DNA Hybridization Sensors Based on Electrochemical Impedance Spectroscopy as a Detection Tool

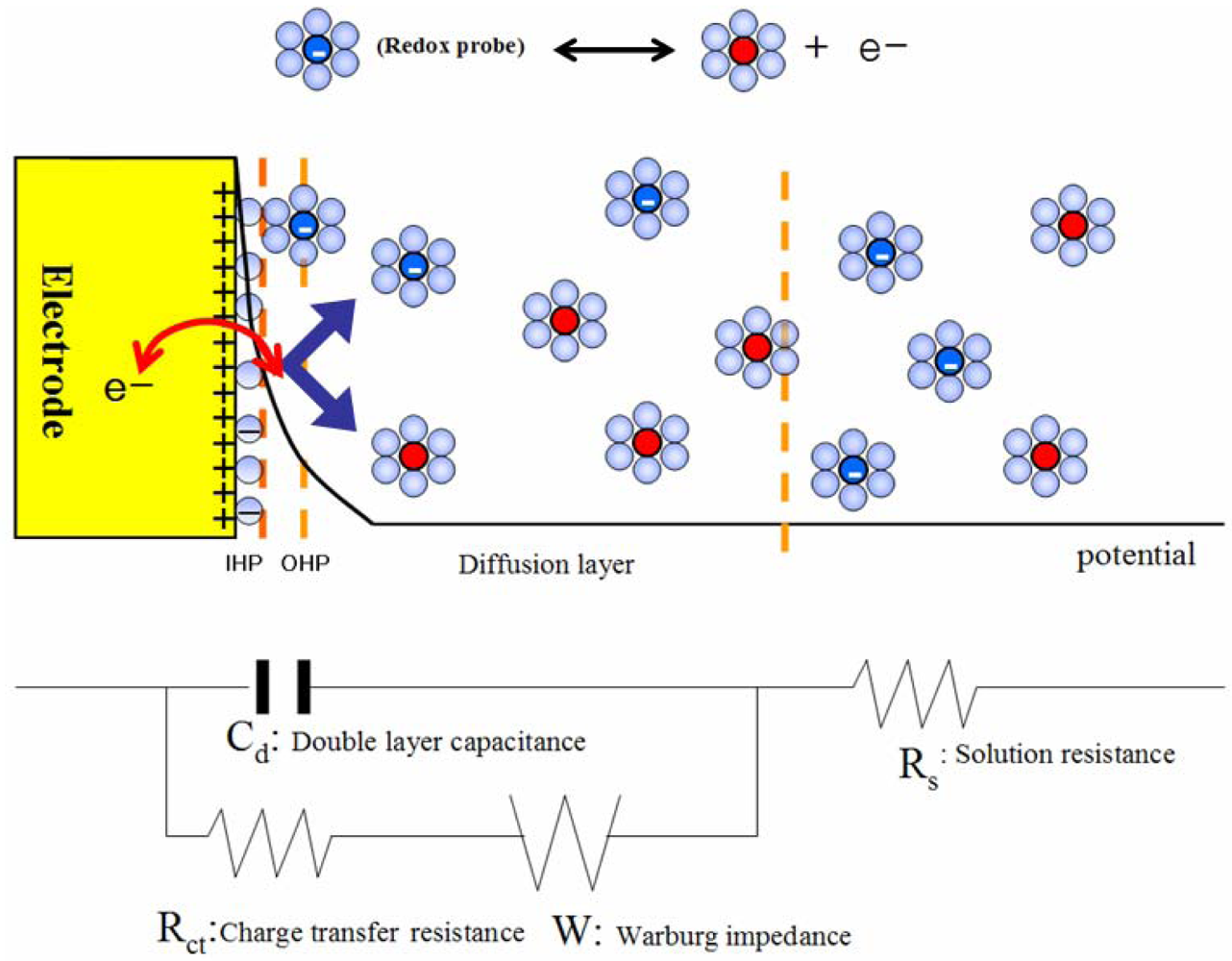{kind=link}
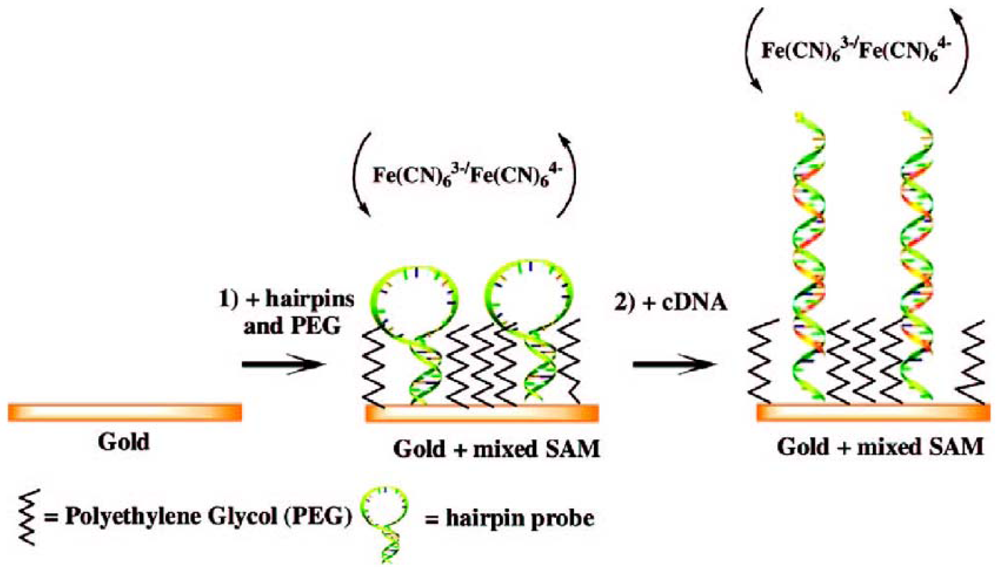{kind=link}
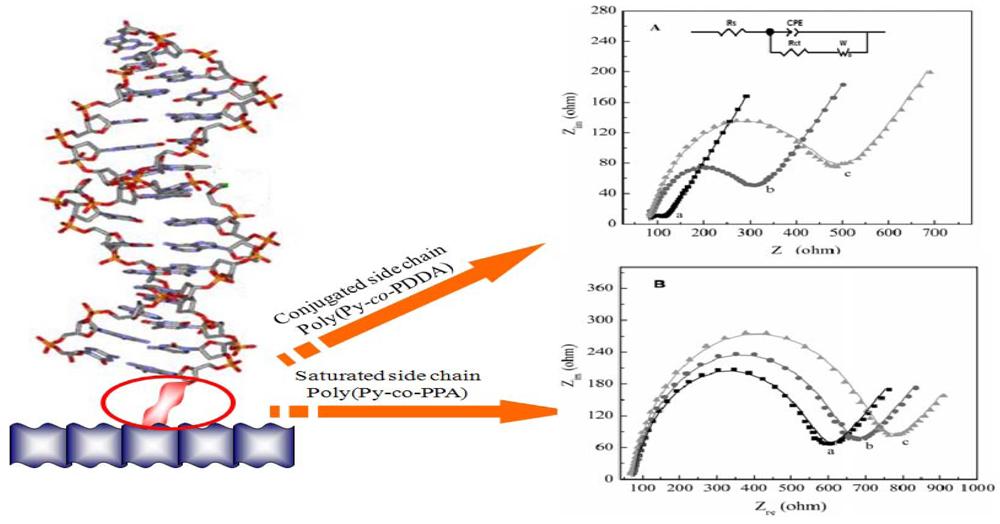{kind=link}
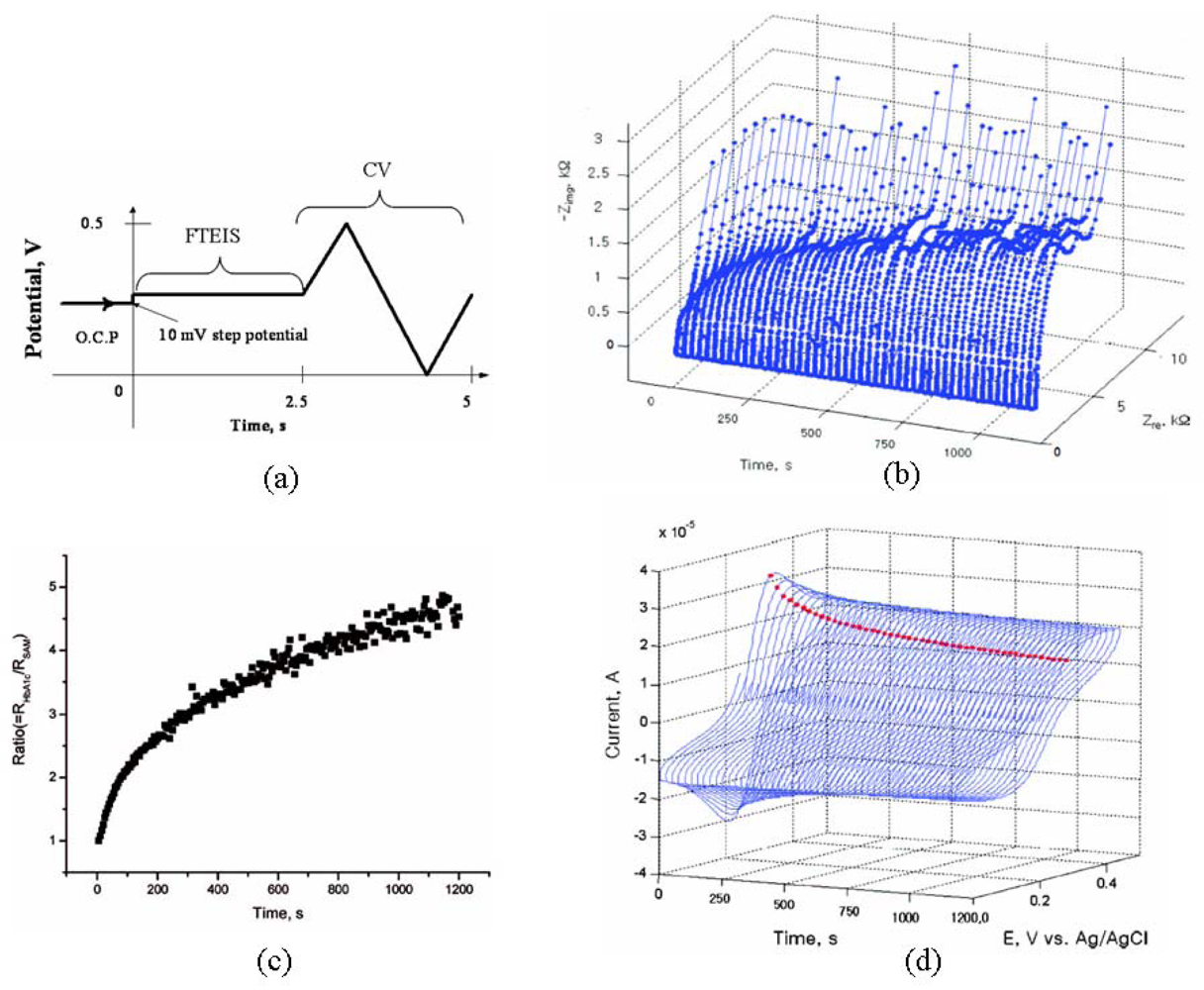{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. DNA Sensors Based on Self-Assembled Monolayers
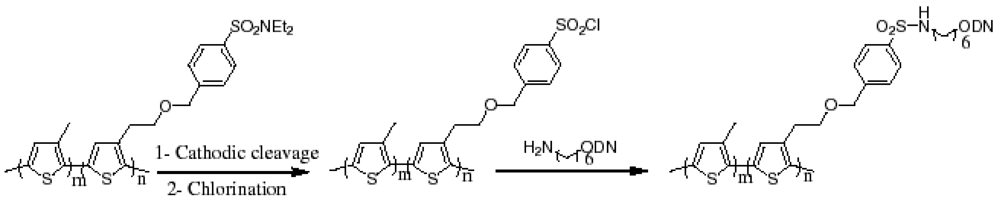
3. DNA Sensors Based on Conducting Polymers
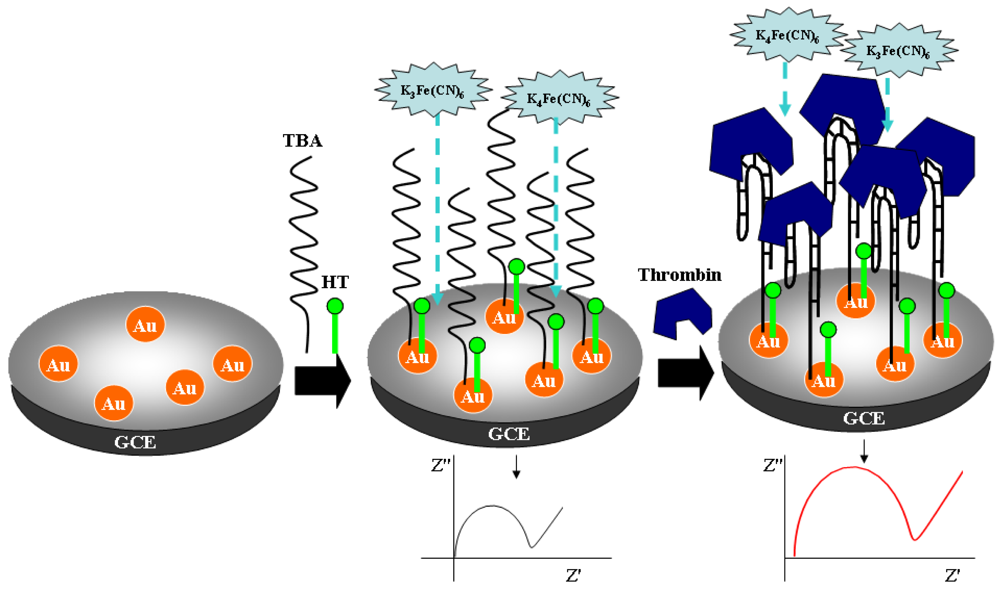
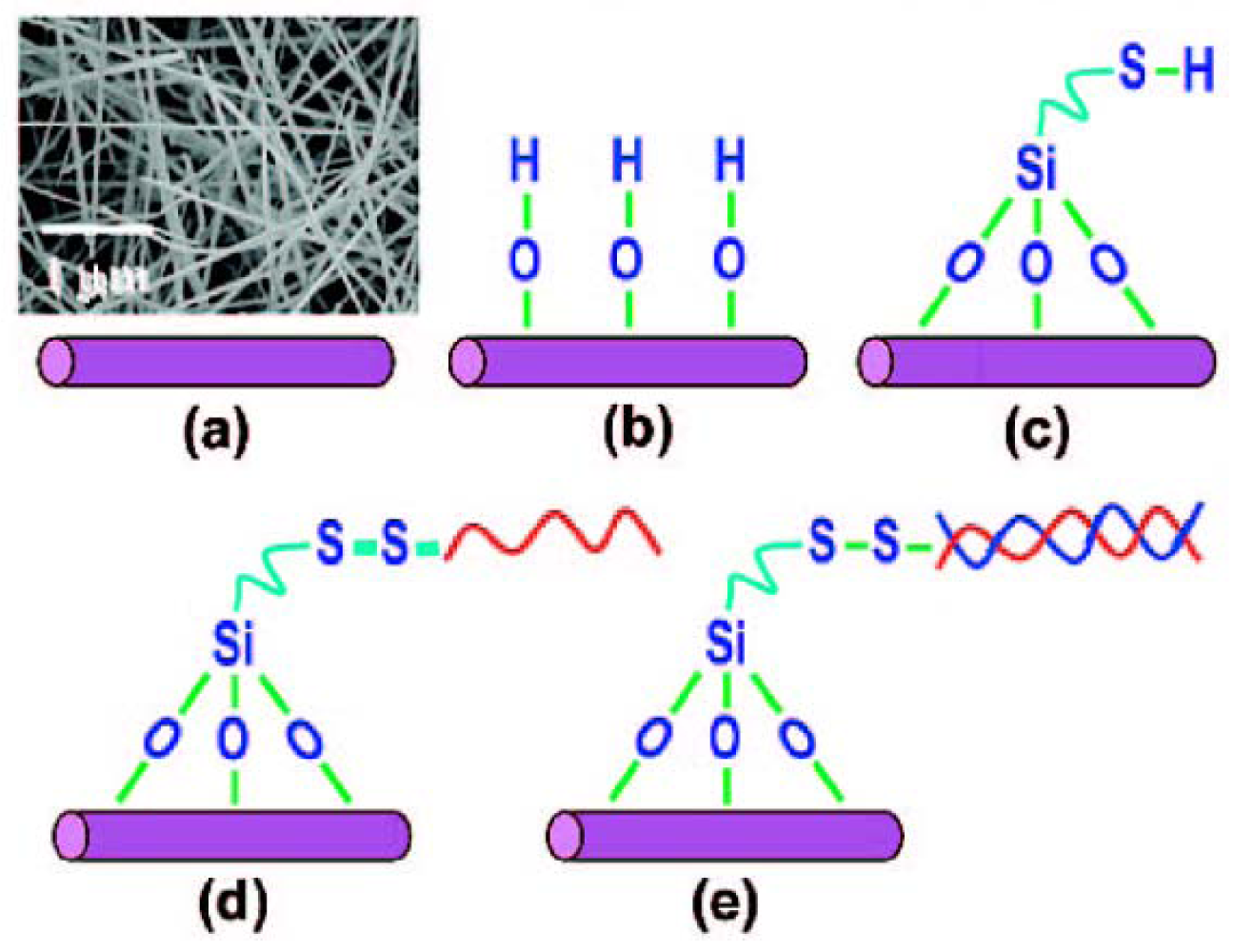
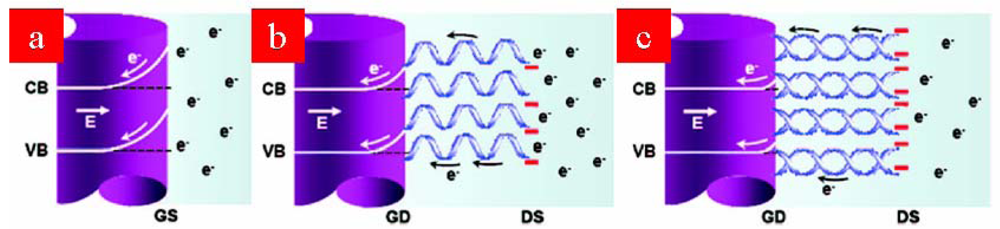
4. DNA Sensors Based on Nanomaterials
5. Amperometric and Voltammetric Detection Versus the EIS Detection
6. Conclusions
Acknowledgments
References and Notes
- Liu, J.; Cao, Z.; Lu, Y. Functional nucleic acid sensors. Chem. Rev. 2009, 109, 1948–1998. [Google Scholar]
- Wilson, J.N.; Yin, N.T.; Kool, E.T. Efficient quenching of oligomeric fluorophores on a DNA backbone. J. Am. Chem. Soc. 2007, 129, 15426–15427. [Google Scholar]
- Yang, R.; Jin, J.; Chen, Y.; Shao, N.; Kang, H.; Xiao, Z.; Tang, Z.; Wu, Y.; Zhu, Z.; Tan, W. Carbon nanotube-quenched fluorescent oligonucleotides: Probes that fluoresce upon hybridization. J. Am. Chem. Soc. 2008, 130, 8351–8358. [Google Scholar]
- Baselt, D.R.; Lee, G.U.; Natesan, M.; Metzger, S.W.; Sheehan, P.E.; Colton, R.J. A biosensor based on magnetoresistance technology. Biosens. Bioelectron 1998, 13, 731–739. [Google Scholar]
- Niu, S.Y.; Li, Q.Y.; Ren, R.; Zhang, S.S. Enzyme-enhanced fluorescence detection of DNA on etched optical fibers. Biosens. Bioelectron 2009, 24, 2943–2946. [Google Scholar]
- Guan, J.-G.; Miao, Y.-Q.; Zhang, Q.-J. Impedimetric biosensors. J. Biosci. Bioeng 2004, 97, 219–226. [Google Scholar]
- Cash, K.J.; Heeger, A.J.; Plaxco, K.W.; Xiao, Y. Optimization of a Reusable, DNA Pseudoknot-Based Electrochemical Sensor for Sequence-Specific DNA Detection in Blood Serum. Anal. Chem. 2009, 81, 656–661. [Google Scholar]
- Zhang, S.S.; Xia, J.P.; Li, X.M. Electrochemical Biosensor for Detection of Adenosine Based on Structure-Switching Aptamer and Amplification with Reporter Probe DNA Modified Au Nanoparticles. Anal. Chem. 2008, 80, 8382–8388. [Google Scholar]
- Zuo, S.H.; Zhang, L.F.; Yuan, H.H.; Lan, M.B.; Lawrance, G.A.; Wei, G. Electrochemical detection of DNA hybridization by using a zirconia modified renewable carbon paste electrode. Bioelectrochemistry 2009, 74, 223–226. [Google Scholar]
- Singh, R.; Prasad, R.; Sumana, G.; Arora, K.; Sood, S.; Gupta, R.K.; Malhotra, B.D. STD sensor based on nucleic acid functionalized nanostructured polyaniline. Biosens. Bioelectron 2009, 24, 2232–2238. [Google Scholar]
- Yang, T.; Zhang, W.; Du, M.; Jiao, K. A PDDA/poly(2,6-pyridinedicarboxylic acid)-CNTs composite film DNA electrochemical sensor and its application for the detection of specific sequences related to PAT gene and NOS gene. Talanta 2008, 75, 987–994. [Google Scholar]
- Shiddiky, M.J.A.; Rahman, M.A.; Cheol, C.S.; Shim, Y.B. Fabrication of disposable sensors for biomolecule detection using hydrazine electrocatalyst. Anal. Biochem 2008, 379, 170–175. [Google Scholar]
- Pournaghi-Azar, M.H.; Alipour, E.; Zununi, S.; Froohandeh, H.; Hejazi, M.S. Direct and rapid electrochemical biosensing of the human interleukin-2 DNA in unpurified polymerase chain reaction (PCR)-amplified real samples. Biosens. Bioelectron 2008, 24, 524–530. [Google Scholar]
- Evtugyn, G.A.; Goldfarb, O.E.; Budnikov, H.C.; Ivanov, A.N.; Vinter, V.G. Amperometric DNA-peroxidase sensor for the detection of pharmaceutical preparations. Sensors 2005, 5, 364–376. [Google Scholar]
- Park, S.M.; Yoo, J.S. Electrochemical impedance spectroscopy for better electrochemical measurements. Anal. Chem. 2003, 75, 455A–461A. [Google Scholar]
- Tang, L.; Zeng, G.; Shen, G.; Li, Y.; Liu, C.; Li, Z.; Luo, J.; Fan, C.; Yang, C. Sensitive detection of lip genes by electrochemical DNA sensor and its application in polymerase chain reaction amplicons from Phanerochaete chrysosporium. Biosens. Bioelectron 2009, 24, 1474–1479. [Google Scholar]
- Patel, M.K.; Solanki, P.R.; Seth, S.; Gupta, S.; Khare, S.; Kumar, A.; Malhotra, B.D. CtrA gene based electrochemical DNA sensor for detection of meningitis. Electrochem. Commun 2009, 11, 969–973. [Google Scholar]
- Katz, E.; Willner, I. Probing Biomolecular Interactions at Conductive and Semiconductive Surfaces by Impedance Spectroscopy: Routes to Impedimetric Immunosensors, DNA-Sensors, and Enzyme Biosensors. Electroanalysis 2003, 15, 913–947. [Google Scholar]
- Berggren, C.; Bjarnason, B.; Johansson, G. Capacitive Biosensors. Electroanalysis 2001, 13, 173–180. [Google Scholar]
- Ito, T.; Hosokawa, K.; Maeda, M. Detection of single-base mismatch at distal end of DNA duplex by electrochemical impedance spectroscopy. Biosens. Bioelectron 2007, 22, 1816–1819. [Google Scholar]
- Li, D.L.; Zou, X.Q.; Shen, Q.; Dong, S.J. Kinetic study of DNA/DNA hybridization with electrochemical impedance spectroscopy. Electrochem. Commun 2007, 9, 191–196. [Google Scholar]
- Long, Y.T.; Li, C.Z.; Kraatz, H.B.; Lee, J.S. AC impedance spectroscopy of native DNA and M-DNA. Biophys. J 2003, 84, 3218–3225. [Google Scholar]
- Long, Y.T.; Li, C.Z.; Sutherland, T.C.; Kraatz, H.B.; Lee, J.S. Electrochemical detection of single-nucleotide mismatches: application of M-DNA. Anal. Chem. 2004, 76, 4059–4065. [Google Scholar]
- Li, X.; Lee, J.S.; Kraatz, H.B. Electrochemical detection of single-nucleotide mismatches using an electrode microarray. Anal. Chem. 2006, 78, 6096–6101. [Google Scholar]
- Li, X.; Zhou, Y.; Sutherland, T.C.; Baker, B.; Lee, J.S.; Kraatz, H.B. Chip-based microelectrodes for detection of single-nucleotide mismatch. Anal. Chem. 2005, 77, 5766–5769. [Google Scholar]
- Wang, Y.; Li, C.; Li, X.; Li, Y.; Kraatz, H.-B. Unlabeled hairpin-DNA probe for the detection of single-nucleotide mismatches by electrochemical impedance spectroscopy. Anal. Chem. 2008, 80, 2255–2260. [Google Scholar]
- Bin, X.; Kraatz, H.-B. Interaction of metal ions and DNA films on gold surfaces: an electrochemical impedance study. Analyst 2009, 134, 1309–1313. [Google Scholar]
- Patolsky, F.; Lichtenstein, A.; Willner, I. Electronic transduction of DNA sensing processes on surfaces: Amplification of DNA detection and analysis of single-base mismatches by tagged liposomes. J. Am. Chem. Soc. 2001, 123, 5194–5205. [Google Scholar]
- Lucarelli, F.; Marrazza, G.; Mascini, M. Enzyme-based impedimetric detection of PCR products using oligonucleotide-modified screen-printed gold electrodes. Biosens. Bioelectron 2005, 20, 2001–2009. [Google Scholar]
- Herne, T.M.; Tarlov, M.J. Characterization of DNA probes immobilized on gold surfaces. J. Am. Chem. Soc. 1997, 119, 8916–8920. [Google Scholar]
- Pan, C.F.; Guo, M.L.; Nie, Z.; Xiao, X.L.; Yao, S.Z. Aptamer-based electrochemical sensor for label-free recognition and detection of cancer cells. Electroanalysis 2009, 21, 1321–1326. [Google Scholar]
- Keighley, S.D.; Estrela, P.; Li, P.; Mighorato, P. Optimization of label-free DNA detection with electrochemical impedance spectroscopy using PNA probes. Biosens. Bioelectron 2008, 24, 906–911. [Google Scholar]
- Kjällman, T.H.M.; Peng, H.; Soeller, C.; Travas-Sejdic, J. Effect of probe density and hybridization temperature on the response of an electrochemical hairpin-DNA sensor. Anal. Chem. 2008, 80, 9460–9466. [Google Scholar]
- Kafka, J.; Pänke, O.; Abendroth, B.; Lisdat, F. A label-free DNA sensor based on impedance spectroscopy. Electrochim. Acta. 2008, 53, 7467–7474. [Google Scholar]
- Snejdarkova, M.; Svobodova, L.; Nikolelis, D.P.; Wang, J.; Hianik, T. Acetylcholine biosensor based on dendrimer layers for pesticides detection. Electroanalysis 2003, 15, 1185–1191. [Google Scholar]
- Svobodová, L.; Šnejdárková, M.; Hianik, T. Properties of glucose biosensors based on dendrimer layers. Effect of enzyme immobilization. Anal. Bioanal. Chem. 2002, 373, 735–741. [Google Scholar]
- Svobodová, L.; Šnejdárková, M.; Tóth, K.; Gyurcsanyi, R.E.; Hianik, T. Properties of mixed alkanethiol-dendrimer layers and their applications in biosensing. Bioelectrochemistry 2004, 63, 285–289. [Google Scholar]
- Zhu, N.; Gu, Y.; Chang, Z.; He, P.; Fang, Y. PAMAM dendrimers-based DNA biosensors for electrochemical detection of DNA hybridization. Electroanalysis 2006, 18, 2107–2114. [Google Scholar]
- Zhu, N.; Gao, H.; Gu, Y.; Xu, Q.; He, P.; Fang, Y. PAMAM dendrimer-enhanced DNA biosensors based on electrochemical impedance spectroscopy. Analyst 2009, 134, 860–866. [Google Scholar]
- Park, J.Y.; Kwon, S.H.; Park, J.W.; Park, S.M. Label-free detection of DNA molecules on the dendron based self-assembled monolayer by electrochemical impedance spectroscopy. Anal. Chim. Acta. 2008, 619, 37–42. [Google Scholar]
- Li, X.; Song, H.; Nakatani, K.; Kraatz, H.-B. Exploiting small molecule binding to DNA for the detection of single-nucleotide mismatches and their base environment. Anal. Chem. 2007, 79, 2552–2555. [Google Scholar]
- Gong, H.; Zhong, T.; Gao, L.; Li, X.; Bi, L.; Kraatz, H.-B. Unlabeled hairpin DNA for electrochemical detection of single-nucleotide mismatches based on MutS-DNA interactions. Anal. Chem. 2009, 81, 8639–8643. [Google Scholar]
- Rahman, M.A.; Kumar, P.; Park, D.S.; Shim, Y.B. Electrochemical sensors based on organic conjugated polymers. Sensors 2008, 8, 118–141. [Google Scholar]
- Minehan, D.S.; Marx, K.A.; Tripathy, S.K. Kinetics of DNA binding to electrically conducting polypyrrole films. Macromolecules 1994, 27, 777–783. [Google Scholar]
- Paleček, E. Past, present and future of nucleic acids electrochemistry. Talanta 2002, 56, 809–819. [Google Scholar]
- Peng, H.; Soeller, C.; Cannell, M.B.; Bowmaker, G.A.; Cooney, R.P.; Travas-Sejdic, J. Electrochemical detection of DNA hybridization amplified by nanoparticles. Biosens. Bioelectron 2006, 21, 1727–1736. [Google Scholar]
- Travas-Sejdic, J.; Peng, H.; Cooney, R.P.; Bowmaker, G.A.; Cannell, M.B.; Soeller, C. Amplification of a conducting polymer-based DNA sensor signal by CdS nanoparticles. Curr. Appl. Phys. 2006, 6, 562–566. [Google Scholar]
- Lassalle, N.; Mailley, P.; Vieil, E.; Livache, T.; Roget, A.; Correia, J.P.; Abrantes, L.M. Electronically conductive polymer grafted with oligonucleotides as electrosensors of DNA: Preliminary study of real time monitoring by in situ techniques. J. Electroanal. Chem. 2001, 509, 48–57. [Google Scholar]
- Delabouglise, D.; Roncali, J.; Lemaire, M.; Garnier, F. Control of the lipophilicity of polypyrrole by 3-alkyl substitution. J. Chem.l Soc. Chem. Commun 1989, 8, 475–477. [Google Scholar]
- Peng, H.; Soeller, C.; Vigar, N.; Kilmartin, P.A.; Cannell, M.B.; Bowmaker, G.A.; Cooney, R.P.; Travas-Sejdic, J. Label-free electrochemical DNA sensor based on functionalised conducting copolymer. Biosens. Bioelectron 2005, 20, 1821–1828. [Google Scholar]
- Travas-Sejdic, J.; Peng, H.; Kilmartin, P.A.; Cannell, M.B.; Bowmaker, G.A.; Cooney, R.P.; Soeller, C. DNA sensor based on functionalized polypyrrole. Synthet. Metal. 2005, 152, 37–40. [Google Scholar]
- Peng, H.; Soeller, C.; Travas-Sejdic, J. Novel conducting polymers for DNA sensing. Macromolecules 2007, 40, 909–914. [Google Scholar]
- Peng, H.; Soeller, C.; Vigar, N.A.; Caprio, V.; Travas-Sejdic, J. Label-free detection of DNA hybridization based on a novel functionalized conducting polymer. Biosens. Bioelectron 2007, 22, 1868–1873. [Google Scholar]
- Lee, T.Y.; Shim, Y.B. Direct DNA hybridization detection based on the oligonucleotide-functionalized conductive polymer. Anal. Chem. 2001, 73, 5629–5632. [Google Scholar]
- Peng, H.; Zhang, L.; Spires, J.; Soeller, C.; Travas-Sejdic, J. Synthesis of a functionalized polythiophene as an active substrate for a label-free electrochemical genosensor. Polymer 2007, 48, 3413–3419. [Google Scholar]
- Spires, J.B.; Peng, H.; Williams, D.E.; Wright, B.E.; Soeller, C.; Travas-Sejdic, J. The effect of the oxidation state of a terthiophene-conducting polymer and of the presence of a redox probe on its gene-sensing properties. Biosens. Bioelectron 2008, 24, 928–933. [Google Scholar]
- Gautier, C.; Esnault, C.; Cougnon, C.; Pilard, J.F.; Casse, N.; Chénais, B. Hybridization-induced interfacial changes detected by non-Faradaic impedimetric measurements compared to Faradaic approach. J. ElectroAnal. Chem. 2007, 610, 227–233. [Google Scholar]
- Gautier, C.; Cougnon, C.; Pilard, J.F.; Casse, N. Label-free detection of DNA hybridization based on EIS investigation of conducting properties of functionalized polythiophene matrix. J. ElectroAnal. Chem. 2006, 587, 276–283. [Google Scholar]
- Gautier, C.; Cougnon, C.; Pilard, J.F.; Casse, N.; Chénais, B.; Laulier, M. Detection and modelling of DNA hybridization by EIS measurements. Mention of a polythiophene matrix suitable for electrochemically controlled gene delivery. Biosens. Bioelectron 2007, 22, 2025–2031. [Google Scholar]
- Davis, F.; Hughes, M.A.; Cossins, A.R.; Higson, S.P.J. Single gene differentiation by DNA-modified carbon electrodes using an AC impedimetric approach. Anal. Chem. 2007, 79, 1153–1157. [Google Scholar]
- Davis, F.; Nabok, A.V.; Higson, S.P.J. Species differentiation by DNA-modified carbon electrodes using an ac impedimetric approach. Biosens. Bioelectron 2005, 20, 1531–1538. [Google Scholar]
- Weng, J.; Zhang, J.; Li, H.; Sun, L.; Lin, C.; Zhang, Q. Label-free DNA sensor by boron-doped diamond electrode using an ac impedimetric approach. Anal. Chem. 2008, 80, 7075–7083. [Google Scholar]
- Gu, H.; Su, X.D.; Loh, K.P. Electrochemical impedance sensing of DNA hybridization on conducting polymer film-modified diamond. J. Phys. Chem. B 2005, 109, 13611–13618. [Google Scholar]
- Zhou, N.; Yang, T.; Jiang, C.; Du, M.; Jiao, K. Highly sensitive electrochemical impedance spectroscopic detection of DNA hybridization based on Au-nano-CNT/PAN(nano) films. Talanta 2009, 77, 1021–1026. [Google Scholar]
- Zhang, W.; Yang, T.; Yin, C.; Li, G.; Jiao, K. An enhanced strategy using poly(m-aminobenzene sulfonic acid) nanofibres for rapid detection of DNA oxidative damage induced by hollow TiO2 nanocubes. Electrochem. Commun 2009, 11, 783–786. [Google Scholar]
- Yang, T.; Zhou, N.; Zhang, Y.; Zhang, W.; Jiao, K.; Li, G. Synergistically improved sensitivity for the detection of specific DNA sequences using polyaniline nanofibers and multi-walled carbon nanotubes composites. Biosens. Bioelectron 2009, 24, 2165–2170. [Google Scholar]
- Wang, J. Nanoparticle-based electrochemical DNA detection. Anal. Chim. Acta. 2003, 500, 247–257. [Google Scholar]
- Xu, Y.; Cai, H.; He, P.G.; Fang, Y.Z. Probing DNA hybridization by impedance measurement based on CdS-oligonucleotide nanoconjugates. Electroanalysis 2004, 16, 150–155. [Google Scholar]
- Zhang, K.; Ma, H.; Zhang, L.; Zhang, Y. Fabrication of a sensitive impedance biosensor of DNA hybridization based on gold nanoparticles modified gold electrode. Electroanalysis 2008, 20, 2127–2133. [Google Scholar]
- Bonanni, A.; Esplandiu, M.J.; del Valle, M. Signal amplification for impedimetric genosensing using gold-streptavidin nanoparticles. Electrochim. Acta. 2008, 53, 4022–4029. [Google Scholar]
- Bonanni, A.; Esplandiu, M.J.; del Valle, M. Impedimetric genosensors employing COOH-modified carbon nanotube screen-printed electrodes. Biosens. Bioelectron 2009, 24, 2885–2891. [Google Scholar]
- He, P.L.; Shen, L.; Cao, Y.H.; Lia, D.F. Ultrasensitive electrochemical detection of proteins by amplification of aptamer-nanoparticle bio bar codes. Anal. Chem. 2007, 79, 8024–8029. [Google Scholar]
- Li, C.Z.; Liu, Y.L.; Luong, J.H.T. Impedance sensing of DNA binding drugs using gold substrates modified with gold nanoparticles. Anal. Chem. 2005, 77, 478–485. [Google Scholar]
- Zhang, K.I.; Ma, H.Y.; Zhang, L.P.; Zhang, Y.Z. Fabrication of a Sensitive Impedance Biosensor of DNA Hybridization Based on Gold Nanoparticles Modified Gold Electrode. Electroanalysis 2008, 20, 2127–2133. [Google Scholar]
- Li, X.X.; Shen, L.H.; Zhang, D.D.; Qi, H.L.; Gao, Q.; Ma, F.; Zhang, C.X. Electrochemical impedance spectroscopy for study of aptamer-thrombin interfacial interactions. Biosens. Bioelectron 2008, 23, 1624–1630. [Google Scholar]
- Deng, C.Y.; Chen, J.H.; Nie, Z.; Wang, M.D.; Chu, X.C.; Chen, X.L.; Xiao, X.L.; Lei, C.Y.; Yao, S.Z. Impedimetric aptasensor with femtomolar sensitivity based on the enlargement of surface-charged gold nanoparticles. Anal. Chem. 2009, 81, 739–745. [Google Scholar]
- Hassen, W.M.; Chaix, C.; Abdelghani, A.; Bessueille, F.; Leonard, D.; Jaffrezic-Renault, N. An impedimetric DNA sensor based on functionalized magnetic nanoparticles for HIV and HBV detection. Sensor. Actuator. B-Chem. 2008, 134, 755–760. [Google Scholar]
- Suehiro, J.; Ikeda, N.; Ohtsubo, A.; Imasaka, K. Fabrication of bio/nano interfaces between biological cells and carbon nanotubes using dielectrophoresis. Microfluid. Nanofluid 2008, 5, 741–747. [Google Scholar]
- Steinhoff, G.; Purrucker, O.; Tanaka, M.; Stutzmann, M.; Eickhoff, M. AlxGa1-xN—a new material system for biosensors. Adv. Funct. Mater 2003, 13, 841–846. [Google Scholar]
- Kang, B.S.; Ren, F.; Wang, L.; Lofton, C.; Tan, W.W.; Pearton, S.J.; Dabiran, A.; Osinsky, A.; Chow, P.P. Electrical detection of immobilized proteins with ungated AlGaNGaN high-electron-mobility Transistors. Appl. Phys. Lett. 2005, 87, 1–3. [Google Scholar]
- Kang, B.S.; Wang, H.T.; Lele, T.P.; Tseng, Y.; Ren, F.; Pearton, S.J.; Johnson, J.W.; Rajagopal, P.; Roberts, J.C.; Piner, E.L.; Linthicum, K.J. Prostate specific antigen detection using AlGaN/GaN high electron mobility transistors. Appl. Phys. Lett. 2007, 91, 112106-1–112106-3. [Google Scholar]
- Chen, C.P.; Ganguly, A.; Wang, C.H.; Hsu, C.W.; Chattopadhyay, S.; Hsu, Y.K.; Chang, Y.C.; Chen, K.H.; Chen, L.C. Label-free dual sensing of DNA molecules using GaN nanowires. Anal. Chem. 2009, 81, 36–42. [Google Scholar]
- Zhu, N.; Chang, Z.; He, P.; Fang, Y. Electrochemical DNA biosensors based on platinum nanoparticles combined carbon nanotubes. Anal. Chim. Acta. 2005, 545, 21–26. [Google Scholar]
- Li, J.; Liu, Q.; Liu, Y.; Liu, S.; Yao, S. DNA biosensor based on chitosan film doped with carbon nanotubes. Anal. Biochem 2005, 346, 107–114. [Google Scholar]
- Daniel, S.; Rao, T.P.; Rao, K.S.; Rani, S.U.; Naidu, G.R.K.; Lee, H.Y.; Kawai, T. A review of DNA functionalized/grafted carbon nanotubes and their characterization. Sensor. Actuator. B-Chem. 2007, 122, 672–682. [Google Scholar]
- Balasubramanian, K.; Burghard, M. Biosensors based on carbon nanotubes. Anal. BioAnal. Chem. 2006, 385, 452–468. [Google Scholar]
- Jiang, C.; Yang, T.; Jiao, K.; Gao, H. A DNA electrochemical sensor with poly-l-lysine/single-walled carbon nanotubes films and its application for the highly sensitive EIS detection of PAT gene fragment and PCR amplification of NOS gene. Electrochim. Acta. 2008, 53, 2917–2924. [Google Scholar]
- Zhang, W.; Yang, T.; Zhuang, X.; Guo, Z.; Jiao, K. An ionic liquid supported CeO2 nanoshuttles-carbon nanotubes composite as a platform for impedance DNA hybridization sensing. Biosens. Bioelectron 2009, 24, 2417–2422. [Google Scholar]
- Drummond, T.G.; Hill, M.G.; Barton, J.K. Electrochemical DNA sensors. Nature Biotech 2003, 21, 1192–1199. [Google Scholar]
- Park, J.Y.; Chang, B.Y.; Nam, H.; Park, S.M. Selective electrochemical sensing of glycated hemoglobin (HbA1c) on thiophene-3-boronic acid self-assembled monolayer covered gold electrodes. Anal. Chem. 2008, 80, 8035–8044. [Google Scholar]
- Park, J.Y.; Lee, Y.S.; Kim, B.H.; Park, S.M. (R)-Lipo-diaza-18-crown-6 self-assembled monolayer as a selective serotonin receptor. Anal. Chem. 2009, 80, 4986–4993. [Google Scholar]
- Yoo, J.-S.; Park, S.-M. An electrochemical impedance measurement technique employing Fourier transform. Anal. Chem. 2000, 72, 2035–2041. [Google Scholar]
- Yoo, J.-S.; Song, I; Lee, J.-H.; Park, S.-M. Real-time impedance measurements during electrochemical experiments and their application to aniline oxidation. Anal. Chem. 2003, 75, 3294–3300. [Google Scholar]
- Park, S.-M.; Yoo, J.-S.; Chang, B.-Y.; Ahn, E.-S. Novel instrumentation in electrochemical impedance spectroscopy and a full description of an electrochemical system. Pure Appl. Chem. 2006, 78, 1069–1080. [Google Scholar]
- Chang, B.-Y.; Park, S.-M. Integrated description of electrode/electrolyte interfaces based on equivalent circuits and its verification using impedance measurements. Anal. Chem. 2006, 78, 1052–1060. [Google Scholar]
- Chang, B.-Y.; Park, S.-M. Fourier transform analysis of chronoamperometric currents obtained during staircase voltammetric experiments. Anal. Chem. 2007, 79, 4892–4899. [Google Scholar]
- Lee, J.-Y.; Park, S.-M. Electrochemistry of Guest Molecules in Thiolated Cyclodextrin Self-Assembled Monolayers : An Implication for Size-Selective Sensor. J. Phys. Chem. B 1998, 102, 9940–9945. [Google Scholar]
- Choi, S.-J.; Choi, B.-G.; Park, S.-M. Electrochemical Sensor for Electrochemically Inactive β-D(+)-Glucose Using α-Cyclodextrin Template Molecules. Anal. Chem. 2002, 74, 1998–2002. [Google Scholar]
- Park, J.-Y.; Kim, B.-C.; Park, S.-M. Molecular recognition of protonated polyamines at calix[4]crown-5 self-assembled monolayer modified electrodes by impedance measurements. Anal. Chem. 2007, 79, 1890–1896. [Google Scholar]
- Park, J.-Y.; Lee, Y.S.; Kim, B.H.; Park, S.-M. Label-free detection of antibody-antigen interactions on (R)-lipo-diaza-18-crown-6 self-assembled monolayer modified gold electrodes. Anal. Chem. 2008, 80, 4986–4993. [Google Scholar]
- Yeo, J; Park, J.-Y.; Bae, W.J.; Lee, Y.S.; Kim, B.H.; Cho, Y.; Park, S.-M. Label-free electrochemical detection of the p53 core domain protein on its antibody immobilized electrode. Anal. Chem. 2009, 81, 4770–4777. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Park, J.-Y.; Park, S.-M. DNA Hybridization Sensors Based on Electrochemical Impedance Spectroscopy as a Detection Tool. Sensors 2009, 9, 9513-9532. https://doi.org/10.3390/s91209513
Park J-Y, Park S-M. DNA Hybridization Sensors Based on Electrochemical Impedance Spectroscopy as a Detection Tool. Sensors. 2009; 9(12):9513-9532. https://doi.org/10.3390/s91209513
Chicago/Turabian StylePark, Jin-Young, and Su-Moon Park. 2009. "DNA Hybridization Sensors Based on Electrochemical Impedance Spectroscopy as a Detection Tool" Sensors 9, no. 12: 9513-9532. https://doi.org/10.3390/s91209513
APA StylePark, J.-Y., & Park, S.-M. (2009). DNA Hybridization Sensors Based on Electrochemical Impedance Spectroscopy as a Detection Tool. Sensors, 9(12), 9513-9532. https://doi.org/10.3390/s91209513