Expression of PHB2 in Rat Brain Cortex Following Traumatic Brain Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
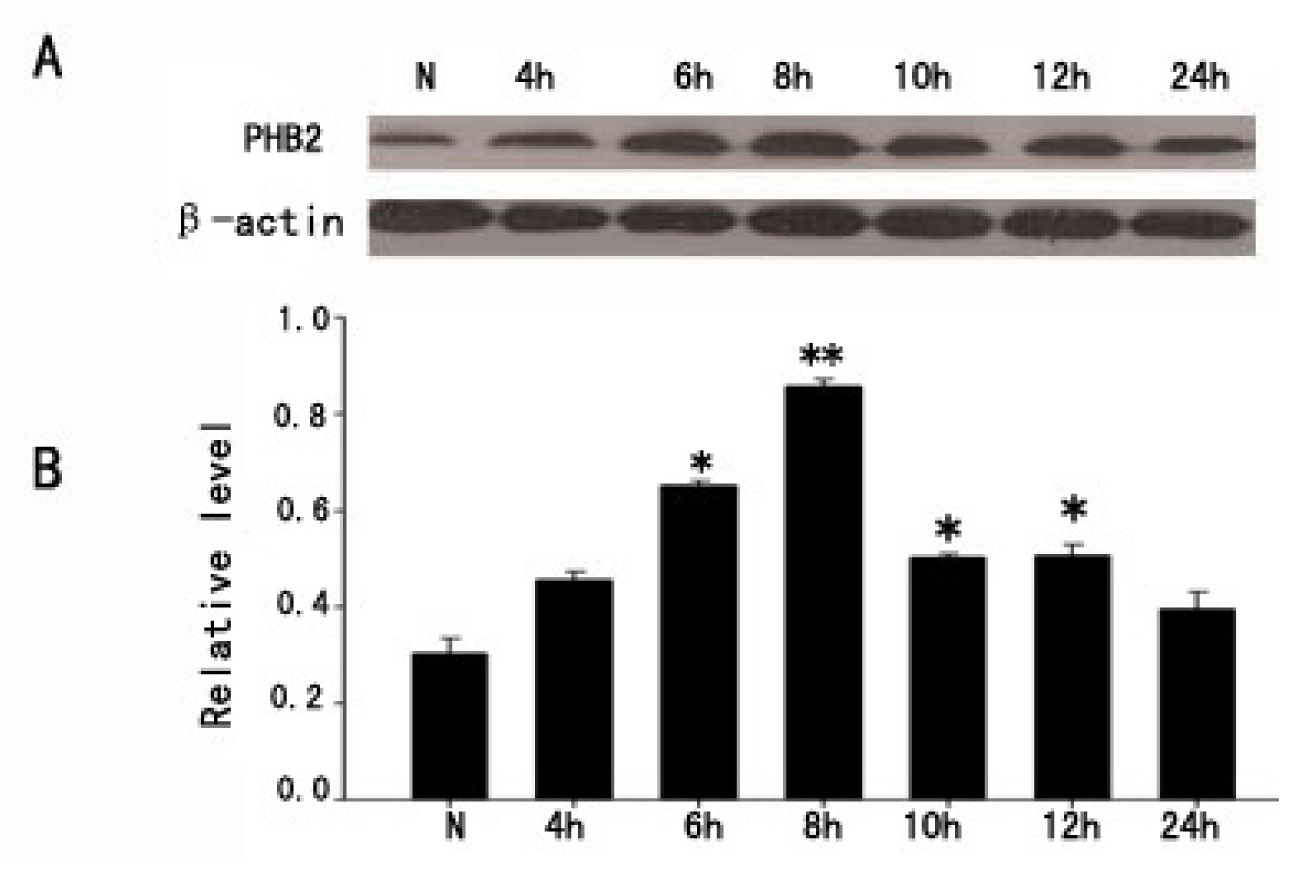
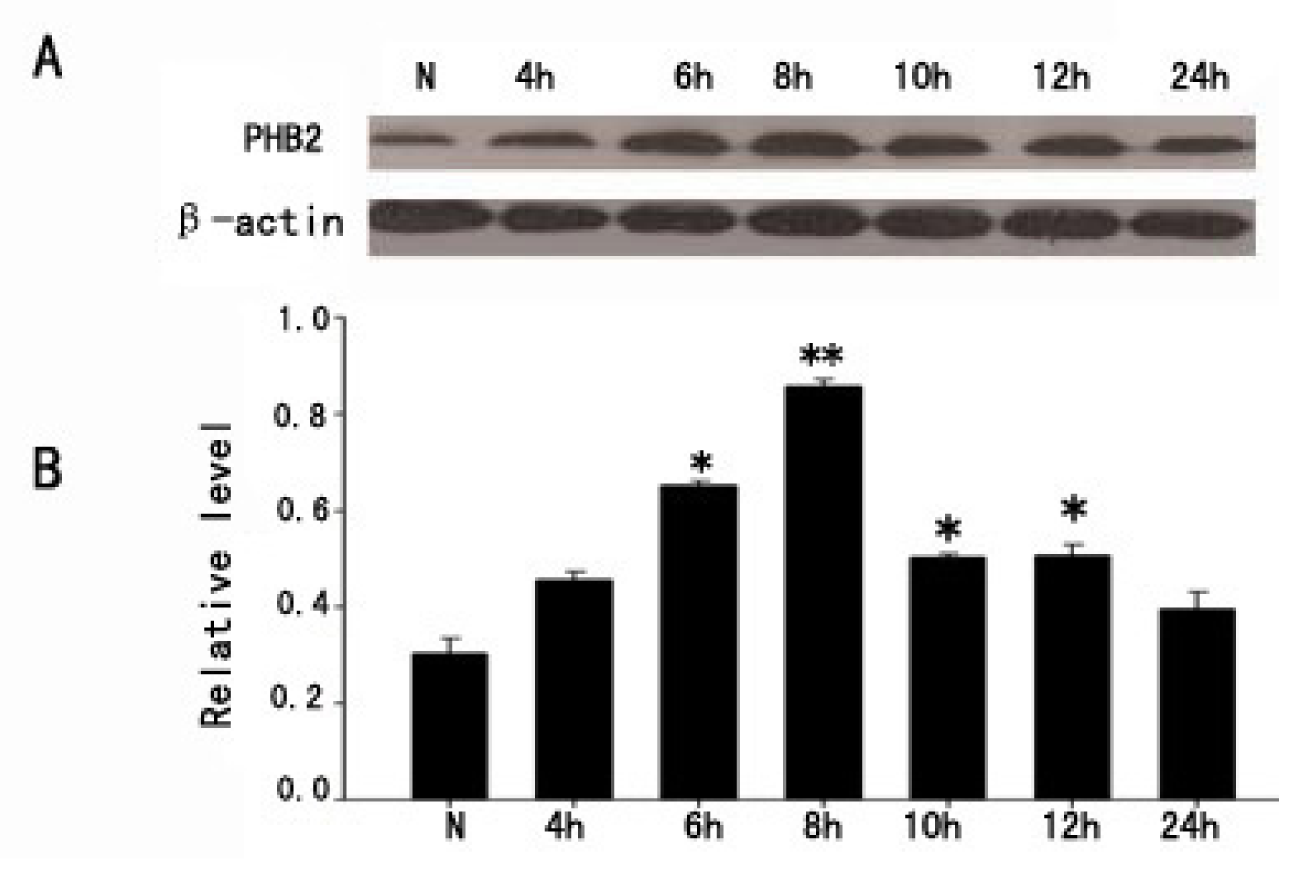
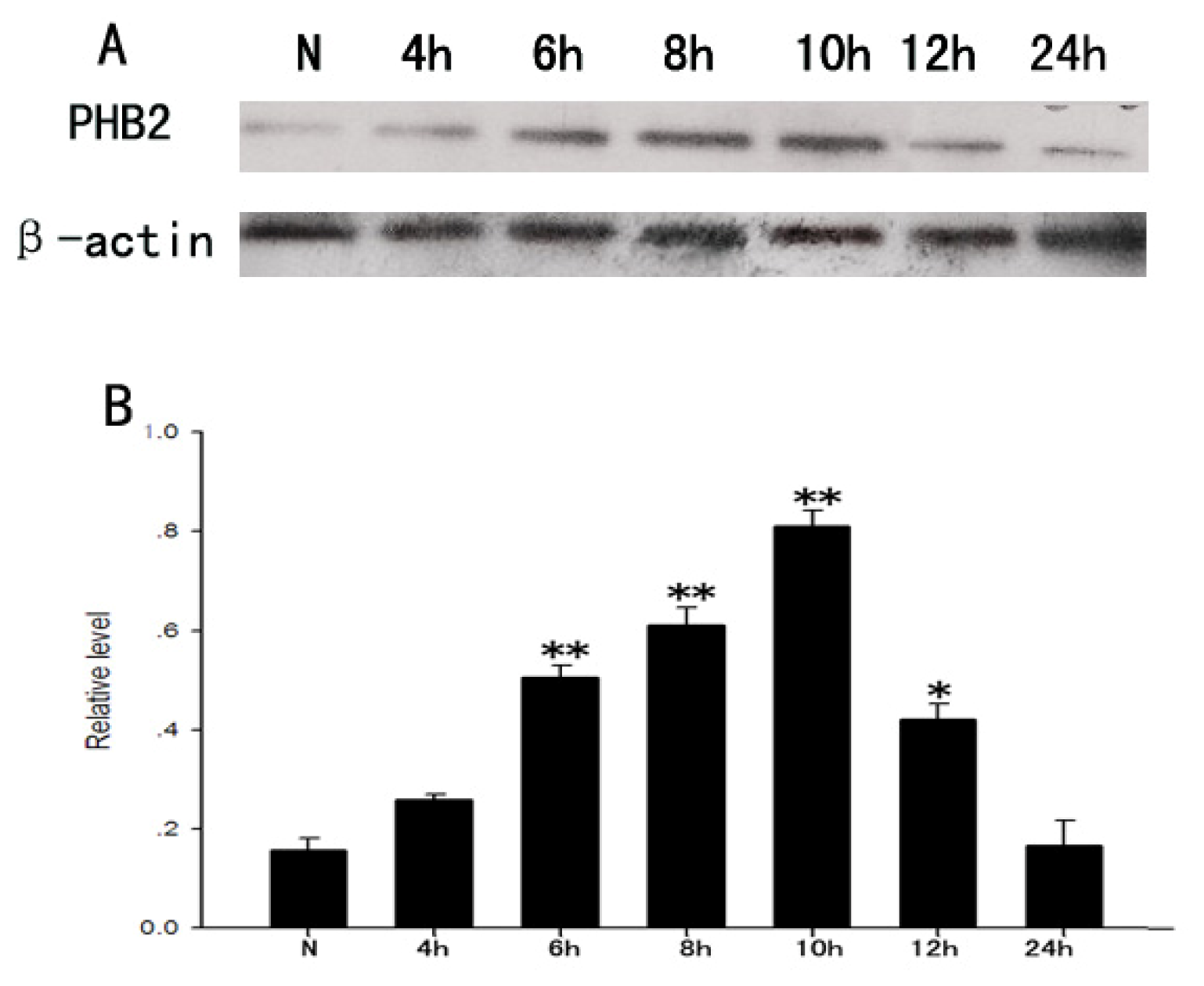
2.1. The Expression of PHB2 in Brain Cortex by Western Blot after TBI
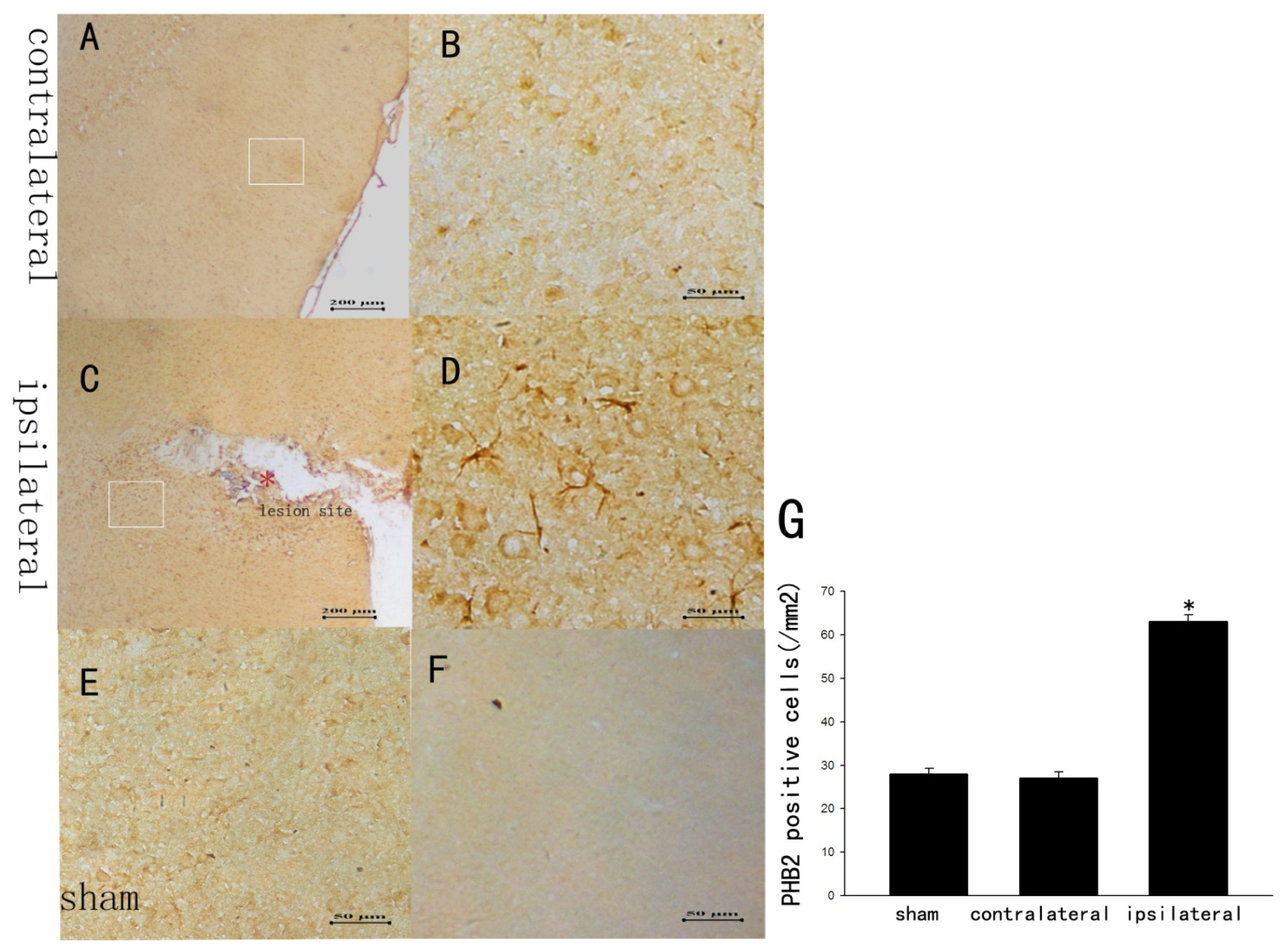
2.2. The Staining Changes of PHB2 Immunoreactivity in Rat Brain Cortex after TBI
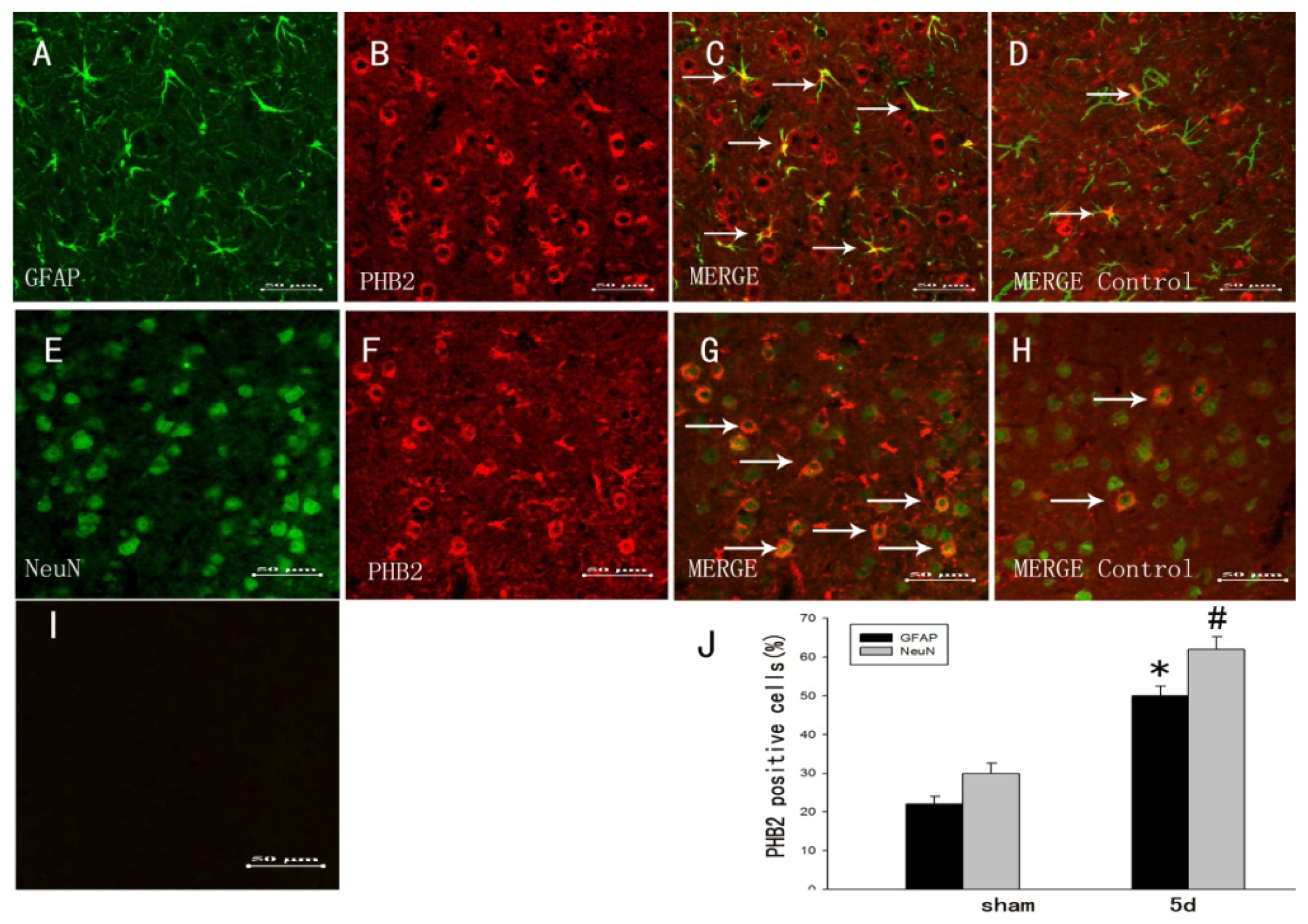
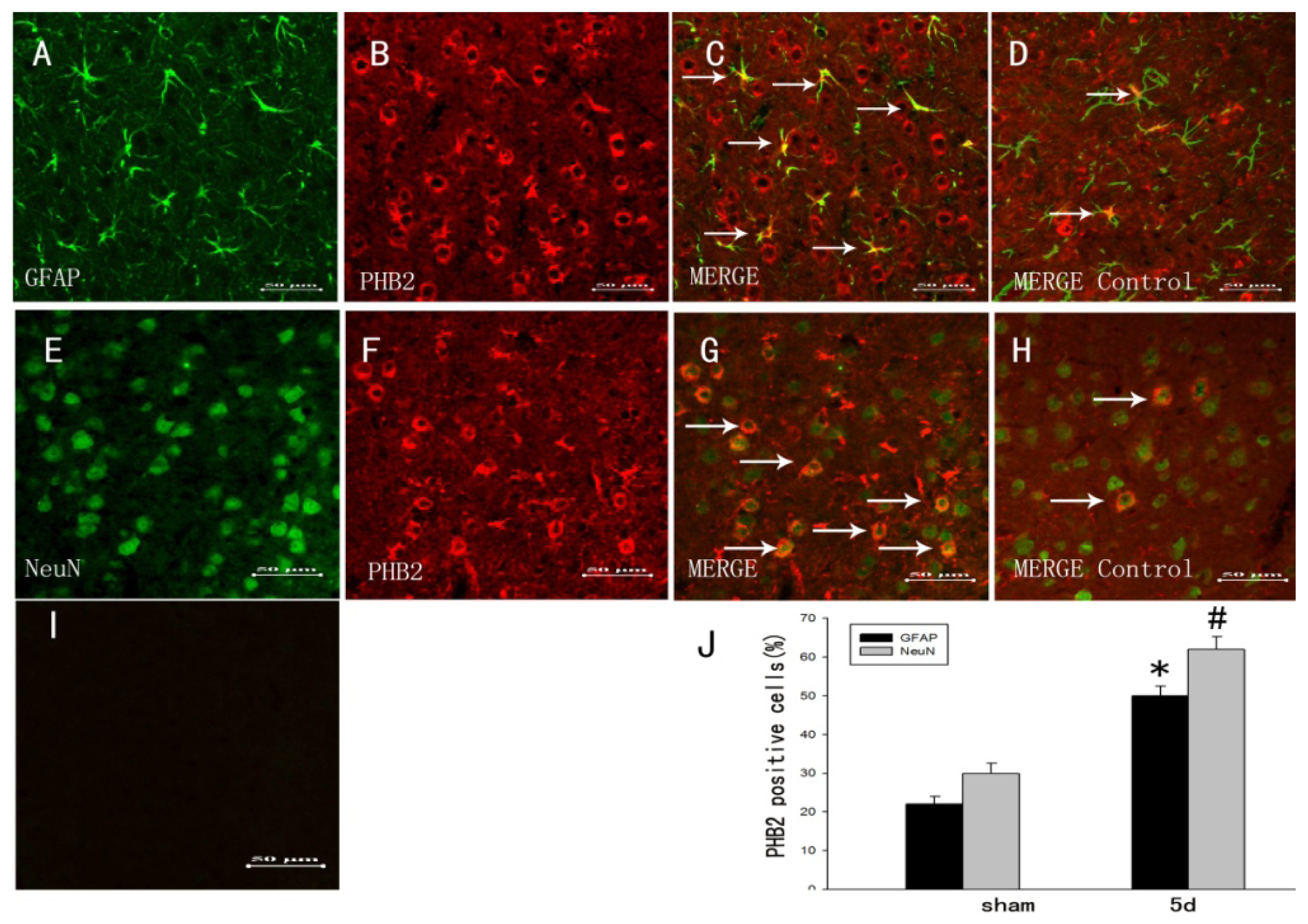
2.3. The Colocalization of PHB2 with Different Cellular Markers by Double Immunofluorescent Staining
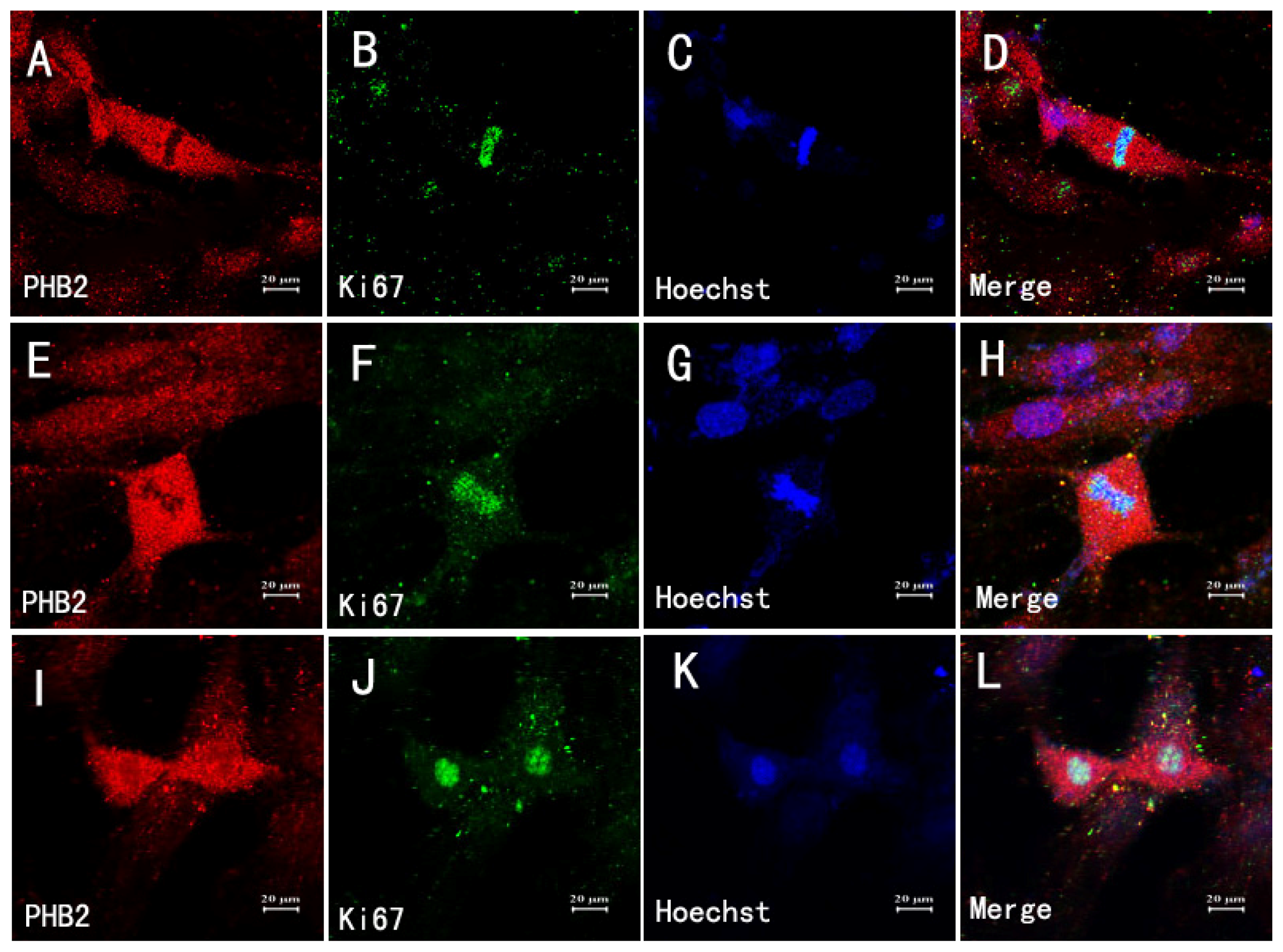
2.4. Association of PHB2 with Survival and Proliferation after TBI
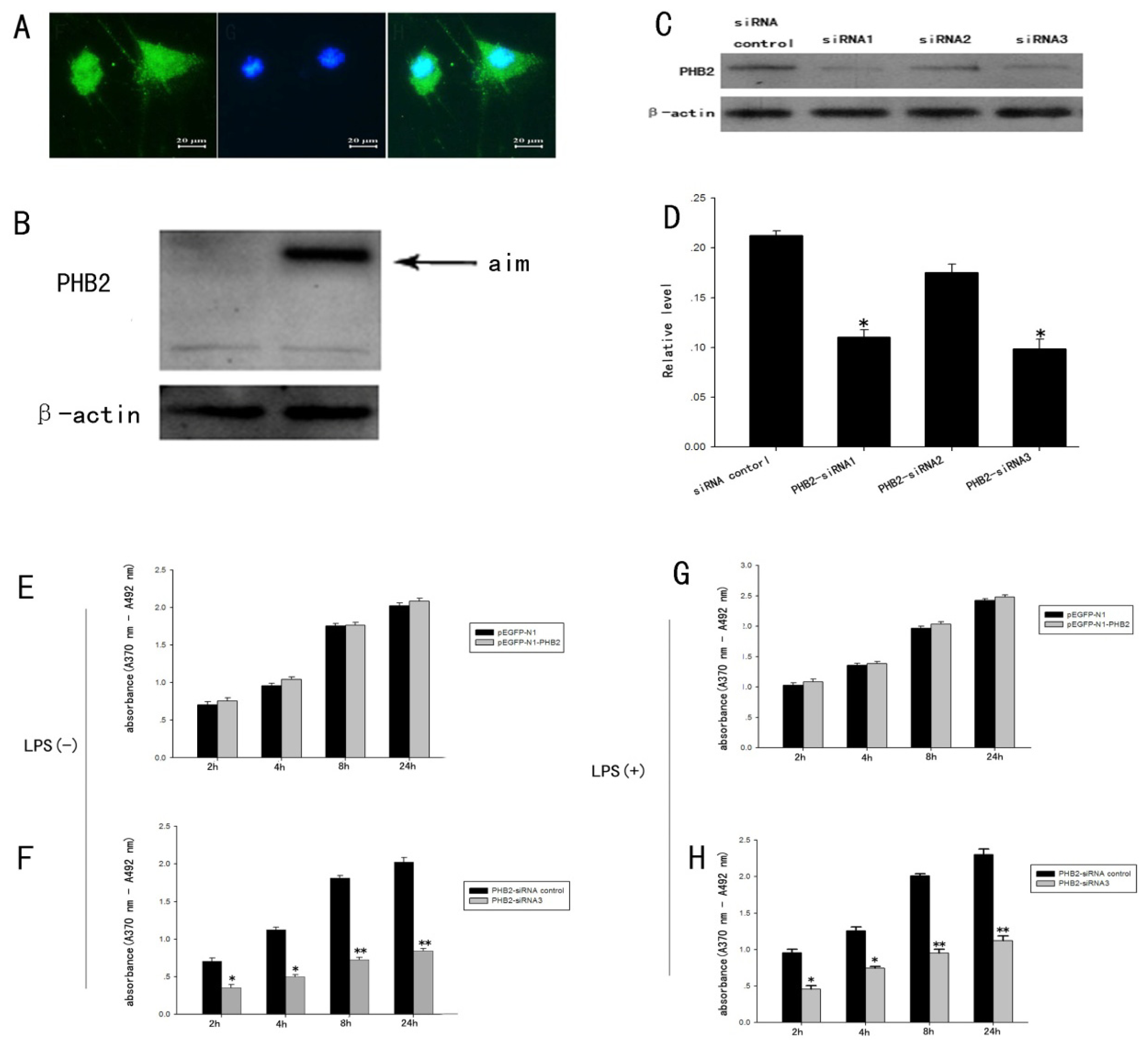
2.5. The Expression Change of PHB2 in the Process of the Proliferation of Astrocytes Stimulated by LPS
2.6. The Influence of PHB2 for Proliferation of Primary Cultured Astrocytes Induced by LPS Confirmed by BrdU ELISA Detection Technology
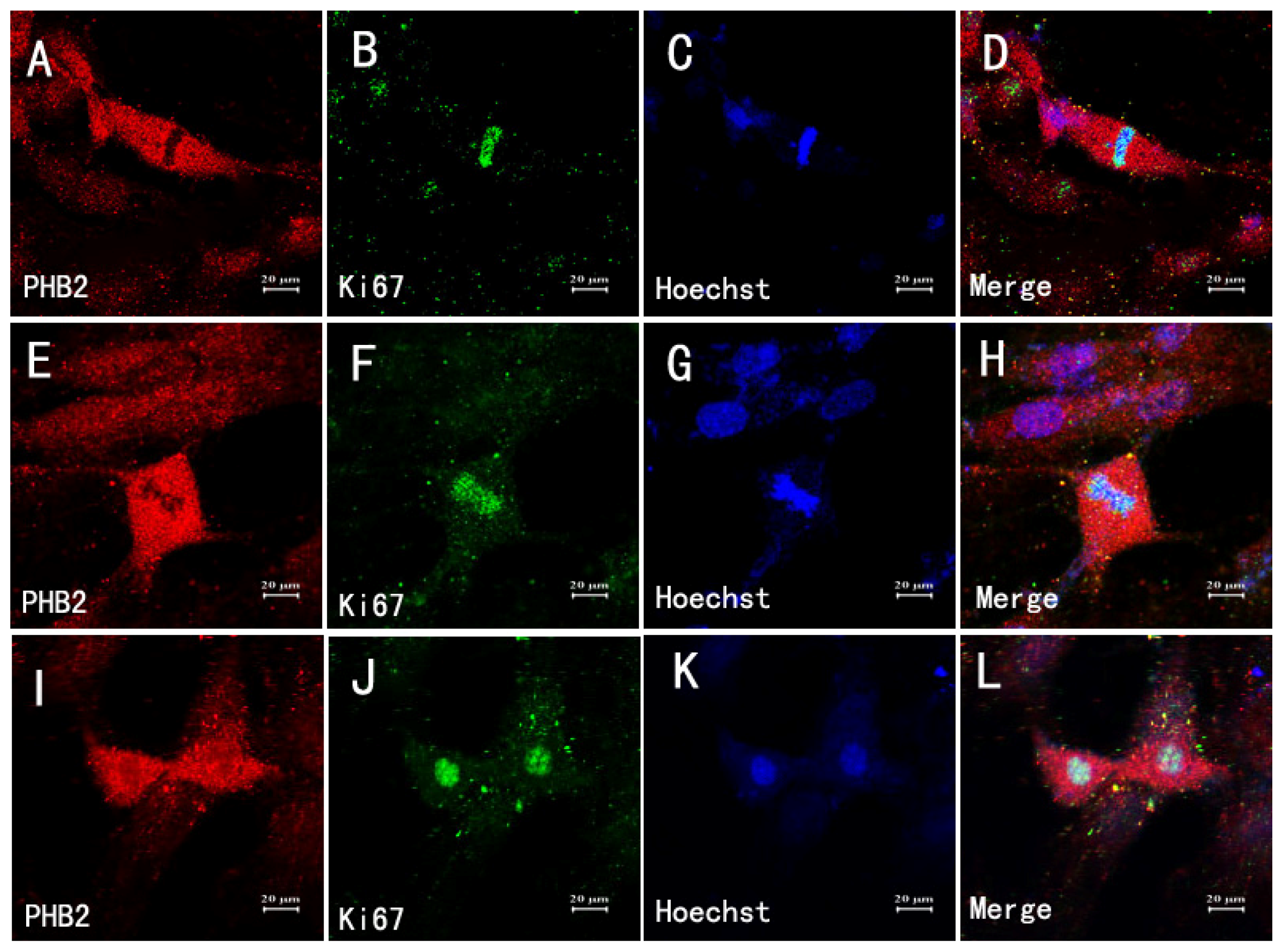
2.7. The Relationship between Astrocytes, Mitosis and PHB2
2.8. The Expression Change of PHB2 in H2O2-Induced PC12 Cell Apoptosis
2.9. The Potential Role of PHB2 during the Process of PC12 Apoptosis Induced by H2O2 Confirmed by Flow Cytometry-Based Annexin V-PE/7-AAD Staining Analysis
2.10. Discussion
3. Experimental Section
3.1. Animals and Surgery
3.2. Western Blot Analysis
3.3. Sections and Double Immunofluorescent Staining
3.4. Immunohistochemistry
3.5. Reverse Transcription (RT)-PCR, Plasmid Construction and Transfections
3.6. Cell Culture
3.7. PHB2 RNA Silencing
3.8. Cell Proliferation ELISA, BrdU
3.9. Flow Cytometry-Based Annexin V-PE/7-ADD Staining
3.10. Quantitative Analysis
3.11. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| CNS | Central nervous system |
| ECL | Enhanced chemiluminescence system |
| PAGE | Polyacrylamide gel electrophoresis |
| BSA | Bovine serum albumin |
| NeuN | Neuronal nuclei |
| GFAP | Glial fibrillary acidic protein |
| PCNA | Proliferating cell nuclear antigen |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| CASPASE-3 | cysteinyl aspartate-specific protease 3 |
| MEFs | mouse embryonic fibroblasts |
References
- Harvey, L.A.; Close, J.C. Traumatic brain injury in older adults: Characteristics, causes and consequences. Injury 2012, 43, 1821–1826. [Google Scholar]
- Cheng, G.; Fu, L.; Zhang, H.Y.; Wang, Y.M.; Zhang, L.M.; Zhang, J.N. The role of mitochondrial calcium uniporter in neuroprotection in traumatic brain injury. Med. Hypotheses 2013, 80, 115–117. [Google Scholar]
- Kumar, A.; Loane, D.J. Neuroinflammation after traumatic brain injury: Opportunities for therapeutic intervention. Brain Behav. Immun 2012, 26, 1191–1201. [Google Scholar]
- McIntosh, T.K.; Smith, D.H.; Meaney, D.F.; Kotapka, M.J.; Gennarelli, T.A.; Graham, D.I. Neuropathological sequelae of traumatic brain injury: Relationship to neurochemical and biomechanical mechanisms. Lab. Invest 1996, 74, 315–342. [Google Scholar]
- Zhang, J.; Jiang, R.; Liu, L.; Watkins, T.; Zhang, F.; Dong, J.F. Traumatic brain injury-associated coagulopathy. J. Neurotrauma 2012, 29, 2597–2605. [Google Scholar]
- Ofek, H.; Defrin, R. The characteristics of chronic central pain after traumatic brain injury. Pain 2007, 131, 330–340. [Google Scholar]
- Blennow, K.; Hardy, J.; Zetterberg, H. The neuropathology and neurobiology of traumatic brain injury. Neuron 2012, 76, 886–899. [Google Scholar]
- Bigler, E.D. Neurobiology and neuropathology underlie the neuropsychological deficits associated with traumatic brain injury. Arch Clin. Neuropsychol 2003, 18, 595–621. [Google Scholar]
- Hart, T.; Brenner, L.; Clark, A.N. Major and minor depression after traumatic brain injury. Arch Phys. Med. Rehabil 2011, 92, 1211–1219. [Google Scholar]
- Laird, M.D.; Vender, J.R.; Dhandapani, K.M. Opposing roles for reactive astrocytes following traumatic brain injury. Neurosignals 2008, 16, 154–164. [Google Scholar]
- Keane, R.W.; Kraydieh, S.; Lotocki, G.; Alonso, O.F.; Aldana, P.; Dietrich, W.D. Apoptotic and antiapoptotic mechanisms after traumatic brain injury. J. Cereb. Blood Flow Metab 2001, 21, 1189–1198. [Google Scholar]
- Bye, N.; Carron, S.; Han, X. Neurogenesis and glial proliferation are stimulated following diffuse traumatic brain injury in adult rats. J. Neurosci. Res 2011, 89, 986–1000. [Google Scholar]
- Coates, P.J.; Jamieson, D.J.; Smart, K.; Prescott, A.R.; Hall, P.A. The prohibitin family of mitochondrial proteins regulate replicative lifespan. Curr. Biol 1997, 7, 607–610. [Google Scholar]
- Marta, A.S.; Nektarios, T. Opposing function of mitochondrial prohibitin in aging. Aging 2010, 2, 1004–1011. [Google Scholar]
- Back, J.W.; Sanz, M.A.; De, J.L. A structure for the yeast prohibitin complex: Structure prediction and evidence from chemical crosslinking and mass spectrometry. Protein Sci 2007, 11, 2471–2478. [Google Scholar]
- Mishra, S.; Murphy, L.C.; Murphy, L.J. The Prohibitins: Emerging roles in diverse functions. J. Cell Mol. Med 2006, 10, 353–363. [Google Scholar]
- Xu, Z.; Wu, J.; Zha, X. Up-regulation of prohibitin 1 is involved in the proliferation and migration of liver cancer cells. Sci. China Life Sci 2001, 54, 121–127. [Google Scholar]
- Coates, P.J.; Nenutil, R.; McGregor, A. Mammalian prohibitin proteins respond to mitochondrial stress and decrease during cellular senescence. Exper. Cell Res 2001, 265, 262–273. [Google Scholar]
- Marta, A.S.; Nektarios, T. Prohibitin and mitochondrial biology. Trends Endocrinol. Metab 2009, 20, 394–401. [Google Scholar]
- Nijtmans, L.G.; Artal, S.M.; Grivell, L.A.; Coates, P.J. The mitochondrial PHB complex: Roles in mitochondrial respiratory complex assembly, ageing and degenerative disease. Cell. Mol. Life Sci 2002, 9, 143–155. [Google Scholar]
- Wang, S.; Nath, N.; Adlam, M.; Chellappan, S. Prohibitin, a potential tumor suppressor, interacts with RB and regulates E2F function. Oncogene 1999, 18, 3501–3510. [Google Scholar]
- Wang, S.; Fusaro, G.; Padmanabhan, J.; Chellappan, S.P. Prohibitin co-localizes with Rb in the nucleus and recruits N-CoR and HDAC1 for transcriptional repression. Oncogene 2002, 21, 8388–8396. [Google Scholar]
- Merkwirth, C.; Dargazanli, S.; Tatsuta, T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev 2008, 22, 476–488. [Google Scholar]
- Frezza, C.; Cipolat, S.; Martins, B.O. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar]
- Duvezin-Caubet, S.; Jagasia, R.; Wagener, J. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J. Biol. Chem 2006, 281, 37972–37979. [Google Scholar]
- Baricault, L.; Segui, B.; Guegand, L. OPA1 cleavage depends on decreased mitochondrial ATP level and bivalent metals. Exp. Cell Res 2007, 313, 3800–3808. [Google Scholar]
- Sun, L.; Liu, L.; Yang, X.J. Akt binds prohibitin 2 and relieves its repression of MyoD and muscle differentiation. J. Cell Sci 2004, 117, 3021–3029. [Google Scholar]
- Mao, Z.; Bonni, A.; Xia, F.; Nadal-Vicens, M.; Greenberg, M.E. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science 1999, 286, 785–790. [Google Scholar]
- Kasashima, K.; Ohta, E.; Kagawa, Y.; Endo, H. Mitochondrial functions and estrogen receptor-dependent nuclear translocation of pleiotropic human Prohibitin 2. J. Biol. Chem 2006, 281, 36401–36410. [Google Scholar]
- Takata, H.; Matsunaga, S.; Morimoto, A. PHB2 protects sister-chromatid cohesion in mitosis. Curr. Biol 2007, 17, 1356–1361. [Google Scholar]
- Fusaro, G.; Dasgupta, P.; Rastogi, S.; Joshi, B.; Chellappan, S. Prohibitin induces the transcriptional activity of p53 and is exported from the nucleus upon apoptotic signaling. J. Biol. Chem 2003, 278, 47853–47861. [Google Scholar]
- Suzuki, Y.; Demoliere, C.; Kitamura, D.; Takeshita, H.; Deuschle, U. Watanabe, HAX-1, a novel intracellular protein, localized on mitochondria, directly associates with HS1, a substrate of Src family tyrosine kinases. J. Immunol 1997, 158, 2736–2744. [Google Scholar]
- Cilenti, L.; Soundarapandian, M.M.; Kyriazis, G.A. Regulation of HAX-1 anti-apoptotic protein by Omi/HtrA2 protease during cell death. J. Biol. Chem 2004, 279, 50295–50230. [Google Scholar]
- Smalla, K.H.; Mikhaylova, M.; Sahin, J.; Bernstein, H.G.; Bogerts, B.; Schmitt, A.; van der Schors, R.; Smit, A.B.; Li, K.W.; Gundelfinger, E.D.; et al. A comparison of the synaptic proteome in human chronic schizophrenia and rat ketamine psychosis suggest that prohibitin is involved in the synaptic pathology of schizophrenia. Mol. Psychiatry 2008, 13, 876–896. [Google Scholar]
- Bernstein, H.G.; Smalla, K.H.; Dürrschmidt, D.; Keilhoff, G.; Dobrowolny, H.; Steiner, J.; Schmitt, A. Increased density of prohibitin-immunoreactive oligodendrocytes in the dorsolateral prefrontal white matter of subjects with schizophrenia suggests extraneuronal roles for the protein in the disease. Neuromol. Med 2012, 14, 270–280. [Google Scholar]
- Shangxun, L.; Bowei, W.; Dan, L. Traumatic brain injury animal model. J. Forensic Med 2011, 27, 286–289. [Google Scholar]
- Swanson, R.A. Astrocyte glutamate uptake during chemical hypoxia in vitro. Neurosci. Lett. 1992, 147, 143–146. [Google Scholar]
- Takuma, K.; Baba, A.; Matsuda, T. Astrocyte apoptosis: Implications for-neuroprotection. Prog. Neurobiol 2004, 72, 111–127. [Google Scholar]
- Lee, M.Y.; Ju, W.K.; Cha, J.H.; Son, B.C.; Chun, M.H.; Kang, J.K.; Park, C.K. Expression of vascular endothelial growth factor mRNA following transient forebrain ischemia in rats. Neurosci. Lett 1999, 265, 107–110. [Google Scholar]
- Ridet, J.L.; Malhotra, S.K.; Privat, A.; Gage, F.H. Reactive astrocytes: Cellular and molecular cues to biological function. Trends Neurosci 1997, 20, 570–577. [Google Scholar]
- Chowdhury, I.; Thompson, W.E.; Welch, C.; Thomas, K.; Matthews, R. Prohibitin (PHB) inhibits apoptosis in rat granulosa cells (GCs) through the extracellular signal-regulated kinase 1/2 (ERK1/2) and the Bcl family of proteins. Apoptosis 2013, 18, 1513–1525. [Google Scholar]
- Fahrig, T.; Gerlach, I.; Horvath, E. A synthetic derivative of the natural product rocaglaol is a potent inhibitor of cytokine-mediated signaling and shows neuroprotective activity in vitro and in animal models of Parkinson’s disease and traumatic brain injury. Mol. Pharmacol 2005, 67, 1544–1555. [Google Scholar]
- Basmadjian, C.; Thuaud, F.; Ribeiro, N.; Désaubry, L. Flavaglines: Potent anticancer drugs that target prohibitins and the helicase eIF4A. Future Med. Chem 2013, 5, 2185–2197. [Google Scholar]
- Ribeiro, N.; Thuaud, F.; Nebigil, C.; Désaubry, L. Recent advances in the biology and chemistry of the flavaglines. Bioorg. Med. Chem 2012, 20, 1857–1864. [Google Scholar]
- Feeney, D.M.; Boyeson, M.G.; Linn, R.T.; Murray, H.M.; Dail, W.G. Responses to cortical injury: I Methodology and local effects of contusions in the rat. Brain Res 1981, 211, 67–77. [Google Scholar]
- Golarai, G.; Greenwood, A.C.; Feeney, D.M.; Connor, J.A. Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury. J. Neurosci 2001, 21, 8523–8537. [Google Scholar]
- McCarthy, K.D.; de Vellis, J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J. Cell. Biol 1980, 85, 890–902. [Google Scholar]



© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xu, T.; Fan, X.; Tan, Y.; Yue, Y.; Chen, W.; Gu, X. Expression of PHB2 in Rat Brain Cortex Following Traumatic Brain Injury. Int. J. Mol. Sci. 2014, 15, 3299-3318. https://doi.org/10.3390/ijms15023299
Xu T, Fan X, Tan Y, Yue Y, Chen W, Gu X. Expression of PHB2 in Rat Brain Cortex Following Traumatic Brain Injury. International Journal of Molecular Sciences. 2014; 15(2):3299-3318. https://doi.org/10.3390/ijms15023299
Chicago/Turabian StyleXu, Ting, Xinjuan Fan, Yuanyuan Tan, Ying Yue, Weijie Chen, and Xingxing Gu. 2014. "Expression of PHB2 in Rat Brain Cortex Following Traumatic Brain Injury" International Journal of Molecular Sciences 15, no. 2: 3299-3318. https://doi.org/10.3390/ijms15023299