Endogenous Protease Nexin-1 Protects against Cerebral Ischemia


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
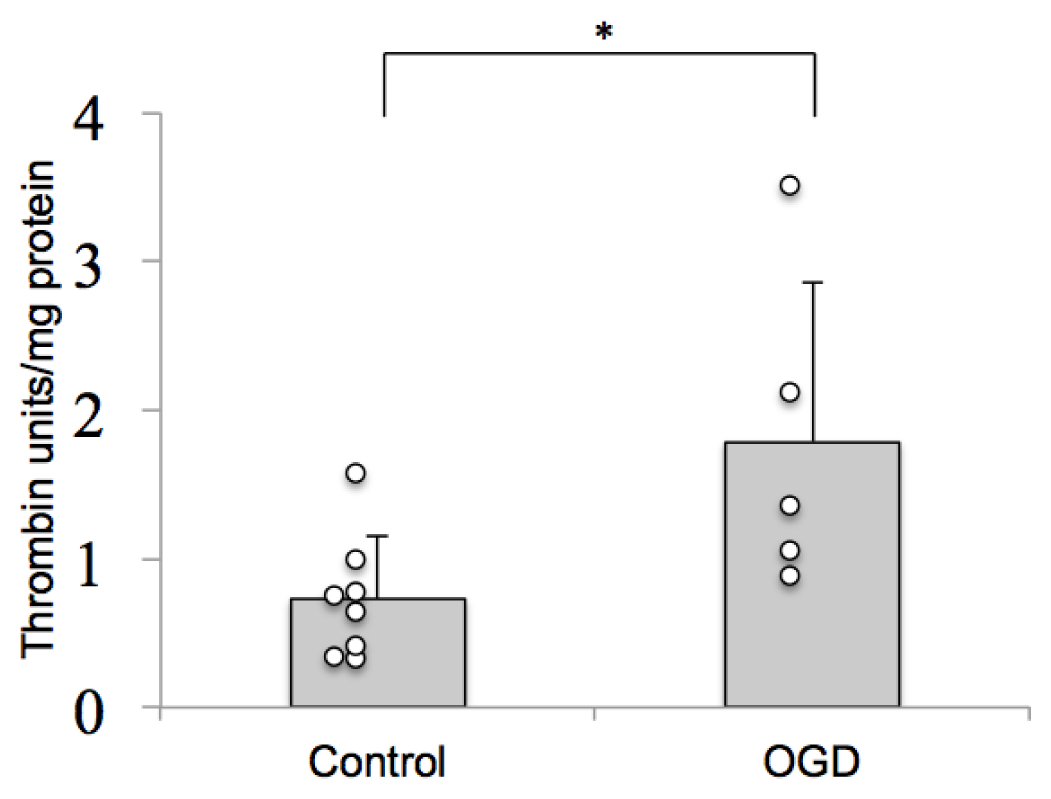
2.1. Thrombin Activity Is Increased in Brain Tissue after Ischemia in Vitro
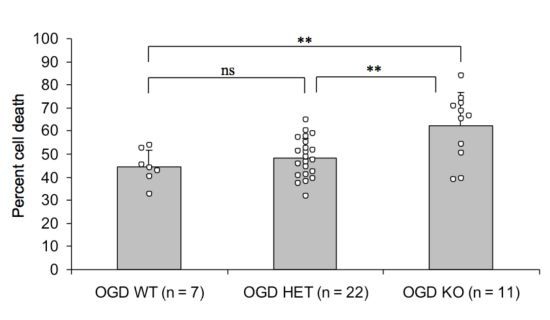
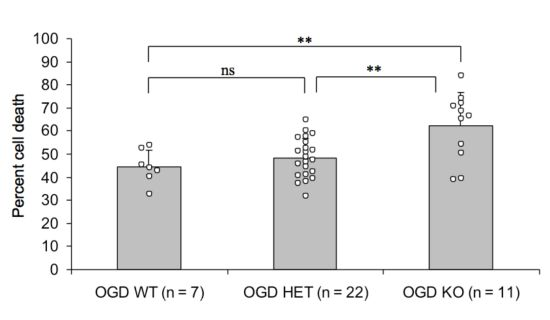
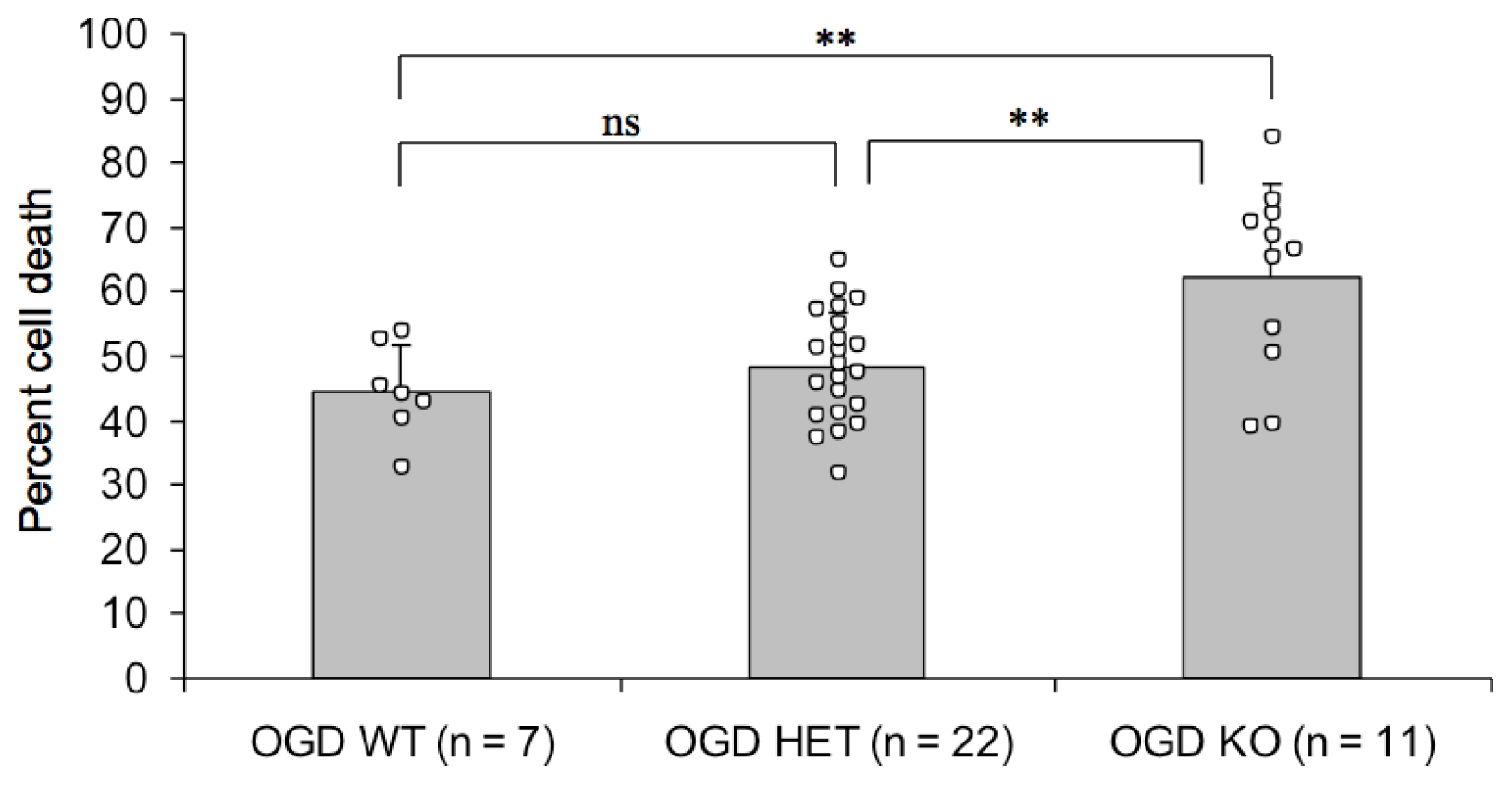
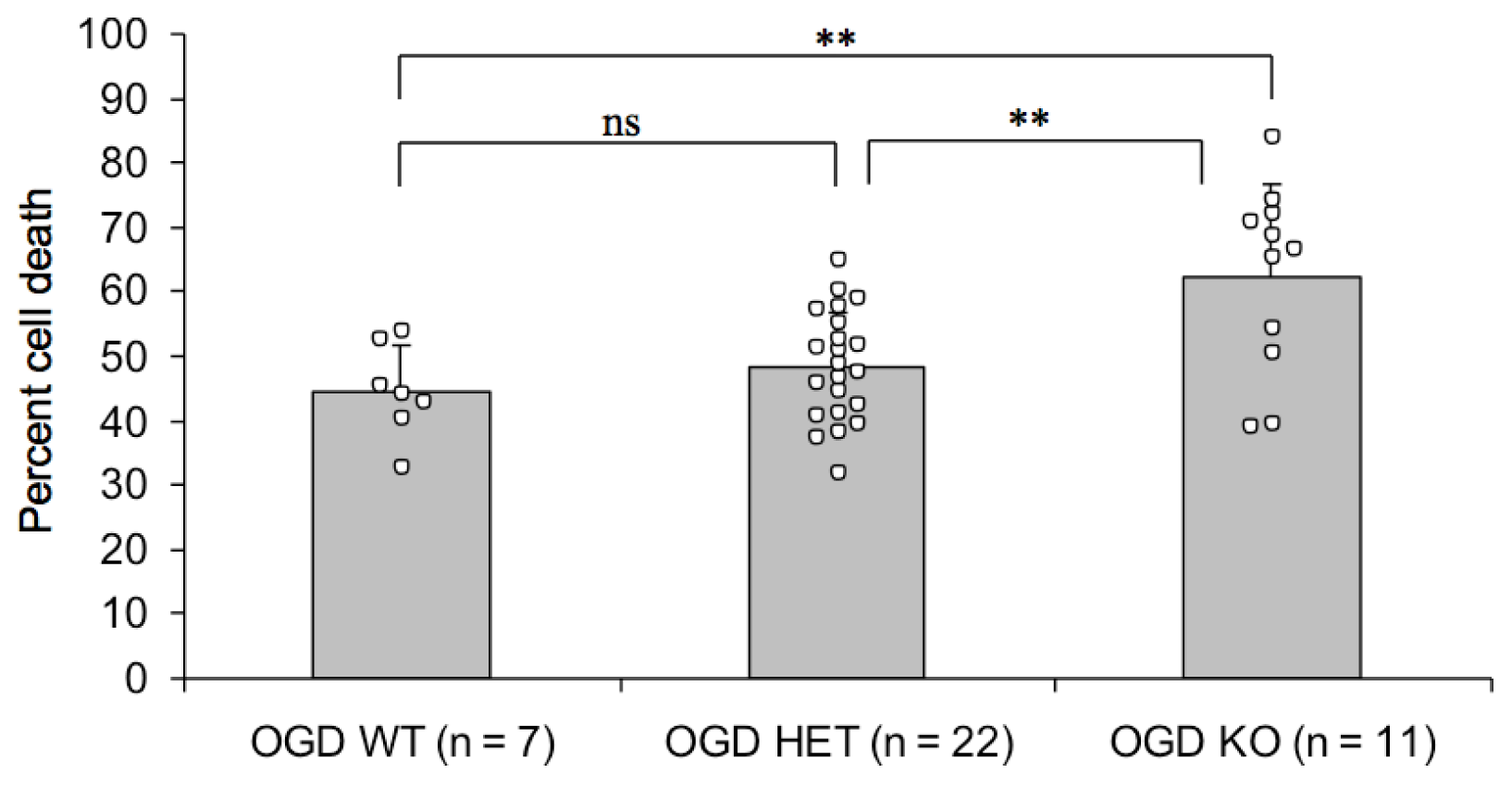
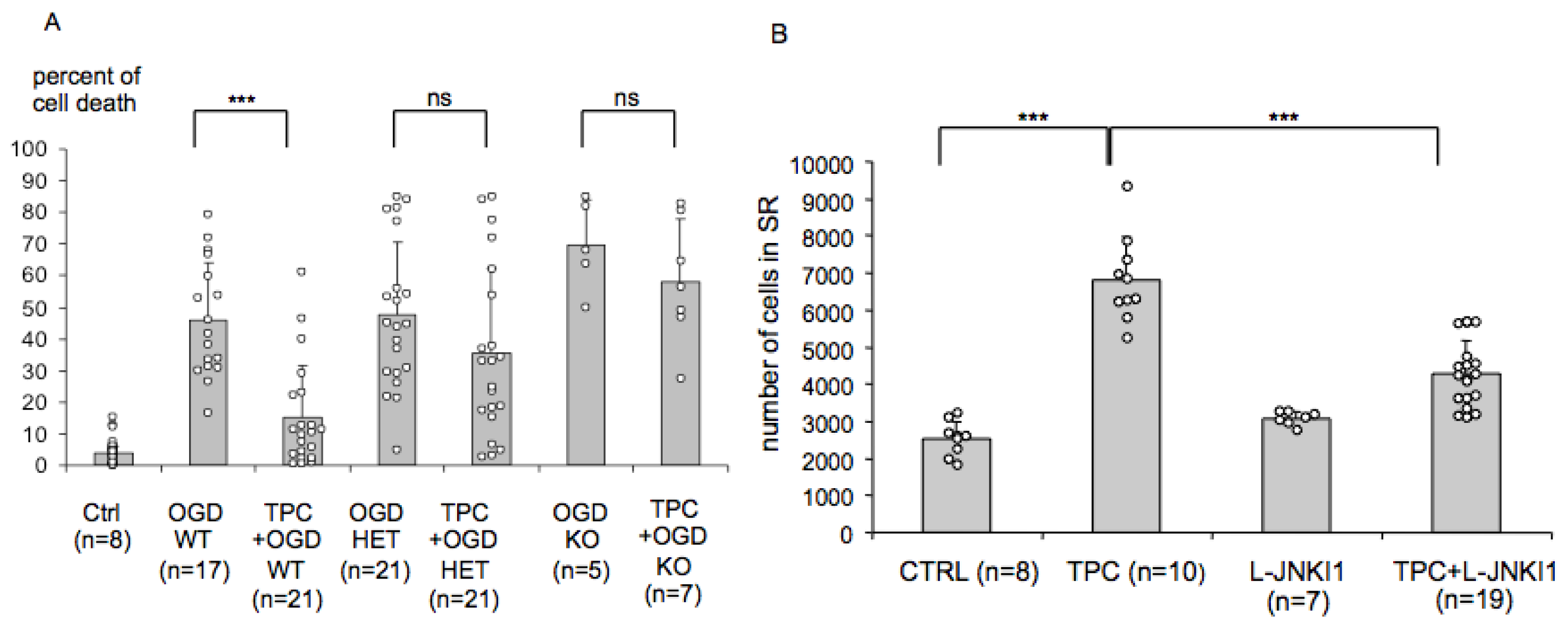
2.2. Endogenous PN-1 Protects against Ischemic Neuronal Death in Hippocampal Slices after OGD
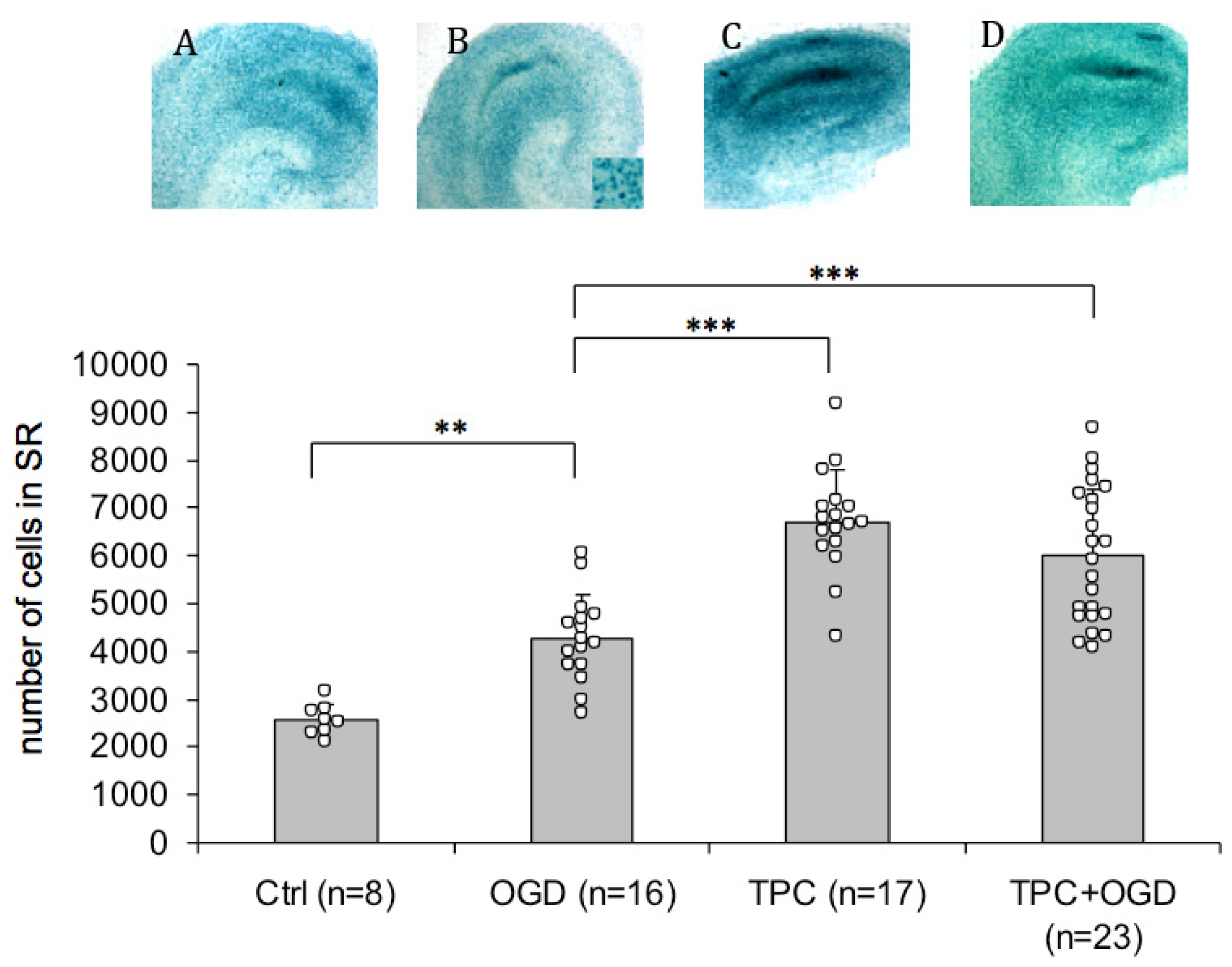
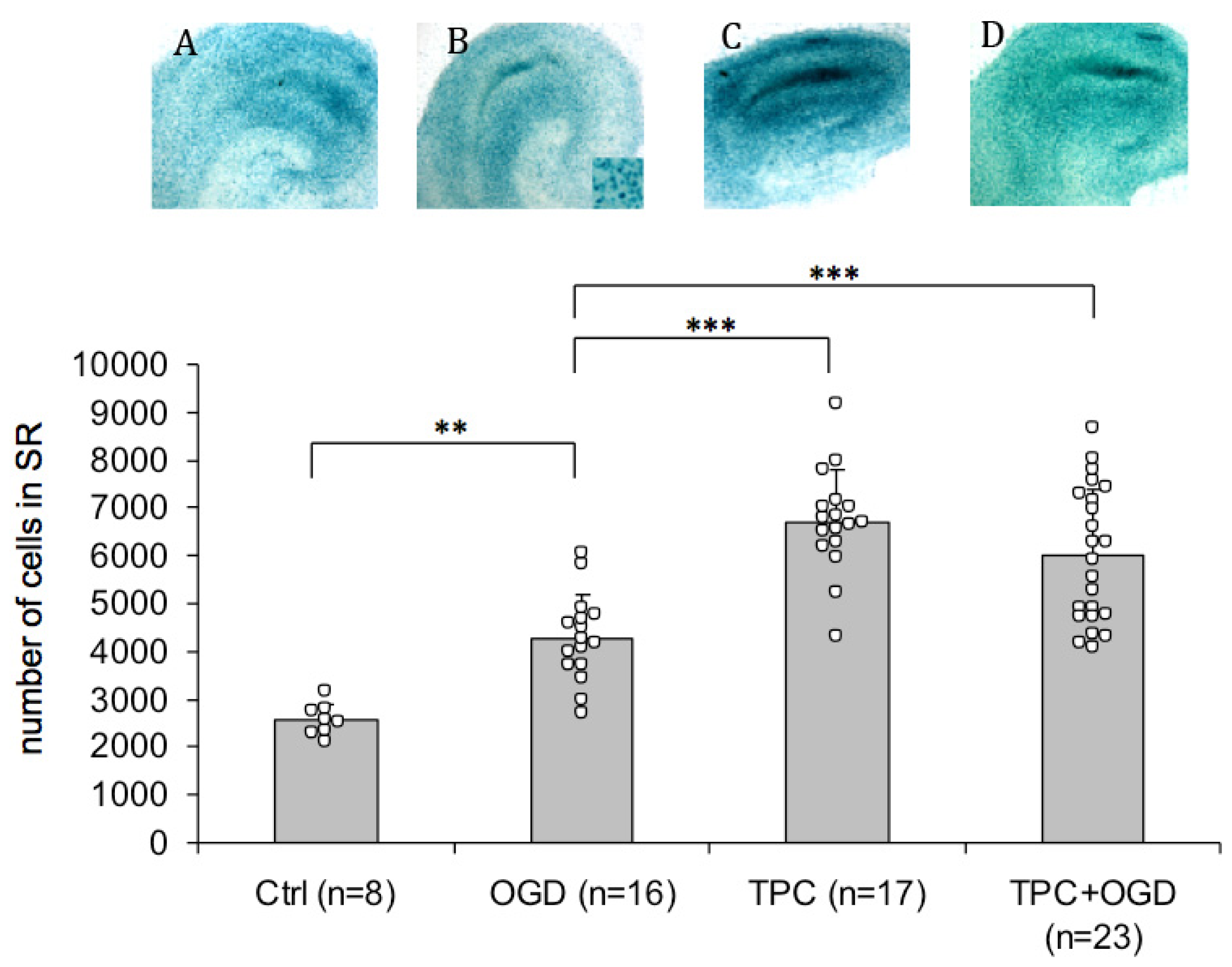
2.3. PN-1 Is Upregulated after OGD and Thrombin Preconditioning
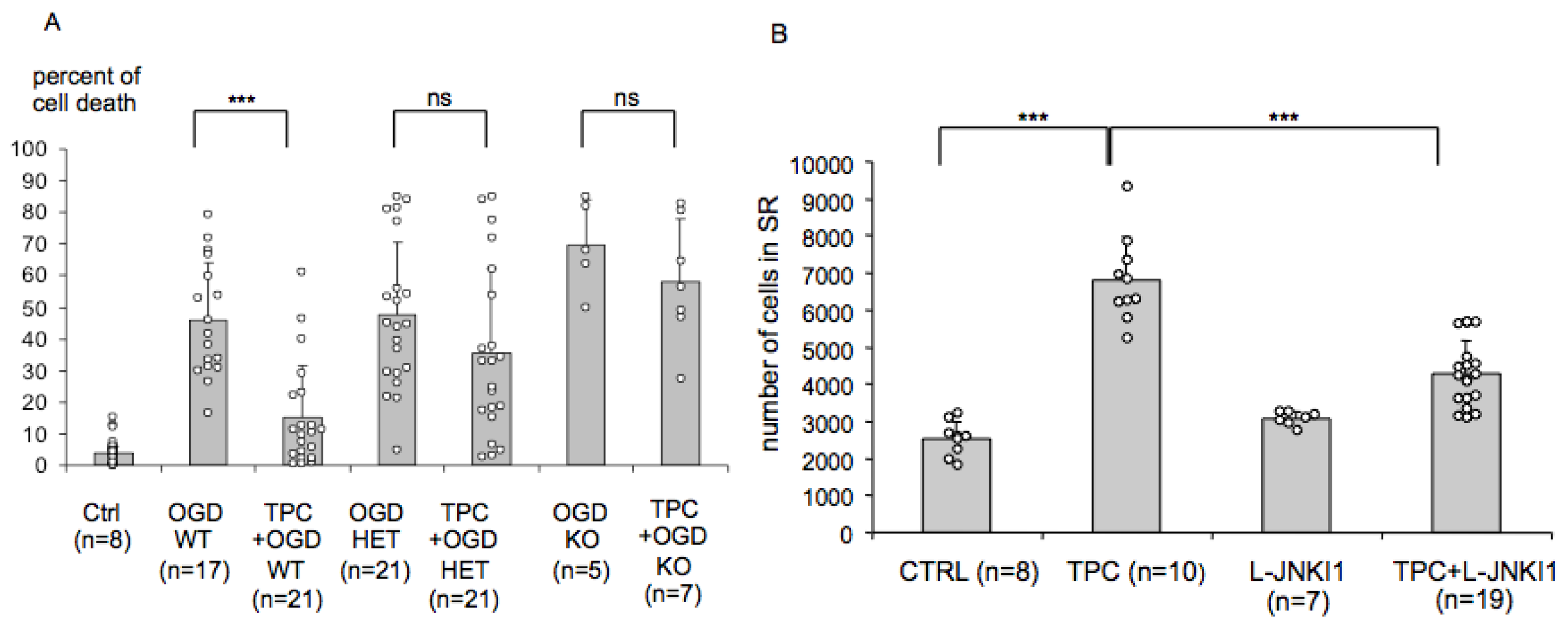
2.4. Endogenous PN-1 Is Necessary for Thrombin Induced Ischemic Tolerance in Hippocampal Slice Cultures
2.5. PN-1 Upregulation in Hippocampal Slices Is Prevented by Inhibiting the c-Jun-N-Terminal Kinase (JNK)
2.6. Discussion
3. Experimental Section
3.1. Animals
3.2. Organotypic Hippocampal Slice Cultures
3.3. Oxygen and Glucose Deprivation (OGD) in Organotypic Hippocampal Slice Cultures
3.4. Chromogenic Thrombin Assay
3.5. β-Galactosidase Enzyme Staining on Hippocampal Slices
3.6. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hacke, W.; Kaste, M.; Bluhmki, E.; Brozman, M.; Davalos, A.; Guidetti, D.; Larrue, V.; Lees, K.R.; Medeghri, Z.; Machnig, T.; et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N. Engl. J. Med 2008, 359, 1317–1329. [Google Scholar]
- Dirnagl, U.; Simon, R.P.; Hallenbeck, J.M. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci 2003, 26, 248–254. [Google Scholar]
- Xi, G.; Keep, R.F.; Hua, Y.; Xiang, J.; Hoff, J.T. Attenuation of thrombin-induced brain edema by cerebral thrombin preconditioning. Stroke 1999, 30, 1247–1255. [Google Scholar]
- Donovan, F.M.; Cunningham, D.D. Signaling pathways involved in thrombin-induced cell protection. J. Biol. Chem 1998, 273, 12746–12752. [Google Scholar]
- Granziera, C.; Thevenet, J.; Price, M.; Wiegler, K.; Magistretti, P.J.; Badaut, J.; Hirt, L. Thrombin-induced ischemic tolerance is prevented by inhibiting c-jun N-terminal kinase. Brain Res 2007, 1148, 217–225. [Google Scholar]
- Price, M.; Badaut, J.; Thevenet, J.; Hirt, L. Activation of c-Jun in the nuclei of neurons of the CA-1 in thrombin preconditioning occurs via PAR-1. J. Neurosci. Res 2010, 88, 1338–1347. [Google Scholar]
- Lee, K.R.; Betz, A.L.; Keep, R.F.; Chenevert, T.L.; Kim, S.; Hoff, J.T. Intracerebral infusion of thrombin as a cause of brain edema. J. Neurosurg 1995, 83, 1045–1050. [Google Scholar]
- Xue, M.; del Bigio, M.R. Acute tissue damage after injections of thrombin and plasmin into rat striatum. Stroke 2001, 32, 2164–2169. [Google Scholar]
- Chen, B.; Friedman, B.; Whitney, M.A.; Winkle, J.A.; Lei, I.F.; Olson, E.S.; Cheng, Q.; Pereira, B.; Zhao, L.; Tsien, R.Y.; et al. Thrombin activity associated with neuronal damage during acute focal ischemia. J. Neurosci 2012, 32, 7622–7631. [Google Scholar]
- Gloor, S.; Odink, K.; Guenther, J.; Nick, H.; Monard, D. A glia-derived neurite promoting factor with protease inhibitory activity belongs to the protease nexins. Cell 1986, 47, 687–693. [Google Scholar]
- Bouton, M.C.; Boulaftali, Y.; Richard, B.; Arocas, V.; Michel, J.B.; Jandrot-Perrus, M. Emerging role of serpinE2/protease nexin-1 in hemostasis and vascular biology. Blood 2012, 119, 2452–2457. [Google Scholar]
- Striggow, F.; Riek, M.; Breder, J.; Henrich-Noack, P.; Reymann, K.G.; Reiser, G. The protease thrombin is an endogenous mediator of hippocampal neuroprotection against ischemia at low concentrations but causes degeneration at high concentrations. Proc. Natl. Acad. Sci. USA 2000, 97, 2264–2269. [Google Scholar]
- Xi, G.; Hua, Y.; Keep, R.F.; Duong, H.K.; Hoff, J.T. Activation of p44/42 mitogen activated protein kinases in thrombin-induced brain tolerance. Brain Res 2001, 895, 153–159. [Google Scholar]
- Jiang, Y.; Wu, J.; Hua, Y.; Keep, R.F.; Xiang, J.; Hoff, J.T.; Xi, G. Thrombin-receptor activation and thrombin-induced brain tolerance. J. Cereb. Blood Flow Metab 2002, 22, 404–410. [Google Scholar]
- Hua, Y.; Keep, R.F.; Hoff, J.T.; Xi, G. Thrombin preconditioning attenuates brain edema induced by erythrocytes and iron. J. Cereb. Blood Flow Metab 2003, 23, 1448–1454. [Google Scholar]
- Karabiyikoglu, M.; Hua, Y.; Keep, R.F.; Ennis, S.R.; Xi, G. Intracerebral hirudin injection attenuates ischemic damage and neurologic deficits without altering local cerebral blood flow. J. Cereb. Blood Flow Metab 2004, 24, 159–166. [Google Scholar]
- De Castro Ribeiro, M.; Badaut, J.; Price, M.; Meins, M.; Bogousslavsky, J.; Monard, D.; Hirt, L. Thrombin in ischemic neuronal death. Exp. Neurol 2006, 198, 199–203. [Google Scholar]
- Borsello, T.; Clarke, P.G.; Hirt, L.; Vercelli, A.; Repici, M.; Schorderet, D.F.; Bogousslavsky, J.; Bonny, C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat. Med 2003, 9, 1180–1186. [Google Scholar]
- Wiegler, K.; Bonny, C.; Coquoz, D.; Hirt, L. The JNK inhibitor XG-102 protects from ischemic damage with delayed intravenous administration also in the presence of recombinant tissue plasminogen activator. Cerebrovasc. Dis 2008, 26, 360–366. [Google Scholar]
- Hirt, L.; Badaut, J.; Thevenet, J.; Granziera, C.; Regli, L.; Maurer, F.; Bonny, C.; Bogousslavsky, J. D-JNKI1, a cell-penetrating c-Jun-N-terminal kinase inhibitor, protects against cell death in severe cerebral ischemia. Stroke 2004, 35, 1738–1743. [Google Scholar]
- Hoffmann, M.C.; Nitsch, C.; Scotti, A.L.; Reinhard, E.; Monard, D. The prolonged presence of glia-derived nexin, an endogenous protease inhibitor, in the hippocampus after ischemia-induced delayed neuronal death. Neuroscience 1992, 49, 397–408. [Google Scholar]
- Nitsch, C.; Scotti, A.L.; Monard, D.; Heim, C.; Sontag, K.H. The glia-derived protease nexin 1 persists for over 1 year in rat brain areas selectively lesioned by transient global ischaemia. Eur. J. Neurosci 1993, 5, 292–297. [Google Scholar]
- Wu, H.; Zhao, R.; Qi, J.; Cong, Y.; Wang, D.; Liu, T.; Gu, Y.; Ban, X.; Huang, Q. The expression and the role of protease nexin-1 on brain edema after intracerebral hemorrhage. J. Neurol. Sci 2008, 270, 172–183. [Google Scholar]
- Luthi, A.; Van der Putten, H.; Botteri, F.M.; Mansuy, I.M.; Meins, M.; Frey, U.; Sansig, G.; Portet, C.; Schmutz, M.; Schroder, M.; et al. Endogenous serine protease inhibitor modulates epileptic activity and hippocampal long-term potentiation. J. Neurosci 1997, 17, 4688–4699. [Google Scholar]
- Kvajo, M.; Albrecht, H.; Meins, M.; Hengst, U.; Troncoso, E.; Lefort, S.; Kiss, J.Z.; Petersen, C.C.; Monard, D. Regulation of brain proteolytic activity is necessary for the in vivo function of NMDA receptors. J. Neurosci 2004, 24, 9734–9743. [Google Scholar]
- Dihanich, M.; Kaser, M.; Reinhard, E.; Cunningham, D.; Monard, D. Prothrombin mRNA is expressed by cells of the nervous system. Neuron 1991, 6, 575–581. [Google Scholar]
- Striggow, F.; Riek-Burchardt, M.; Kiesel, A.; Schmidt, W.; Henrich-Noack, P.; Breder, J.; Krug, M.; Reymann, K.G.; Reiser, G. Four different types of protease-activated receptors are widely expressed in the brain and are up-regulated in hippocampus by severe ischemia. Eur. J. Neurosci 2001, 14, 595–608. [Google Scholar]
- Ubl, J.J.; Vohringer, C.; Reiser, G. Co-existence of two types of [Ca2+]i-inducing rotease-activated receptors (PAR-1 and PAR-2) in rat astrocytes and C6 glioma cells. Neuroscience 1998, 86, 597–609. [Google Scholar]
- Choi, B.H.; Suzuki, M.; Kim, T.; Wagner, S.L.; Cunningham, D.D. Protease nexin-1. Localization in the human brain suggests a protective role against extravasated serine proteases. Am. J. Pathol 1990, 137, 741–747. [Google Scholar]
- Sokolova, E.; Reiser, G. Prothrombin/thrombin and the thrombin receptors PAR-1 and PAR-4 in the brain: Localization, expression and participation in neurodegenerative diseases. Thromb. Haemost 2008, 100, 576–581. [Google Scholar]
- Thevenet, J.; Angelillo-Scherrer, A.; Price, M.; Hirt, L. Coagulation factor xa activates thrombin in ischemic neural tissue. J. Neurochem 2009, 111, 828–836. [Google Scholar]
- Weinstein, J.R.; Gold, S.J.; Cunningham, D.D.; Gall, C.M. Cellular localization of thrombin receptor mRNA in rat brain: Expression by mesencephalic dopaminergic neurons and codistribution with prothrombin mRNA. J. Neurosci 1995, 15, 2906–2919. [Google Scholar]
- Hengst, U.; Albrecht, H.; Hess, D.; Monard, D. The phosphatidylethanolamine-binding protein is the prototype of a novel family of serine protease inhibitors. J. Biol. Chem 2001, 276, 535–540. [Google Scholar]
- Bonny, C.; Oberson, A.; Negri, S.; Sauser, C.; Schorderet, D.F. Cell-permeable peptide inhibitors of JNK: Novel blockers of beta-cell death. Diabetes 2001, 50, 77–82. [Google Scholar]
- Xi, G.; Reiser, G.; Keep, R.F. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: Deleterious or protective? J. Neurochem 2003, 84, 3–9. [Google Scholar]
- De Castro Ribeiro, M.; Hirt, L.; Bogousslavsky, J.; Regli, L.; Badaut, J. Time course of aquaporin expression after transient focal cerebral ischemia in mice. J. Neurosci. Res 2006, 83, 1231–1240. [Google Scholar]
- Hirt, L.; Ternon, B.; Price, M.; Mastour, N.; Brunet, J.F.; Badaut, J. Protective role of early aquaporin 4 induction against postischemic edema formation. J. Cereb. Blood Flow Metab 2009, 29, 423–433. [Google Scholar]
- Donovan, F.M.; Pike, C.J.; Cotman, C.W.; Cunningham, D.D. Thrombin induces apoptosis in cultured neurons and astrocytes via a pathway requiring tyrosine kinase and RhoA activities. J. Neurosci 1997, 17, 5316–5326. [Google Scholar]
- Pike, C.J.; Vaughan, P.J.; Cunningham, D.D.; Cotman, C.W. Thrombin attenuates neuronal cell death and modulates astrocyte reactivity induced by beta-amyloid in vitro. J. Neurochem 1996, 66, 1374–1382. [Google Scholar]
- Vaughan, P.J.; Pike, C.J.; Cotman, C.W.; Cunningham, D.D. Thrombin receptor activation protects neurons and astrocytes from cell death produced by environmental insults. J. Neurosci 1995, 15, 5389–5401. [Google Scholar]
- Wang, Y.; Luo, W.; Reiser, G. The role of calcium in protease-activated receptor-induced secretion of chemokine GRO/CINC-1 in rat brain astrocytes. J. Neurochem 2007, 103, 814–819. [Google Scholar]
- Wang, Y.; Luo, W.; Reiser, G. Proteinase-activated receptor-1 and -2 induce the release of chemokine GRO/CINC-1 from rat astrocytes via differential activation of JNK isoforms, evoking multiple protective pathways in brain. Biochem. J 2007, 401, 65–78. [Google Scholar]
- Wang, Y.; Luo, W.; Stricker, R.; Reiser, G. Protease-activated receptor-1 protects rat astrocytes from apoptotic cell death via JNK-mediated release of the chemokine GRO/CINC-1. J. Neurochem 2006, 98, 1046–1060. [Google Scholar]
- Badaut, J.; Hirt, L.; Price, M.; de Castro Ribeiro, M.; Magistretti, P.J.; Regli, L. Hypoxia/hypoglycemia preconditioning prevents the loss of functional electrical activity in organotypic slice cultures. Brain Res 2005, 1051, 117–122. [Google Scholar]
- Murer, V.; Spetz, J.F.; Hengst, U.; Altrogge, L.M.; de, A.A.; Monard, D. Male fertility defects in mice lacking the serine protease inhibitor protease nexin-1. Proc. Natl. Acad. Sci. USA 2001, 98, 3029–3033. [Google Scholar]
- Sinnreich, M.; Meins, M.; Niclou, S.P.; Suidan, H.S.; Monard, D. Prothrombin overexpressed in post-natal neurones requires blood factors for activation in the mouse brain. J. Neurochem 2004, 88, 1380–1388. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mirante, O.; Price, M.; Puentes, W.; Castillo, X.; Benakis, C.; Thevenet, J.; Monard, D.; Hirt, L. Endogenous Protease Nexin-1 Protects against Cerebral Ischemia. Int. J. Mol. Sci. 2013, 14, 16719-16731. https://doi.org/10.3390/ijms140816719
Mirante O, Price M, Puentes W, Castillo X, Benakis C, Thevenet J, Monard D, Hirt L. Endogenous Protease Nexin-1 Protects against Cerebral Ischemia. International Journal of Molecular Sciences. 2013; 14(8):16719-16731. https://doi.org/10.3390/ijms140816719
Chicago/Turabian StyleMirante, Osvaldo, Melanie Price, Wilfredo Puentes, Ximena Castillo, Corinne Benakis, Jonathan Thevenet, Denis Monard, and Lorenz Hirt. 2013. "Endogenous Protease Nexin-1 Protects against Cerebral Ischemia" International Journal of Molecular Sciences 14, no. 8: 16719-16731. https://doi.org/10.3390/ijms140816719