Planas, J.V.; Fuste, B.; Beltran, S.; Novoa, B.; Figueras, A. Transcriptomics of in vitro immune-stimulated hemocytes from the Manila clam Ruditapes philippinarum using high-throughput sequencing. PLoS One 2012, 7, e35009. [Google Scholar]
- Rosani, U.; Varotto, L.; Rossi, A.; Roch, P.; Novoa, B.; Figueras, A.; Pallavicini, A.; Venier, P. Massively parallel amplicon sequencing reveals isotype-specific variability of antimicrobial peptide transcripts in Mytilus galloprovincialis. PLoS One 2011, 6, e26680. [Google Scholar]
- Dondero, F.; Piacentini, L.; Marsano, F.; Rebelo, M.; Vergani, L.; Venier, P.; Viarengo, A. Gene transcription profiling in pollutant exposed mussels (Mytilus spp.) using a new low-density oligonucleotide microarray. Gene 2006, 376, 24–36. [Google Scholar] [CrossRef]
- Venier, P.; de Pitta, C.; Pallavicini, A.; Marsano, F.; Varotto, L.; Romualdi, C.; Dondero, F.; Viarengo, A.; Lanfranchi, G. Development of mussel mRNA profiling: Can gene expression trends reveal coastal water pollution? Mutat. Res. 2006, 602, 121–134. [Google Scholar] [CrossRef]
- Zapata, M.; Tanguy, A.; David, E.; Moraga, D.; Riquelme, C. Transcriptomic response of Argopecten purpuratus post-larvae to copper exposure under experimental conditions. Gene 2009, 442, 37–46. [Google Scholar] [CrossRef]
- Brown, M.; Davies, I.M.; Moffat, C.F.; Craft, J.A. Application of SSH and a macroarray to investigate altered gene expression in Mytilus edulis in response to exposure to benzo[α]pyrene. Mar. Environ. Res. 2006, 62, S128–S135. [Google Scholar] [CrossRef]
- Tanguy, A.; Boutet, I.; Laroche, J.; Moraga, D. Molecular identification and expression study of differentially regulated genes in the Pacific oyster Crassostrea gigas in response to pesticide exposure. FEBS J. 2005, 272, 390–403. [Google Scholar] [CrossRef]
- Navarro, A.; Campos, B.; Barata, C.; Pina, B. Transcriptomic seasonal variations in a natural population of zebra mussel (Dreissena polymorpha). Sci.Total Environ. 2013, 454–455, 482–489. [Google Scholar] [CrossRef]
- Meng, J.; Zhu, Q.; Zhang, L.; Li, C.; Li, L.; She, Z.; Huang, B.; Zhang, G. Genome and transcriptome analyses provide insight into the euryhaline adaptation mechanism of Crassostrea gigas. PLoS One 2013, 8, e58563. [Google Scholar]
- Pante, E.; Rohfritsch, A.; Becquet, V.; Belkhir, K.; Bierne, N.; Garcia, P. SNP detection from de novo transcriptome sequencing in the bivalve Macoma balthica: Marker development for evolutionary studies. PLoS One 2012, 7, e52302. [Google Scholar]
- Huan, P.; Wang, H.; Liu, B. Transcriptomic analysis of the clam Meretrix meretrix on different larval stages. Mar. Biotechnol. (N.Y.) 2012, 14, 69–78. [Google Scholar] [CrossRef]
- Coppe, A.; Bortoluzzi, S.; Murari, G.; Marino, I.A.; Zane, L.; Papetti, C. Sequencing and characterization of striped venus transcriptome expand resources for clam fishery genetics. PLoS One 2012, 7, e44185. [Google Scholar]
- Qin, J.; Huang, Z.; Chen, J.; Zou, Q.; You, W.; Ke, C. Sequencing and de novo analysis of Crassostrea angulata (Fujian oyster) from 8 different developing phases using 454 GSFlx. PLoS One 2012, 7, e43653. [Google Scholar]
- Shi, Y.; Yu, C.; Gu, Z.; Zhan, X.; Wang, Y.; Wang, A. Characterization of the pearl oyster (Pinctada martensii) mantle transcriptome unravels biomineralization genes. Mar. Biotechnol. (N.Y.) 2013, 15, 175–187. [Google Scholar] [CrossRef]
- Rodríguez-Juíz, A.M.; Torrado, M.; Méndez, J. Genome-size variation in bivalve molluscs determined by flow cytometry. Mar. Biol. 1996, 126, 489–497. [Google Scholar] [CrossRef]
- Ruiz-Lara, S.; Prats, E.; Sainz, J.; Cornudella, L. Cloning and characterization of a highly conserved satellite DNA from the mollusc Mytilus edulis. Gene 1992, 117, 237–242. [Google Scholar]
- Philipp, E.E.; Kraemer, L.; Melzner, F.; Poustka, A.J.; Thieme, S.; Findeisen, U.; Schreiber, S.; Rosenstiel, P. Massively parallel RNA sequencing identifies a complex immune gene repertoire in the lophotrochozoan Mytilus edulis. PLoS One 2012, 7, e33091. [Google Scholar] [CrossRef] [Green Version]
- Egas, C.; Pinheiro, M.; Gomes, P.; Barroso, C.; Bettencourt, R. The transcriptome of Bathymodiolus azoricus gill reveals expression of genes from endosymbionts and free-living deep-sea bacteria. Mar. Drugs 2012, 10, 1765–1783. [Google Scholar] [CrossRef]
- Venier, P.; Varotto, L.; Rosani, U.; Millino, C.; Celegato, B.; Bernante, F.; Lanfranchi, G.; Novoa, B.; Roch, P.; Figueras, A.; et al. Insights into the innate immunity of the Mediterranean mussel Mytilus galloprovincialis. BMC Genomics 2011, 12, 69. [Google Scholar] [CrossRef]
- Milan, M.; Coppe, A.; Reinhardt, R.; Cancela, L.M.; Leite, R.B.; Saavedra, C.; Ciofi, C.; Chelazzi, G.; Patarnello, T.; Bortoluzzi, S.; et al. Transcriptome sequencing and microarray development for the Manila clam, Ruditapes philippinarum: Genomic tools for environmental monitoring. BMC Genomics 2011, 12, 234. [Google Scholar] [CrossRef] [Green Version]
- De Lorgeril, J.; Zenagui, R.; Rosa, R.D.; Piquemal, D.; Bachere, E. Whole transcriptome profiling of successful immune response to Vibrio infections in the oyster Crassostrea gigas by digital gene expression analysis. PLoS One 2011, 6, e23142. [Google Scholar]
- Chapman, R.W.; Mancia, A.; Beal, M.; Veloso, A.; Rathburn, C.; Blair, A.; Holland, A.F.; Warr, G.W.; Didinato, G.; Sokolova, I.M.; et al. The transcriptomic responses of the eastern oyster, Crassostrea virginica, to environmental conditions. Mol. Ecol. 2011, 20, 1431–1449. [Google Scholar] [CrossRef]
- Canesi, L.; Negri, A.; Barmo, C.; Banni, M.; Gallo, G.; Viarengo, A.; Dondero, F. The organophosphate Chlorpyrifos interferes with the responses to 17beta-estradiol in the digestive gland of the marine mussel Mytilus galloprovincialis. PLoS One 2011, 6, e19803. [Google Scholar]
- Lockwood, B.L.; Sanders, J.G.; Somero, G.N. Transcriptomic responses to heat stress in invasive and native blue mussels (genus Mytilus): Molecular correlates of invasive success. J. Exp. Biol. 2010, 213, 3548–3558. [Google Scholar] [CrossRef]
- Lockwood, B.L.; Somero, G.N. Transcriptomic responses to salinity stress in invasive and native blue mussels (genus Mytilus). Mol. Ecol. 2011, 20, 517–529. [Google Scholar] [CrossRef]
- Shendure, J. The beginning of the end for microarrays? Nat. Methods 2008, 5, 585–587. [Google Scholar] [CrossRef]
- Francis, W.R.; Christianson, L.M.; Kiko, R.; Powers, M.L.; Shaner, N.C.; Haddock, S.H. A comparison across non-model animals suggests an optimal sequencing depth for de novo transcriptome assembly. BMC Genomics 2013, 14, 167. [Google Scholar] [CrossRef] [Green Version]
- Ghiselli, F.; Milani, L.; Chang, P.L.; Hedgecock, D.; Davis, J.P.; Nuzhdin, S.V.; Passamonti, M. De novo assembly of the Manila clam Ruditapes philippinarum transcriptome provides new insights into expression bias, mitochondrial doubly uniparental inheritance and sex determination. Mol. Biol. Evol. 2012, 29, 771–786. [Google Scholar] [CrossRef]
- Yue, X.; Wang, H.; Huang, X.; Wang, C.; Chai, X.; Liu, B. Single nucleotide polymorphisms in i-type lysozyme gene and their correlation with vibrio-resistance and growth of clam Meretrix meretrix based on the selected resistance stocks. Fish. Shellfish Immunol. 2012, 33, 559–568. [Google Scholar] [CrossRef]
- Gerdol, M.; De Moro, G.; Manfrin, C.; Venier, P.; Pallavicini, A. Big defensins and mytimacins, new AMP families of the Mediterranean mussel Mytilus galloprovincialis. Dev. Comp. Immunol. 2012, 36, 390–399. [Google Scholar] [CrossRef]
- Feder, M.E.; Walser, J.C. The biological limitations of transcriptomics in elucidating stress and stress responses. J. Evol. Biol. 2005, 18, 901–910. [Google Scholar] [CrossRef]
- Dowd, W.W. Challenges for biological interpretation of environmental proteomics data in non-model organisms. Integr. Comp. Biol. 2012, 52, 705–720. [Google Scholar] [CrossRef]
- Lambert, J.P.; Ethier, M.; Smith, J.C.; Figeys, D. Proteomics: From gel based to gel free. Anal. Chem. 2005, 77, 3771–3787. [Google Scholar] [CrossRef]
- Shepard, J.L.; Bradley, B.P. Protein expression signatures and lysosomal stability in Mytilus edulis exposed to graded copper concentrations. Mar. Environ. Res. 2000, 50, 457–463. [Google Scholar] [CrossRef]
- Leung, P.T.; Wang, Y.; Mak, S.S.; Ng, W.C.; Leung, K.M. Differential proteomic responses in hepatopancreas and adductor muscles of the green-lipped mussel Perna viridis to stresses induced by cadmium and hydrogen peroxide. Aquat. Toxicol. 2011, 105, 49–61. [Google Scholar] [CrossRef]
- Apraiz, I.; Mi, J.; Cristobal, S. Identification of proteomic signatures of exposure to marine pollutants in mussels (Mytilus edulis). Mol. Cell. Proteomics 2006, 5, 1274–1285. [Google Scholar] [CrossRef]
- Kingtong, S.; Kellner, K.; Bernay, B.; Goux, D.; Sourdaine, P.; Berthelin, C.H. Proteomic identification of protein associated to mature spermatozoa in the Pacific oyster Crassostrea gigas. J. Proteomics 2013, 82, 81–91. [Google Scholar] [CrossRef]
- Corporeau, C.; Vanderplancke, G.; Boulais, M.; Suquet, M.; Quere, C.; Boudry, P.; Huvet, A.; Madec, S. Proteomic identification of quality factors for oocytes in the Pacific oyster Crassostrea gigas. J. Proteomics 2012, 75, 5554–5563. [Google Scholar] [CrossRef]
- López, J.L.; Mosquera, E.; Fuentes, J.; Marina, A.; Vázquez, J.; Álvarez, G. Two-dimensional gel electrophoresis of Mytilus galloprovincialis: differences in protein expression between intertidal and cultured mussels. Mar. Ecol. Progr. Ser. 2001, 224, 149–156. [Google Scholar] [CrossRef]
- Tomanek, L.; Zuzow, M.J. The proteomic response of the mussel congeners Mytilus galloprovincialis and M.trossulus to acute heat stress: Implications for thermal tolerance limits and metabolic costs of thermal stress. J. Exp. Biol. 2010, 213, 3559–3574. [Google Scholar] [CrossRef]
- Tomanek, L.; Zuzow, M.J.; Ivanina, A.V.; Beniash, E.; Sokolova, I.M. Proteomic response to elevated PCO2 level in eastern oysters, Crassostrea virginica: Evidence for oxidative stress. J. Exp. Biol. 2011, 214, 1836–1844. [Google Scholar]
- Robertson, D.G.; Watkins, P.B.; Reily, M.D. Metabolomics in toxicology: Preclinical and clinical applications. Toxicol. Sci. 2005, 120, S146–S170. [Google Scholar] [CrossRef]
- Kwon, Y.K.; Jung, Y.S.; Park, J.C.; Seo, J.; Choi, M.S.; Hwang, G.S. Characterizing the effect of heavy metal contamination on marine mussels using metabolomics. Mar. Pollut. Bull. 2012, 64, 1874–1879. [Google Scholar]
- Wu, H.; Wang, W.X. Tissue-specific toxicological effects of cadmium in green mussels (Perna viridis): Nuclear magnetic resonance-based metabolomics study. Environ. Toxicol. Chem. 2010, 30, 806–812. [Google Scholar]
- Tuffnail, W.; Mills, G.A.; Cary, P.; Greenwood, R. An environmental H-1 NMR metabolomic study of the exposure of the marine mussel Mytilus edulis to atrazine, lindane, hypoxia and starvation. Metabolomics 2009, 5, 33–43. [Google Scholar]
- Liu, X.; Zhang, L.; You, L.; Yu, J.; Zhao, J.; Li, L.; Wang, Q.; Li, F.; Li, C.; Liu, D.; et al. Differential toxicological effects induced by mercury in gills from three pedigrees of Manila clam Ruditapes philippinarum by NMR-based metabolomics. Ecotoxicology 2010, 20, 177–186. [Google Scholar]
- Wu, H.; Liu, X.; Zhao, J.; Yu, J. NMR-based metabolomic investigations on the differential responses in adductor muscles from two pedigrees of Manila clam Ruditapes philippinarum to Cadmium and Zinc. Mar. Drugs 2011, 9, 1566–1579. [Google Scholar]
- Zhang, L.; Liu, X.; You, L.; Zhou, D.; Wu, H.; Li, L.; Zhao, J.; Feng, J.; Yu, J. Metabolic responses in gills of Manila clam Ruditapes philippinarum exposed to copper using NMR-based metabolomics. Mar. Environ. Res. 2011, 72, 33–39. [Google Scholar]
- Zhang, L.; Liu, X.; You, L.; Zhou, D.; Wang, Q.; Li, F.; Cong, M.; Li, L.; Zhao, J.; Liu, D.; et al. Benzo(a)pyrene-induced metabolic responses in Manila clam Ruditapes philippinarum by proton nuclear magnetic resonance ((1)H NMR) based metabolomics. Environ. Toxicol. Pharmacol. 2011, 32, 218–225. [Google Scholar]
- Metzker, M.L. Sequencing technologies—the next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar]
- Klironomos, F.D.; Berg, J.; Collins, S. How epigenetic mutations can affect genetic evolution: Model and mechanism. Bioessays 2013, 35, 571–578. [Google Scholar]
- Gavery, M.R.; Roberts, S.B. DNA methylation patterns provide insight into epigenetic regulation in the Pacific oyster (Crassostrea gigas). BMC Genomics 2010, 11, 483. [Google Scholar]
- Eirín-López, J.M.; Ruiz, M.F.; González-Tizón, A.M.; Martínez, A.; Sánchez, L.; Méndez, J. Molecular evolutionary characterization of the mussel Mytilus histone multigene family: First record of a tandemly repeated unit of five histone genes containing an H1 subtype with “orphon” features. J. Mol. Evol. 2004, 58, 131–144. [Google Scholar]
- González-Romero, R.; Ausió, J.; Méndez, J.; Eirín-López, J.M. Early evolution of histone genes: Prevalence of an “orphon” H1 lineage in protostomes and birth-and-death process in the H2A family. J. Mol. Evol. 2008, 66, 505–518. [Google Scholar]
- González-Romero, R.; Ausió, J.; Méndez, J.; Eirín-López, J.M. Histone genes of the razor clam Solen marginatus unveil new aspects of linker histone evolution in protostomes. Genome 2009, 52, 597–607. [Google Scholar]
- González-Romero, R.; Rivera-Casas, C.; Frehlick, L.J.; Méndez, J.; Ausió, J.; Eirín-López, J.M. Histone H2A (H2A.X and H2A.Z) variants in molluscs: Molecular characterization and potential implications for chromatin dynamics. PLoS One 2012, 7, e30006. [Google Scholar]
- Gonzalez-Romero, R.; Rivera-Casas, C.; Fernandez-Tajes, J.; Ausio, J.; Méndez, J.; Eirín-López, J.M. Chromatin specialization in bivalve molluscs: A leap forward for the evaluation of okadaic acid genotoxicity in the marine environment. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2012, 155, 175–181. [Google Scholar]
- NCBI Genome. Available online: http://www.ncbi.nlm.nih.gov/genome/ (accessed on 3 March 2013).
- RefSeq. Available online: http://www.ncbi.nlm.nih.gov/refseq/ (accessed on 3 March 2013).
- Gene Expression Omnibus. Available online: http://www.ncbi.nlm.nih.gov/geo/ (accessed on 4 March 2013).
- Genbank. Available online: http://www.ncbi.nlm.nih.gov/genbank/ (accessed on 3 March 2013).
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; Yefanov, A.; et al. NCBI GEO: archive for functional genomics data sets-update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar]
- SRA. Available online: http://www.ncbi.nlm.nih.gov/sra/ (accessed on 6 March 2013).
- TSA. Available online: http://www.ncbi.nlm.nih.gov/genbank/tsa/ (accessed on 6 March 2013).
- Murgarella, M.; Novoa, B.; Figueras, A.; Posada, D.; Canchaya, C. Proceedings of the International Society of Fire Service Instructors (ISFSI), First Insights into the Genome of Mytilus Galloprovincialis: De Novo Sequencing of a Non-Model Marine Organism. Vigo, Spain, 25–28 June 2013.
- Banni, M.; Negri, A.; Mignone, F.; Boussetta, H.; Viarengo, A.; Dondero, F. Gene expression rhythms in the mussel Mytilus galloprovincialis (Lam.) across an annual cycle. PLoS One 2011, 6, e18904. [Google Scholar]
- Chaney, M.L.; Gracey, A.Y. Mass mortality in Pacific oysters is associated with a specific gene expression signature. Mol. Ecol. 2011, 20, 2942–2954. [Google Scholar]
- Dheilly, N.M.; Lelong, C.; Huvet, A.; Favrel, P. Development of a Pacific oyster (Crassostrea gigas) 31,918-feature microarray: Identification of reference genes and tissue-enriched expression patterns. BMC Genomics 2011, 12, 468. [Google Scholar]
- Fleury, E.; Huvet, A. Microarray analysis highlights immune response of pacific oysters as a determinant of resistance to summer mortality. Mar. Biotechnol. (N.Y.) 2012, 14, 203–217. [Google Scholar]
- Fleury, E.; Moal, J.; Boulo, V.; Daniel, J.Y.; Mazurais, D.; Henaut, A.; Corporeau, C.; Boudry, P.; Favrel, P.; Huvet, A. Microarray-based identification of gonad transcripts differentially expressed between lines of Pacific oyster selected to be resistant or susceptible to summer mortality. Mar. Biotechnol. (N.Y.) 2010, 12, 326–339. [Google Scholar]
- Gardner, L.D.; Mills, D.; Wiegand, A.; Leavesley, D.; Elizur, A. Spatial analysis of biomineralization associated gene expression from the mantle organ of the pearl oyster Pinctada maxima. BMC Genomics 2011, 12, 455. [Google Scholar]
- Place, S.P.; Menge, B.A.; Hofmann, G.E. Transcriptome profiles link environmental variation and physiological response of Mytilus californianus between Pacific tides. Funct. Ecol. 2012, 26, 144–155. [Google Scholar] [CrossRef]
- Varotto, L.; Domeneghetti, S.; Rosani, U.; Manfrin, C.; Cajaraville, M.P.; Raccanelli, S.; Pallavicini, A.; Venier, P. DNA damage and transcriptional changes in the gills of Mytilus galloprovincialis exposed to nanomolar doses of combined metal salts (Cd, Cu, Hg). PLoS One 2013, 8, e54602. [Google Scholar] [CrossRef]
- Kennish, M.J. Practical Handbook of Estuarine and Marine Pollution; CRC Press: Boca Raton, FL, USA, 1996; p. 544. [Google Scholar]
- McKillen, D.J.; Chen, Y.A.; Chen, C.; Jenny, M.J.; Trent, H.F., III; Robalino, J.; McLean, D.C., Jr.; Gross, P.S.; Chapman, R.W.; Warr, G.W.; et al. Marine genomics: A clearing-house for genomic and transcriptomic data of marine organisms. BMC Genomic 2005, 6, 34. [Google Scholar]
- OIST Marine Genomics Unit. Available online: http://marinegenomics.oist.jp/ (accessed on 27 February 2013).
- Venier, P.; de Pitta, C.; Bernante, F.; Varotto, L.; de Nardi, B.; Bovo, G.; Roch, P.; Novoa, B.; Figueras, A.; Pallavicini, A.; et al. MytiBase: A knowledgebase of mussel (M. galloprovincialis) transcribed sequences. BMC Genomics 2005, 10, 72. [Google Scholar]
- Fleury, E.; Huvet, A.; Lelong, C.; de Lorgeril, J.; Boulo, V.; Gueguen, Y.; Bachere, E.; Tanguy, A.; Moraga, D.; Fabioux, C.; et al. Generation and analysis of a 29,745 unique Expressed Sequence Tags from the Pacific oyster (Crassostrea gigas) assembled into a publicly accessible database: The GigasDatabase. BMC Genomics 2009, 10, 341. [Google Scholar] [CrossRef] [Green Version]
- Mytibase. Available online: http://mussel.cribi.unipd.it (accessed on 27 February 2013).
- Public Sigenae Contig Browser. Available online: http://public-contigbrowser.sigenae.org:9090/Crassostrea_gigas/index.html (accessed on 27 February 2013).
- Ruphibase. Available online: http://compgen.bio.unipd.it/ruphibase (accessed on 28 February 2013).
- Chameleabase. Available online: http://compgen.bio.unipd.it/chameleabase (accessed on 27 February 2013).
- Deep Sea Vent. Bathymodiolus Azoricus. Available online: http://transcriptomics.biocant.pt:8080/deepSeaVent (accessed on 25 February 2013).
- CHROMEVALOAdb. Available online: http://chromevaloa.com (accessed on 28 February 2013).
- Suárez-Ulloa, V.; Fernández-Tajes, J.; Eirín-López, J.M. Florida International University: Miami, FL, USA., 2013; Unpublished work.
- Meistertzheim, A.L.; Tanguy, A.; Moraga, D.; Thebault, M.T. Identification of differentially expressed genes of the Pacific oyster Crassostrea gigas exposed to prolonged thermal stress. FEBS J. 2007, 274, 6392–6402. [Google Scholar] [CrossRef]
- David, E.; Tanguy, A.; Pichavant, K.; Moraga, D. Response of the Pacific oyster Crassostrea gigas to hypoxia exposure under experimental conditions. FEBS J. 2005, 272, 5635–5652. [Google Scholar] [CrossRef]
- Araya, M.T.; Markham, F.; Mateo, D.R.; McKenna, P.; Johnson, G.R.; Berthe, F.C.; Siah, A. Identification and expression of immune-related genes in hemocytes of soft-shell clams, Mya arenaria, challenged with Vibrio splendidus. Fish. Shellfish Immunol. 2010, 29, 557–564. [Google Scholar] [CrossRef]
- Morga, B.; Renault, T.; Faury, N.; Chollet, B.; Arzul, I. Cellular and molecular responses of haemocytes from Ostrea edulis during in vitro infection by the parasite Bonamia ostreae. Int.J. Parasitol. 2011, 41, 755–764. [Google Scholar] [CrossRef]
- Gerdol, M. University of Trieste: Trieste, Italy, 2013; Unpublished work.
- Diaz de Cerio, O.; Hands, E.; Humble, J.; Cajaraville, M.P.; Craft, J.A.; Cancio, I. Construction and characterization of a forward subtracted library of blue mussels Mytilus edulis for the identification of gene transcription signatures and biomarkers of styrene exposure. Mar. Pollut. Bull. 2013, 71, 230–239. [Google Scholar]
- Dondero, F.; Banni, M.; Negri, A.; Boatti, L.; Dagnino, A.; Viarengo, A. Interactions of a pesticide/heavy metal mixture in marine bivalves: A transcriptomic assessment. BMC Genomics 2011, 12, 195. [Google Scholar]
- Liu, F.; Wang, W.X. Proteome pattern in oysters as a diagnostic tool for metal pollution. J. Hazard. Mater. 2012, 239–240, 241–248. [Google Scholar] [CrossRef]
- Gomes, T.; Pereira, C.G.; Cardoso, C.; Bebianno, M.J. Differential protein expression in mussels Mytilus galloprovincialis exposed to nano and ionic Ag. Aquat.Toxicol. 2013, 136–137, 79–90. [Google Scholar] [CrossRef]
- Tomanek, L. Environmental proteomics of the mussel Mytilus: Implications for tolerance to stress and change in limits of biogeographic ranges in response to climate change. Integr. Comp. Biol. 2012, 52, 648–664. [Google Scholar]
- Puerto, M.; Campos, A.; Prieto, A.; Camean, A.; de Almeida, A.M.; Coelho, A.V.; Vasconcelos, V. ifferential protein expression in two bivalve species; Mytilus galloprovincialis and Corbicula fluminea; exposed to Cylindrospermopsis raciborskii cells. Aquat. Toxicol. 2011, 101, 109–116. [Google Scholar] [CrossRef]
- Nzoughet, J.K.; Hamilton, J.T.; Botting, C.H.; Douglas, A.; Devine, L.; Nelson, J.; Elliott, C.T. Proteomics identification of azaspiracid toxin biomarkers in blue mussels, Mytilus edulis. Mol. Cell. Proteomics 2009, 8, 1811–1822. [Google Scholar]
- Tomanek, L.; Zuzow, M.J.; Hitt, L.; Serafini, L.; Valenzuela, J.J. Proteomics of hyposaline stress in blue mussel congeners (genus Mytilus): Implications for biogeographic range limits in response to climate change. J. Exp. Biol. 2012, 215, 3905–3916. [Google Scholar]
- Fields, P.A.; Cox, K.M.; Karch, K.R. Latitudinal variation in protein expression after heat stress in the salt marsh mussel Geukensia demissa. Integr.Comp.Biol. 2012, 52, 636–647. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Suárez-Ulloa, V.; Fernández-Tajes, J.; Manfrin, C.; Gerdol, M.; Venier, P.; Eirín-López, J.M. Bivalve Omics: State of the Art and Potential Applications for the Biomonitoring of Harmful Marine Compounds. Mar. Drugs 2013, 11, 4370-4389. https://doi.org/10.3390/md11114370
Suárez-Ulloa V, Fernández-Tajes J, Manfrin C, Gerdol M, Venier P, Eirín-López JM. Bivalve Omics: State of the Art and Potential Applications for the Biomonitoring of Harmful Marine Compounds. Marine Drugs. 2013; 11(11):4370-4389. https://doi.org/10.3390/md11114370
Chicago/Turabian StyleSuárez-Ulloa, Victoria, Juan Fernández-Tajes, Chiara Manfrin, Marco Gerdol, Paola Venier, and José M. Eirín-López. 2013. "Bivalve Omics: State of the Art and Potential Applications for the Biomonitoring of Harmful Marine Compounds" Marine Drugs 11, no. 11: 4370-4389. https://doi.org/10.3390/md11114370