Transition Pathway and Its Free-Energy Profile: A Protocol for Protein Folding Simulations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
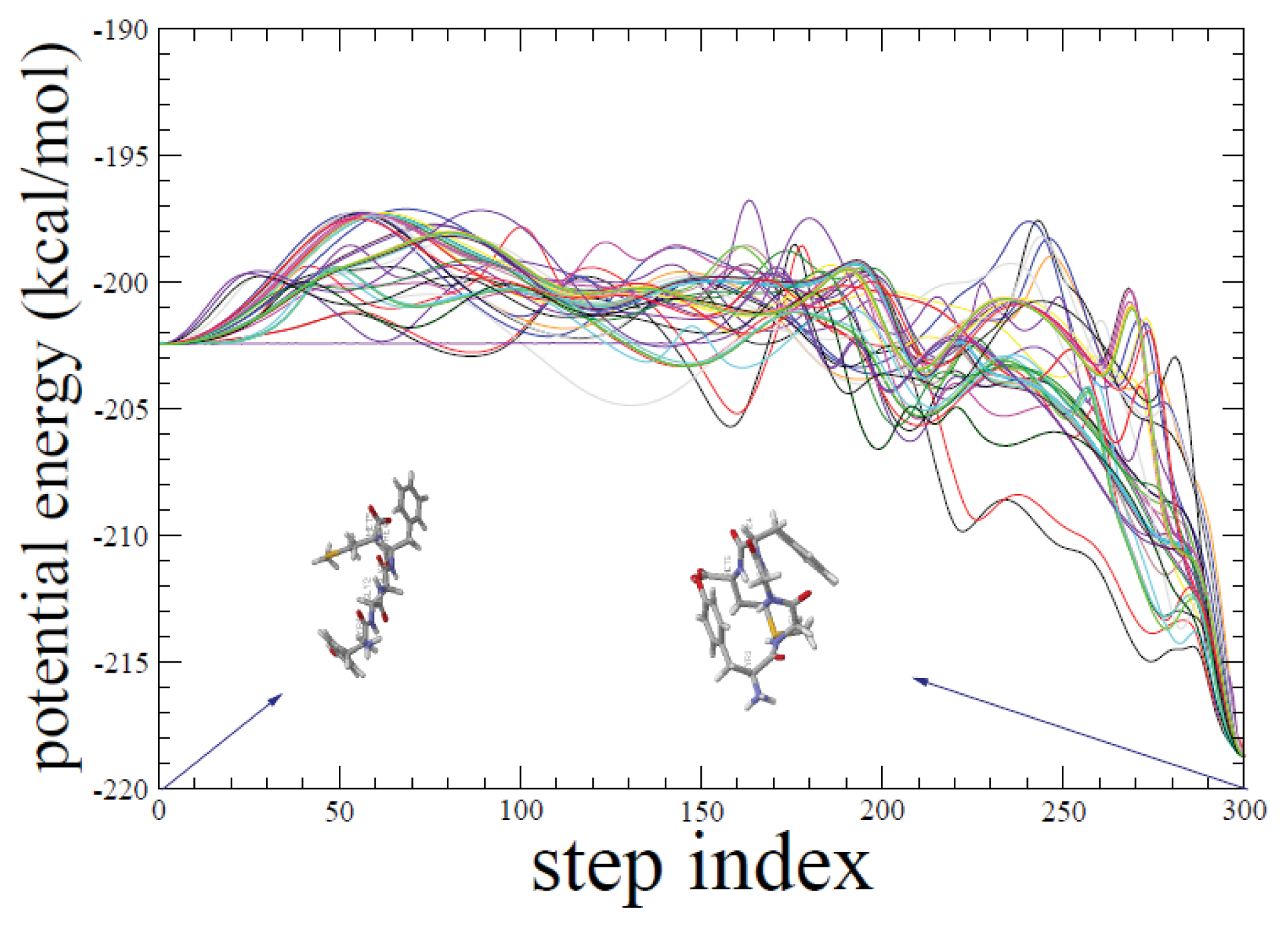
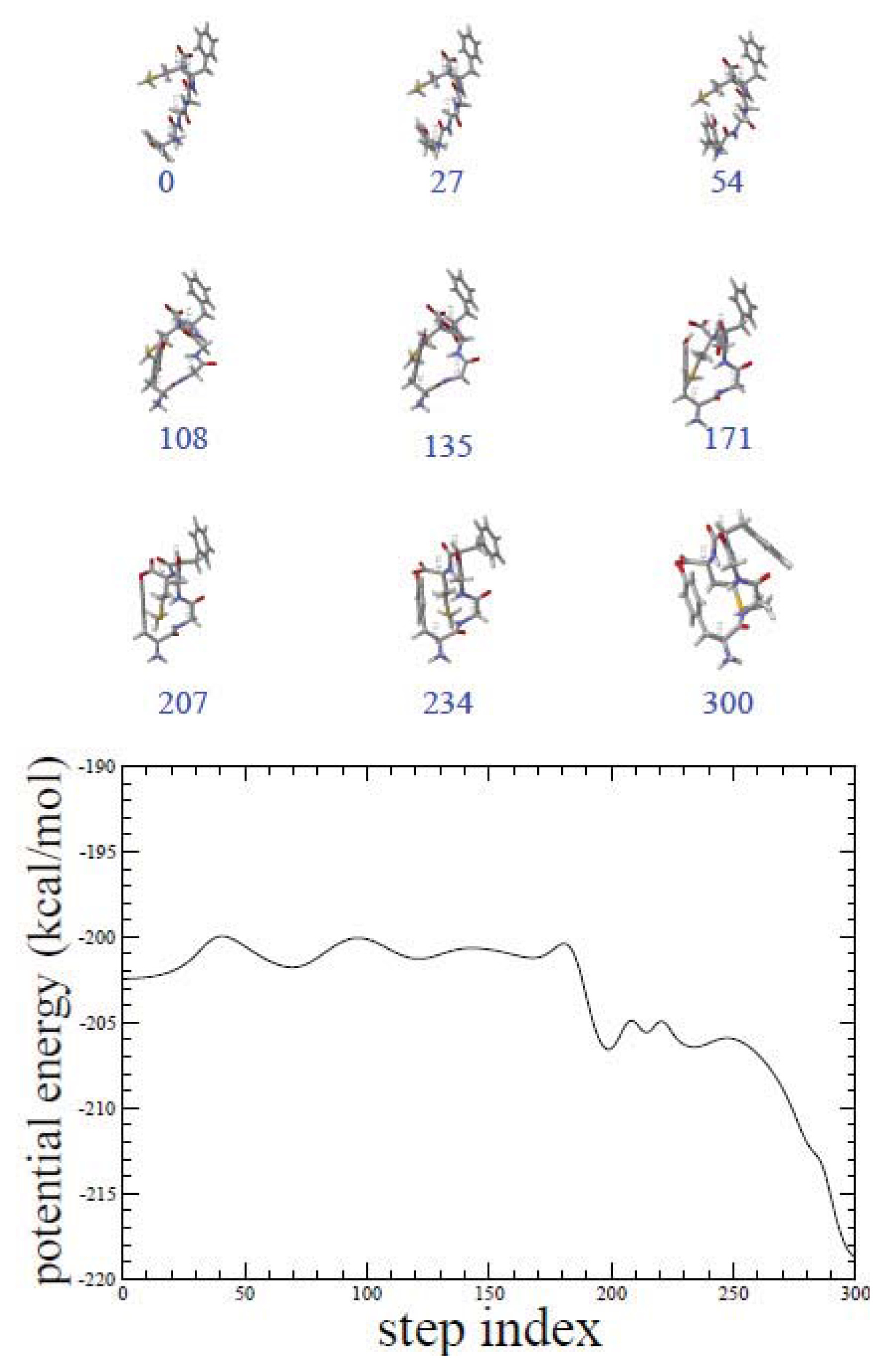
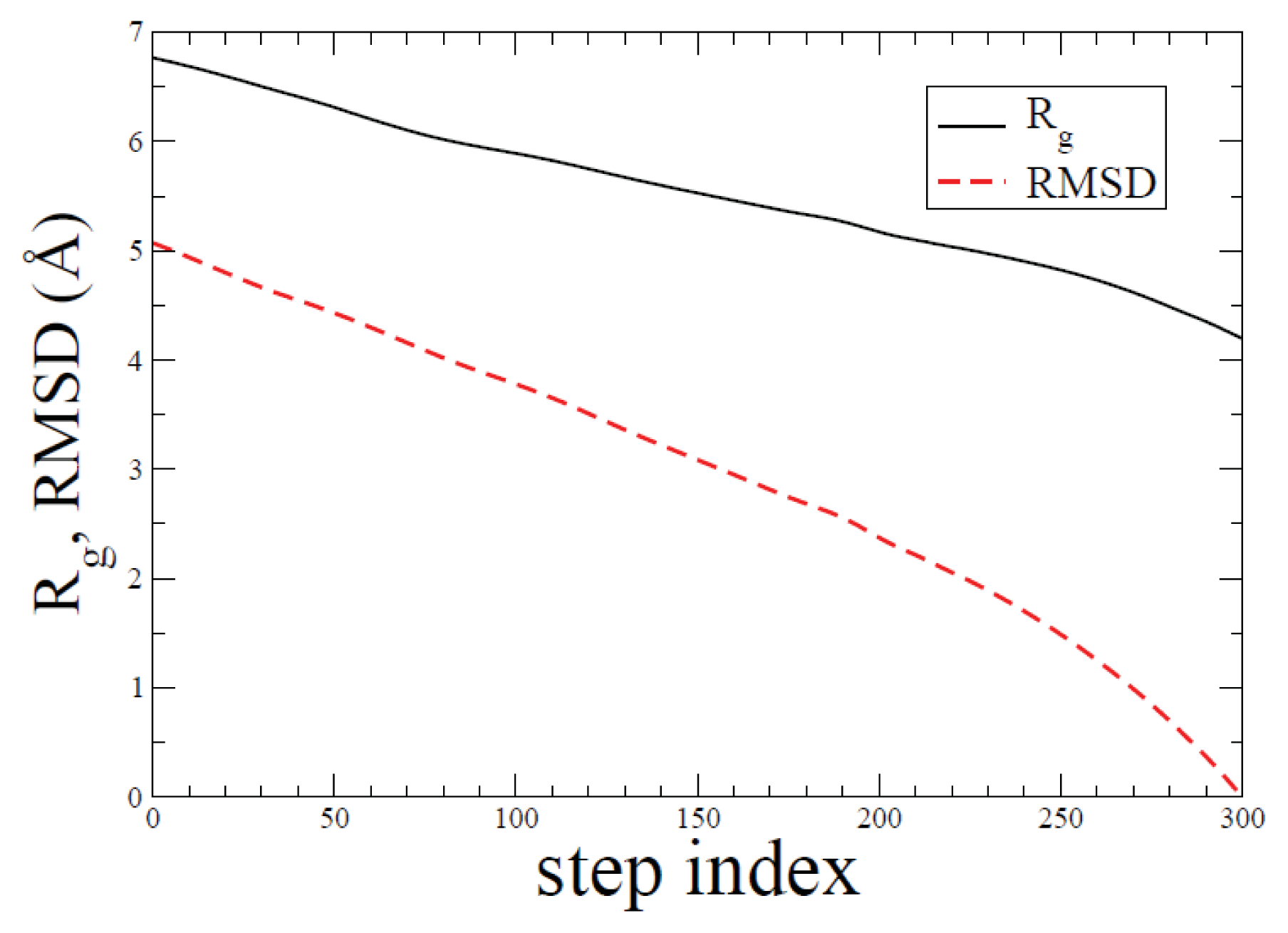
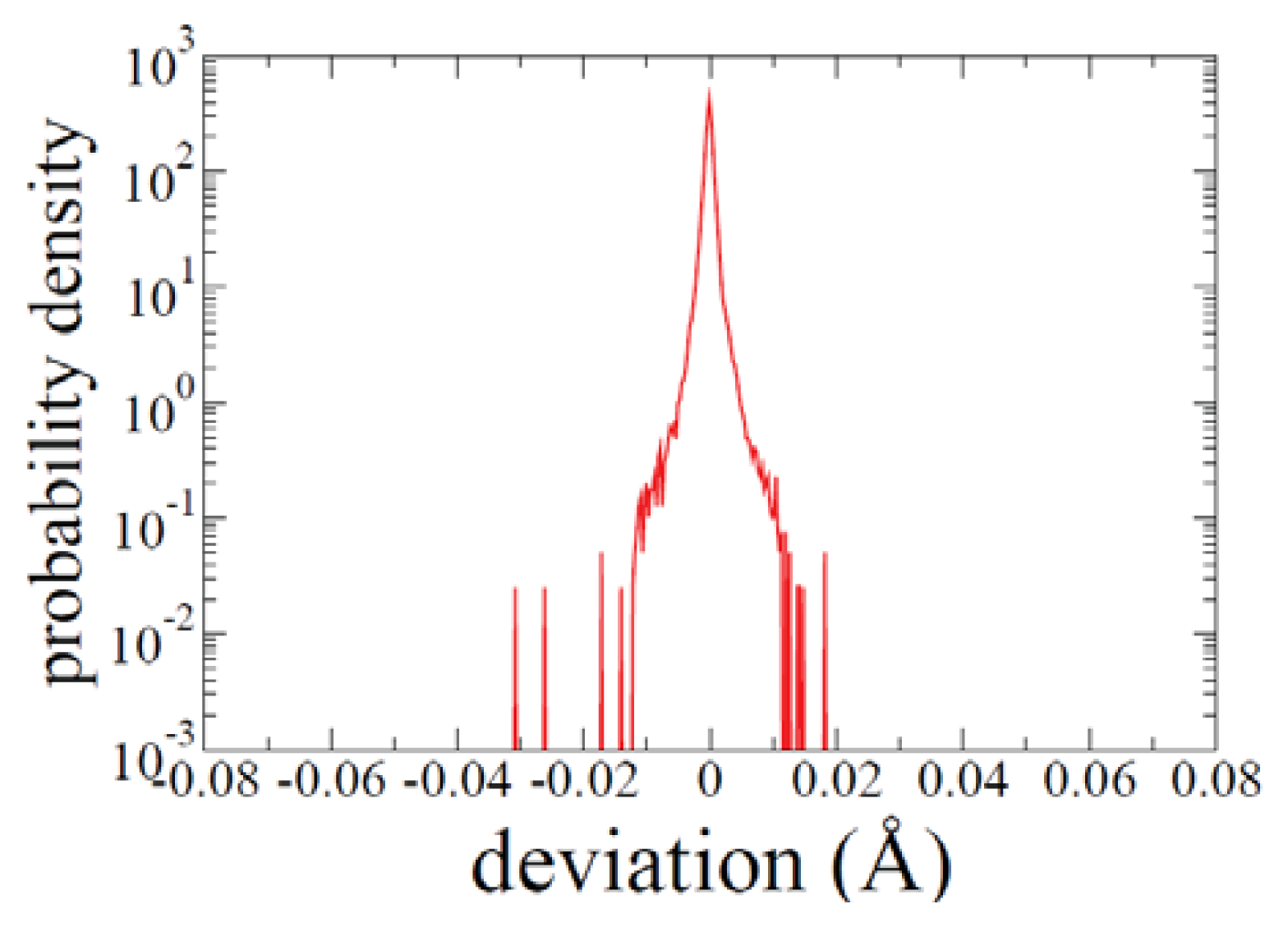
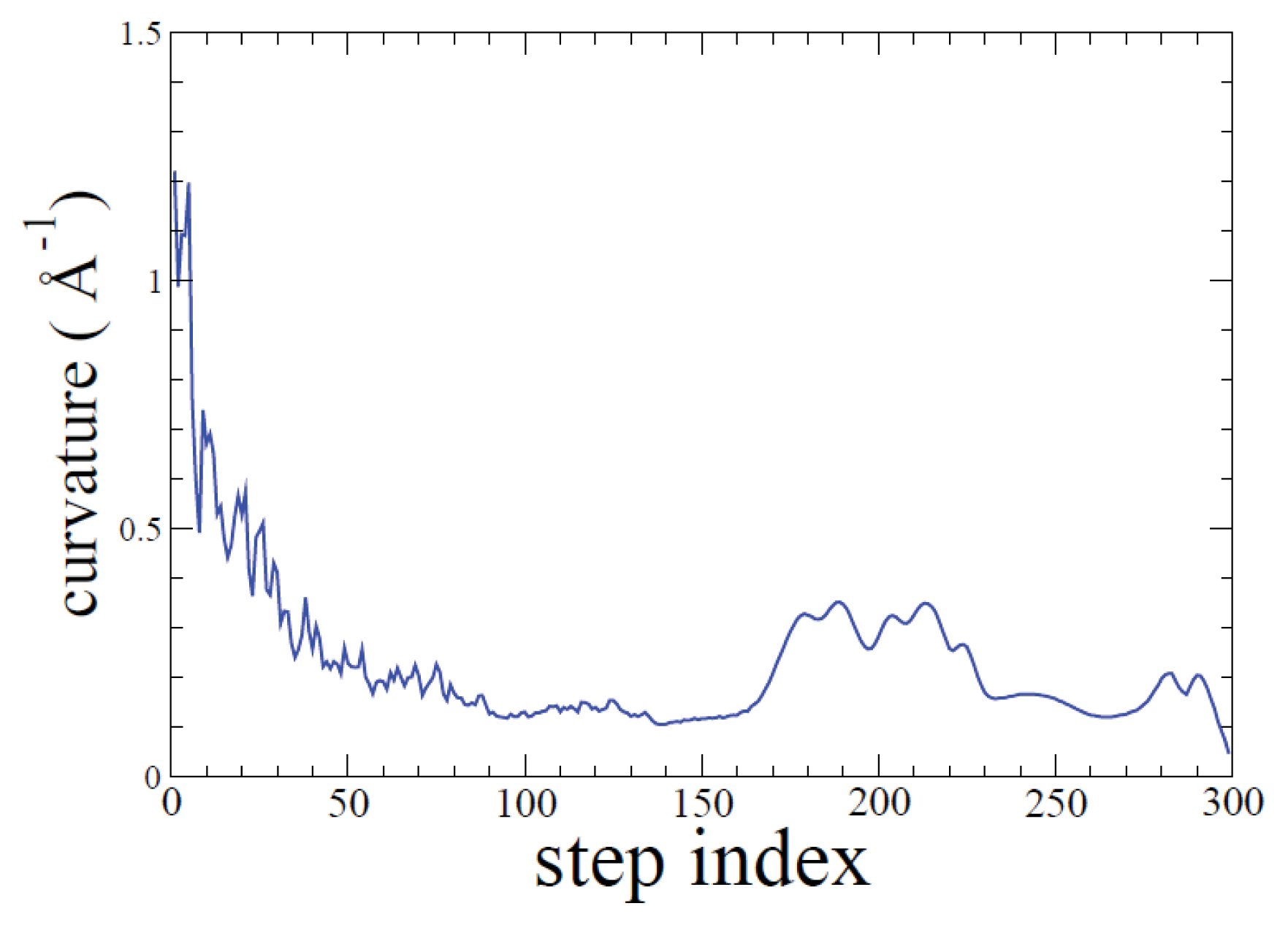
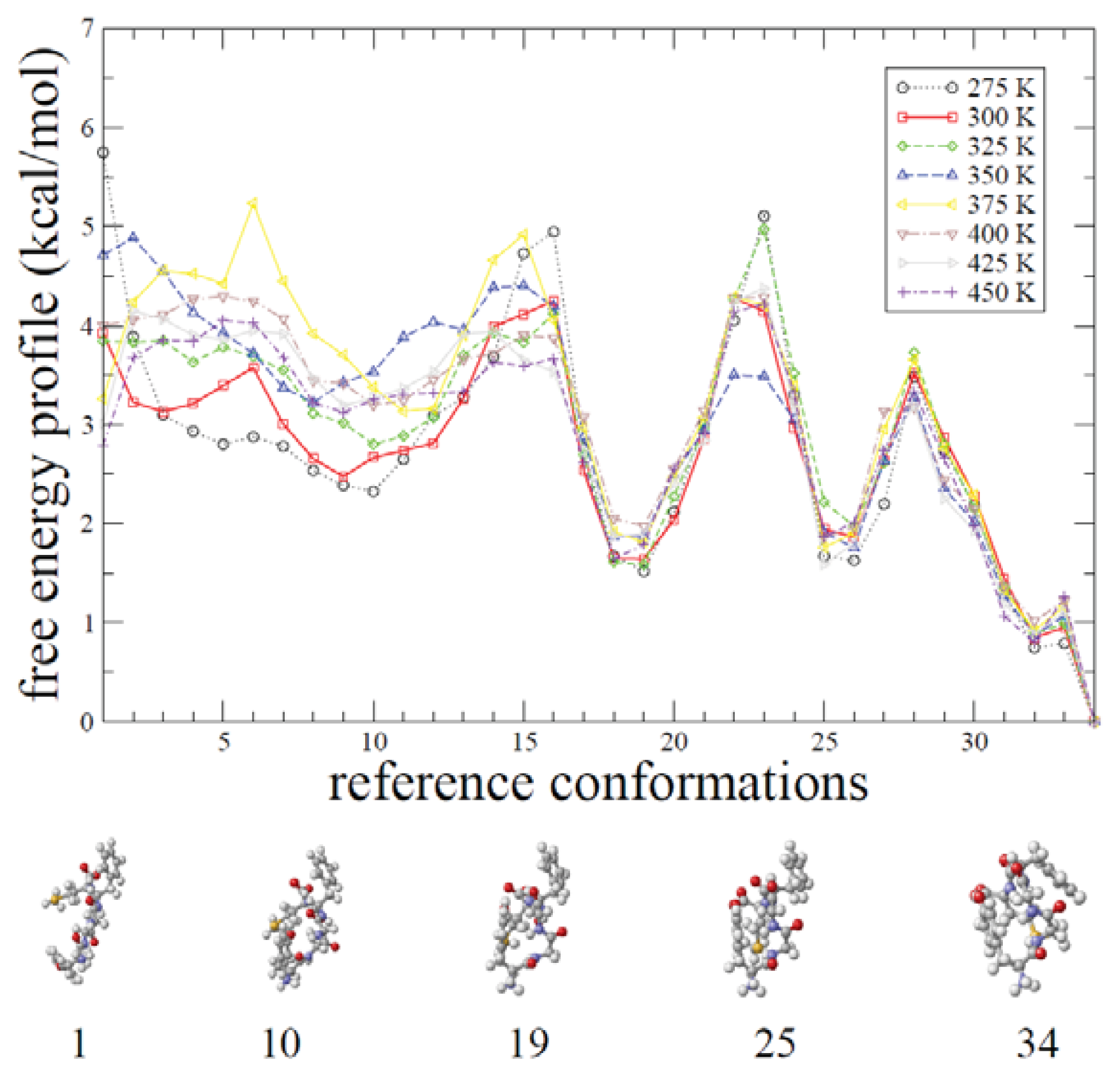
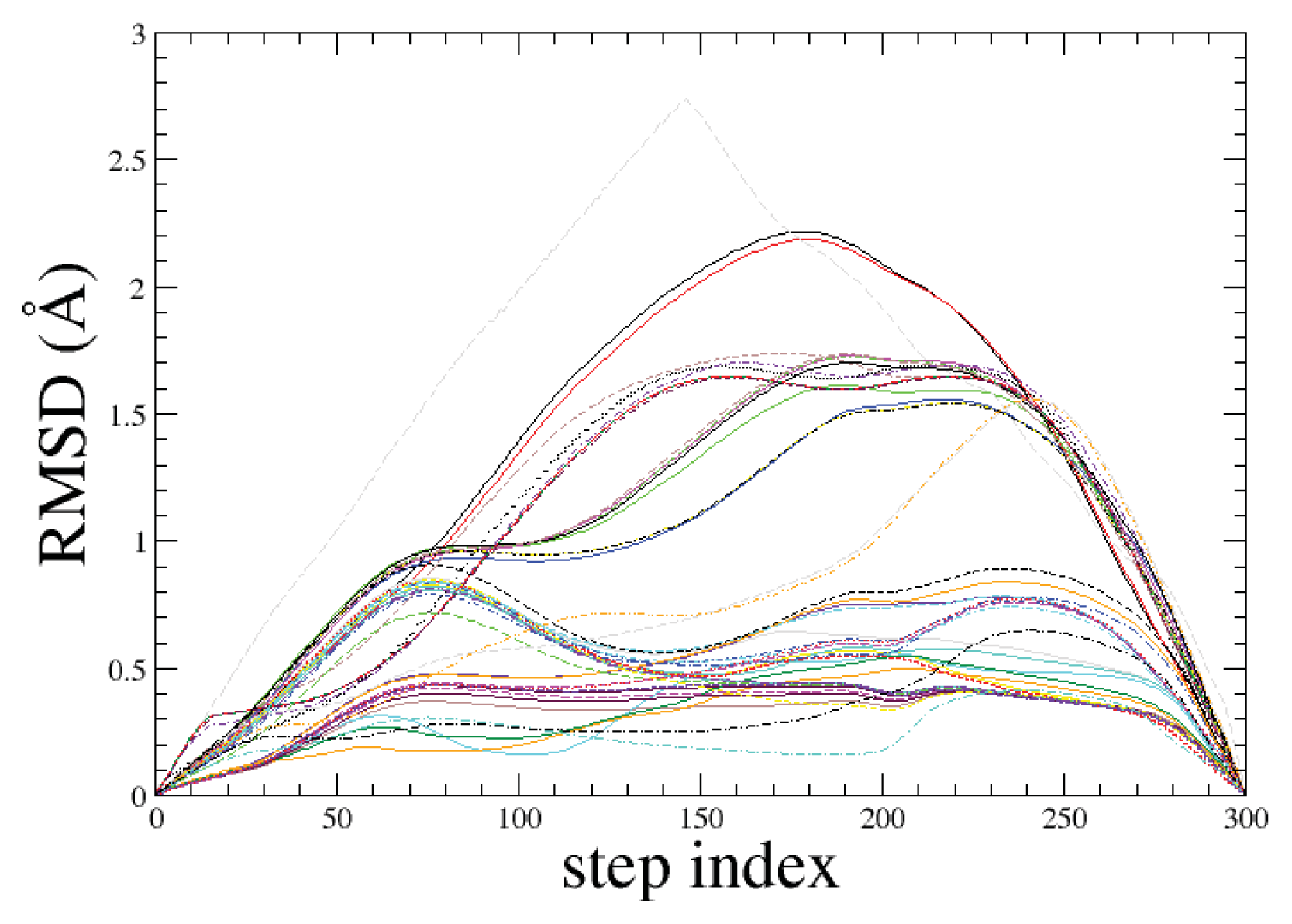
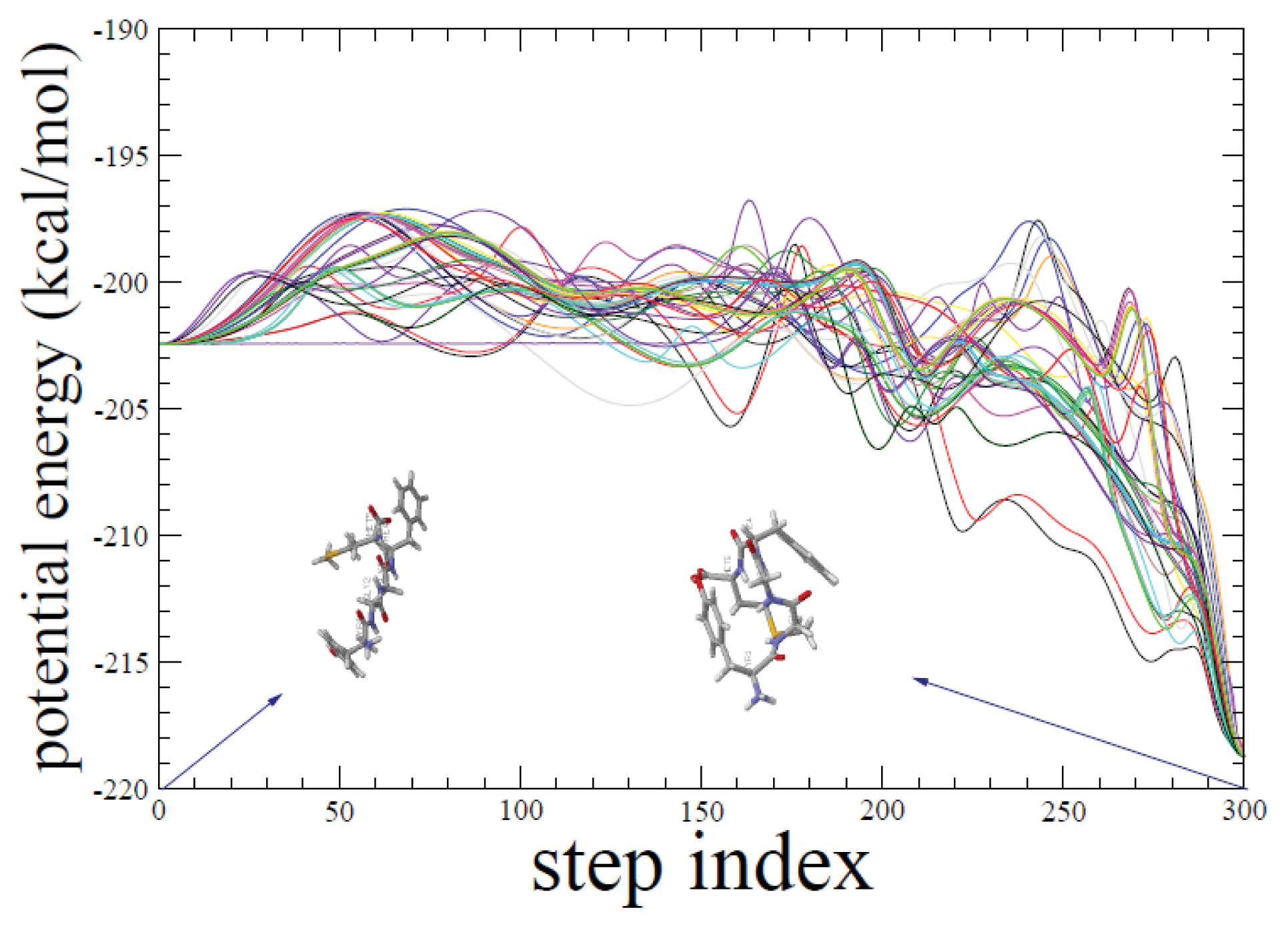
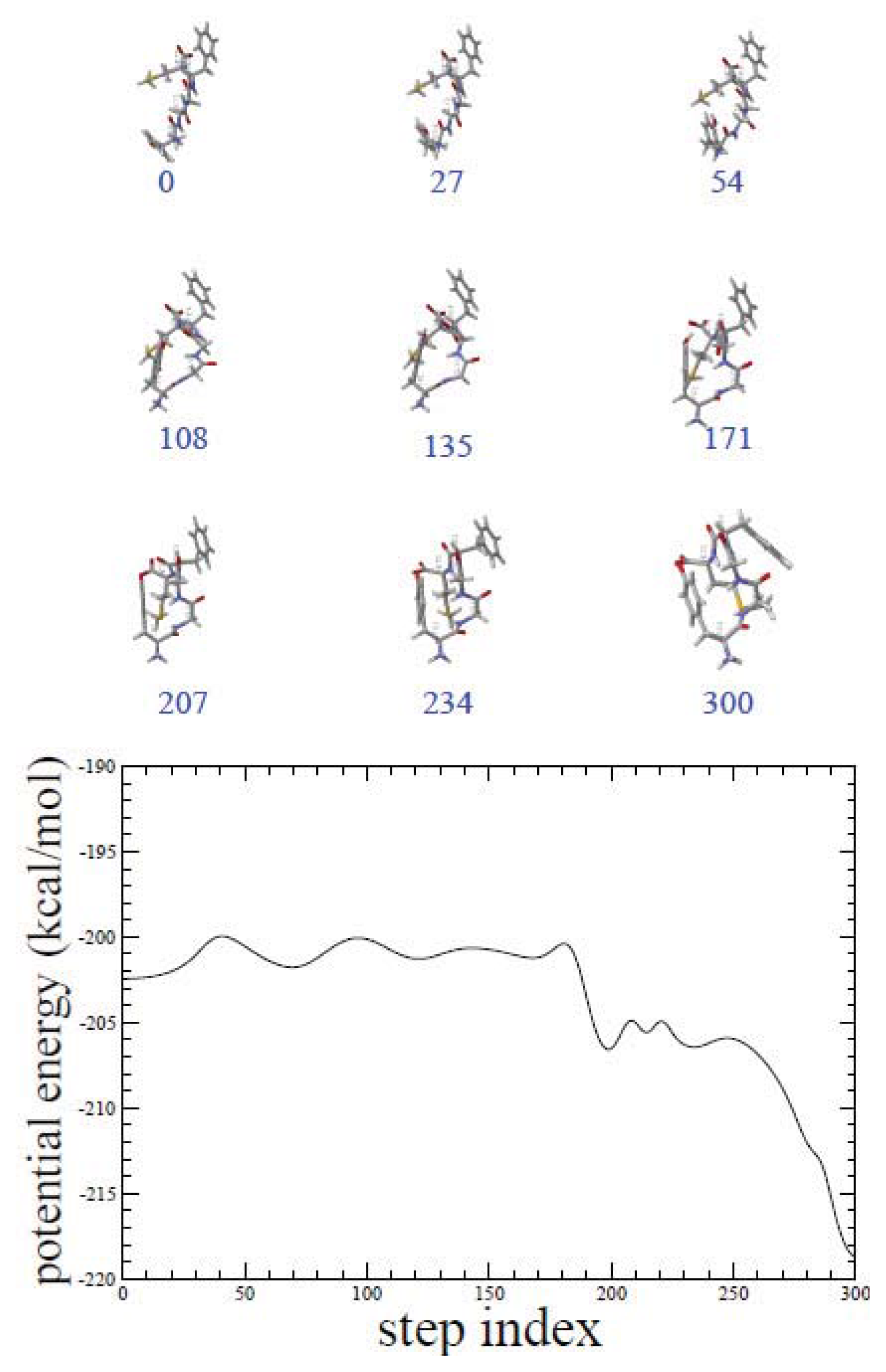
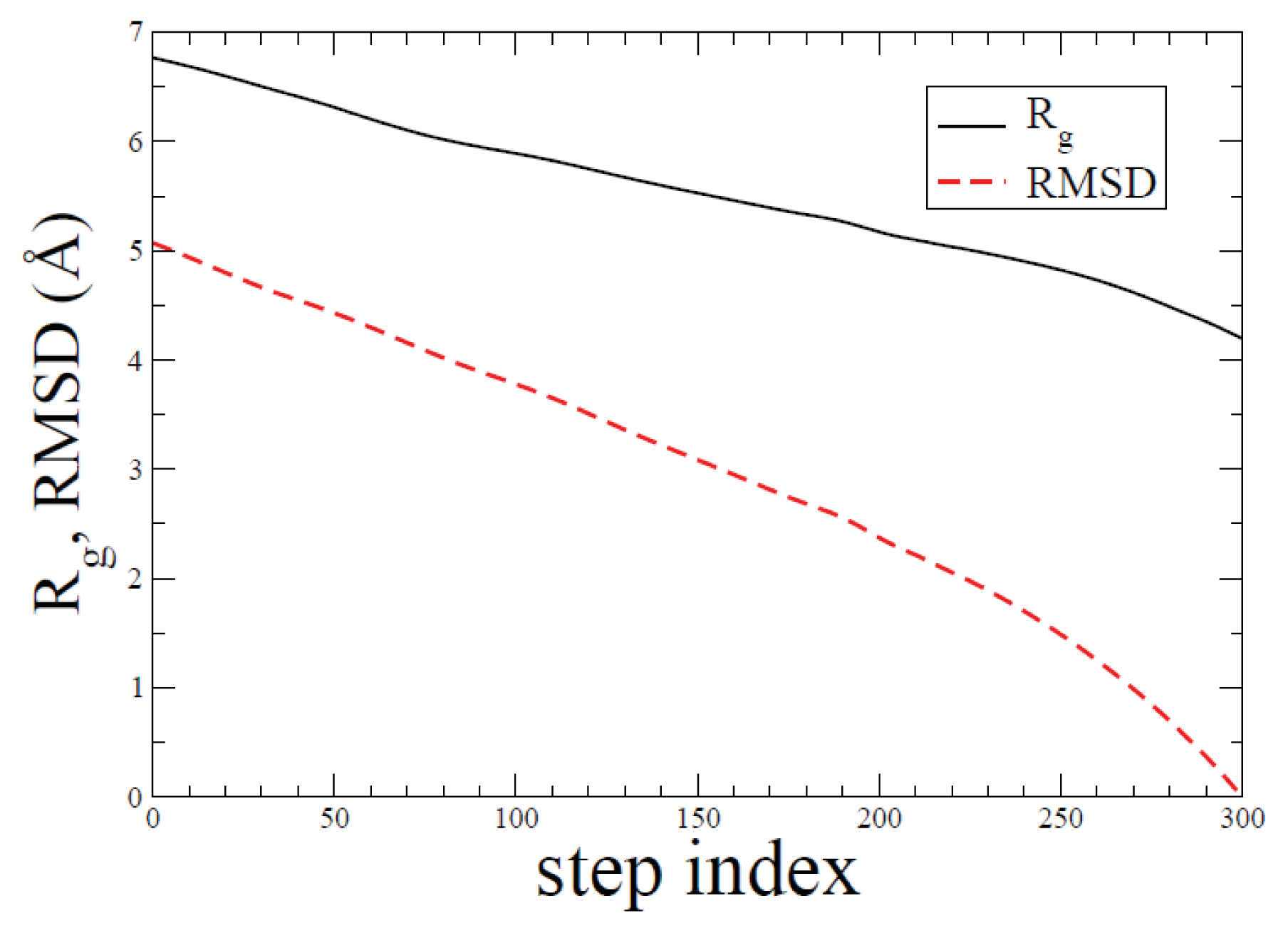
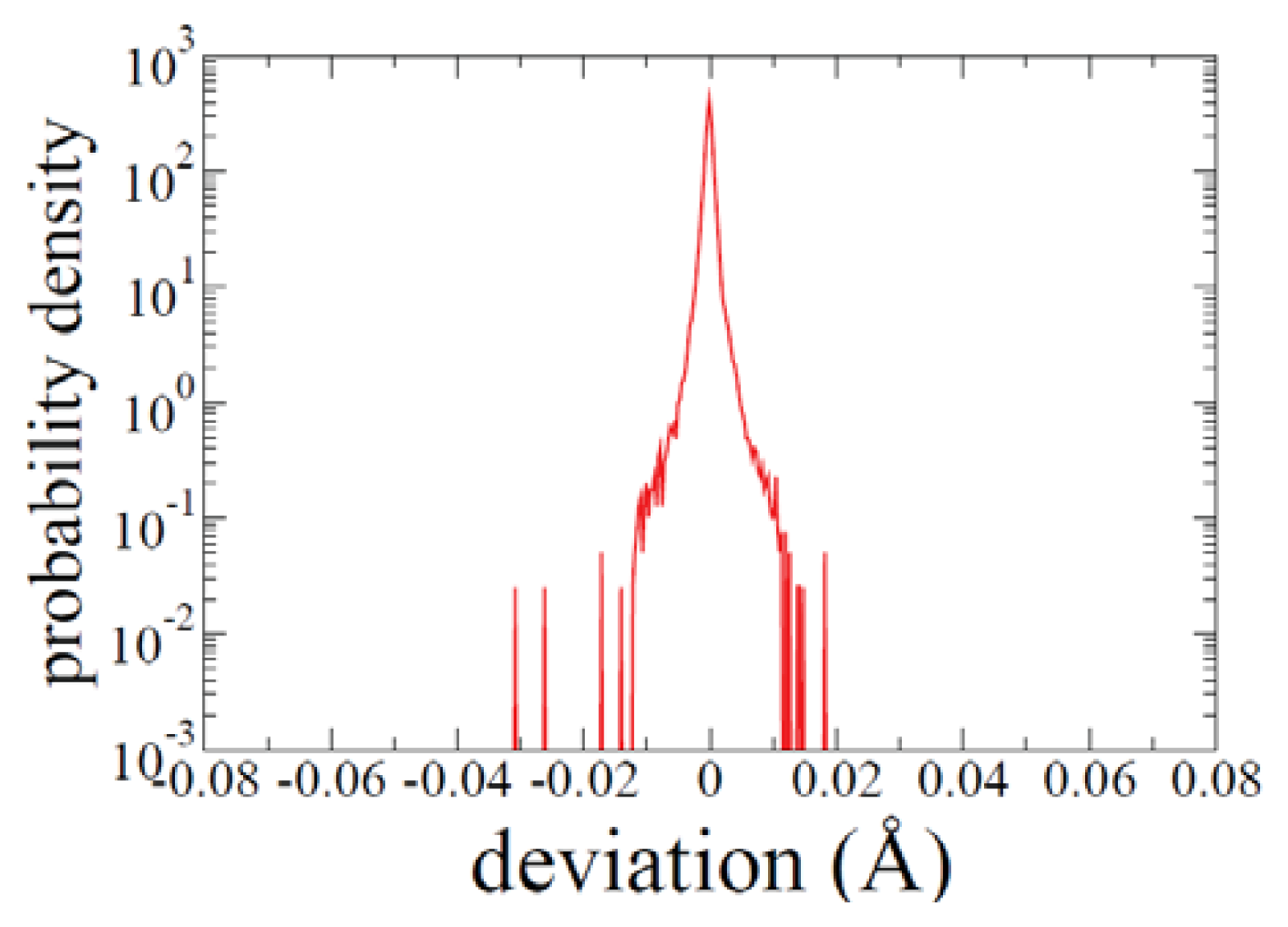
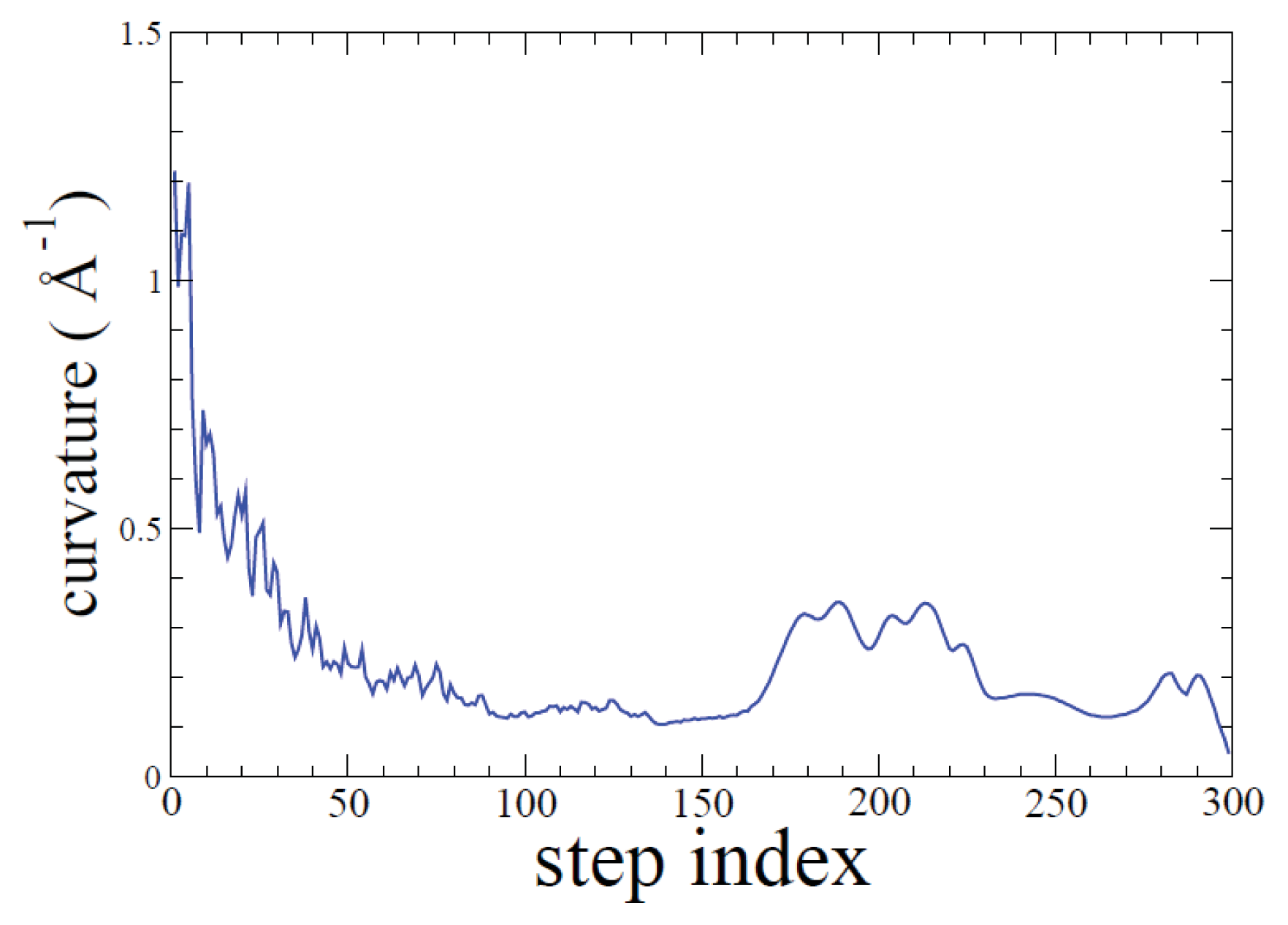
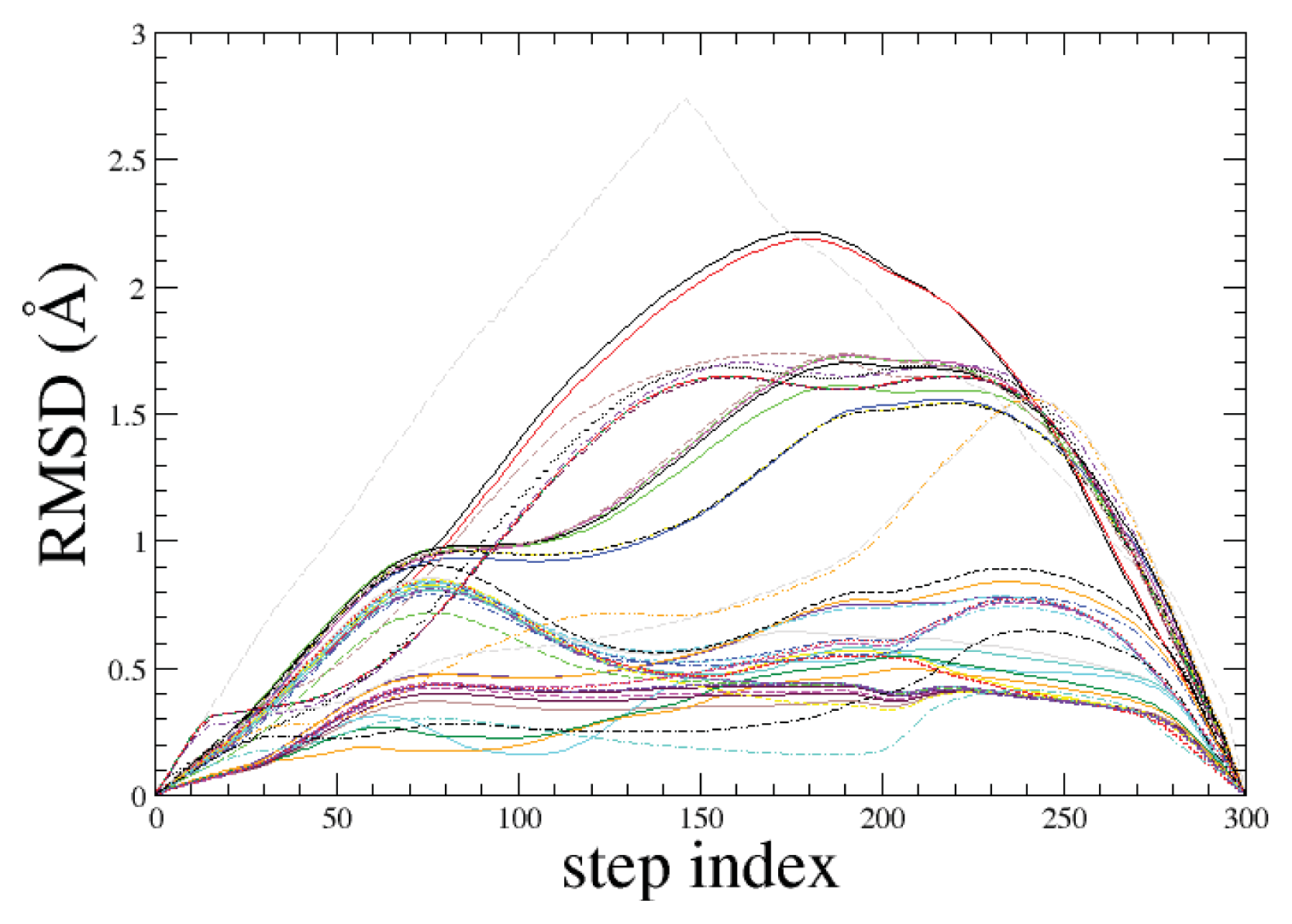
2.1. ADMD Simulations: Pathway Analysis and Reaction Coordinate
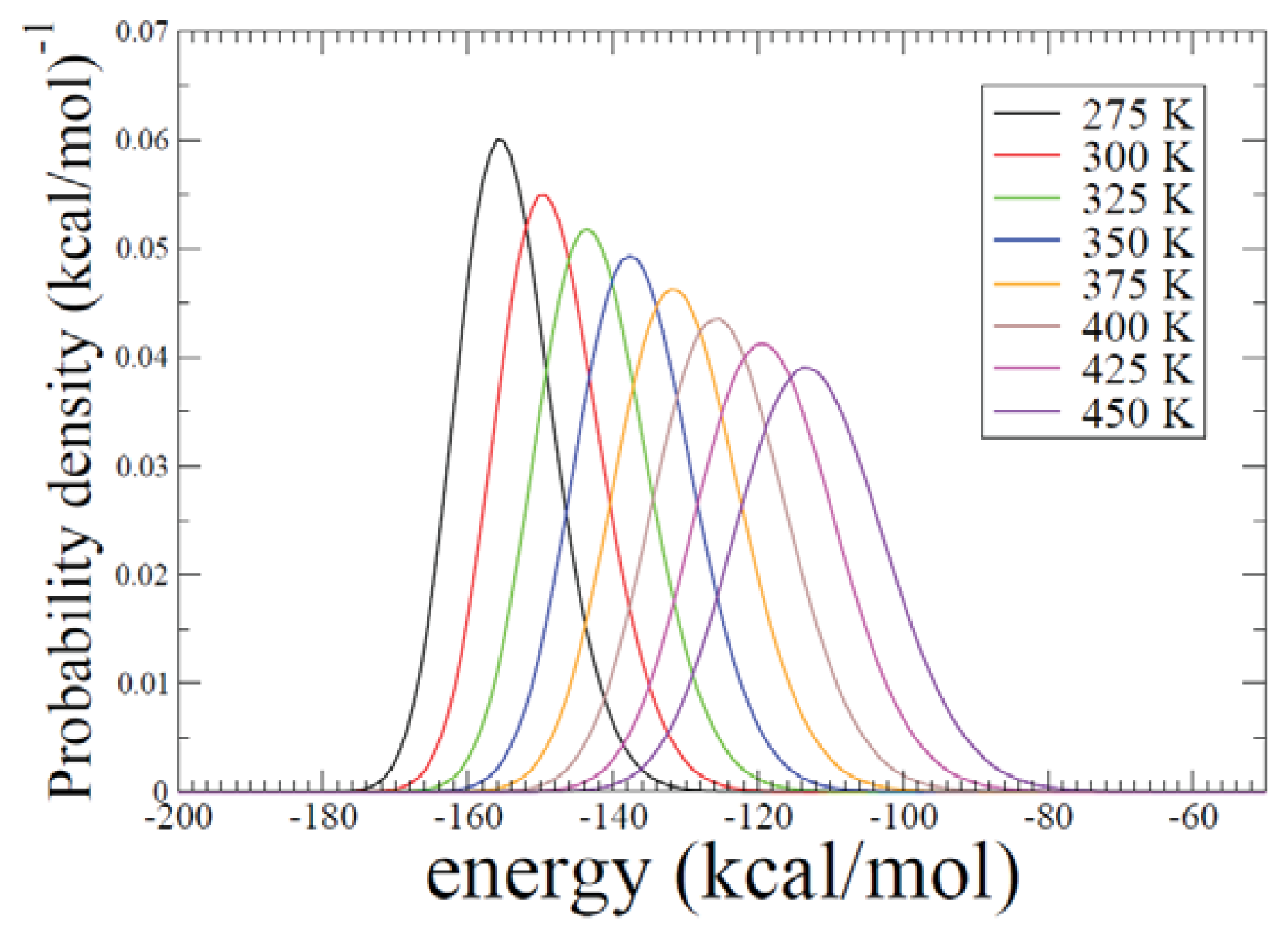
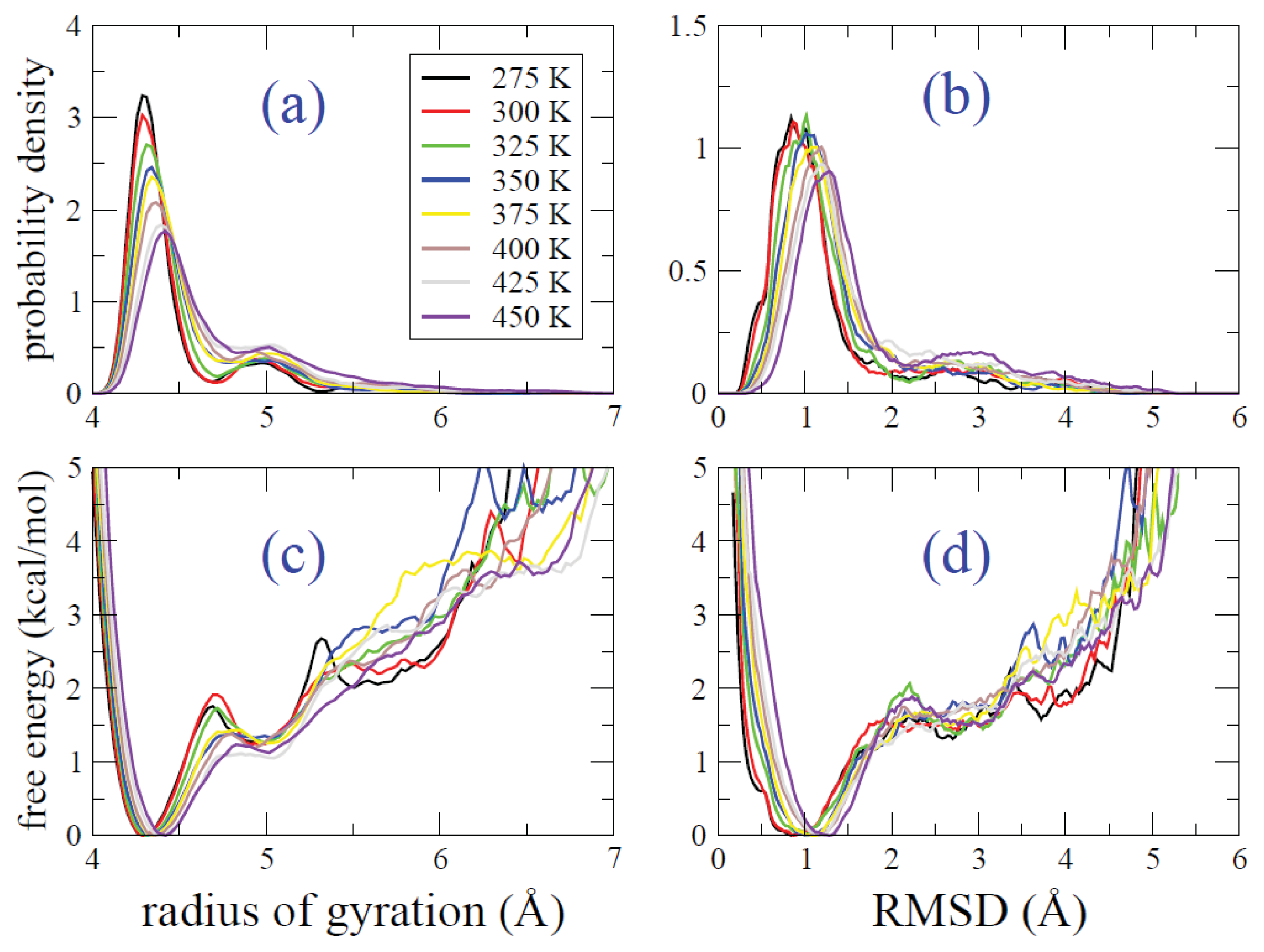
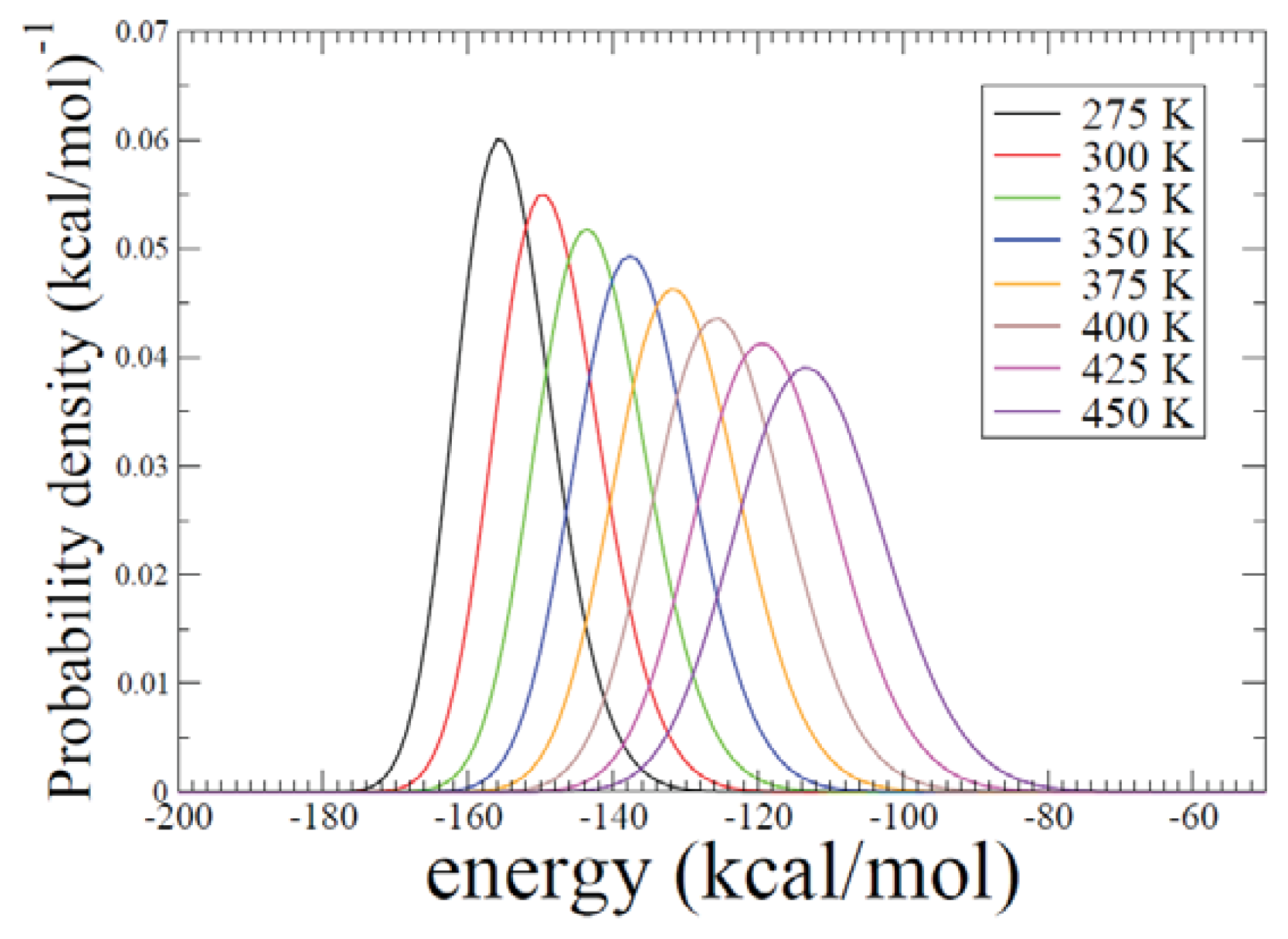
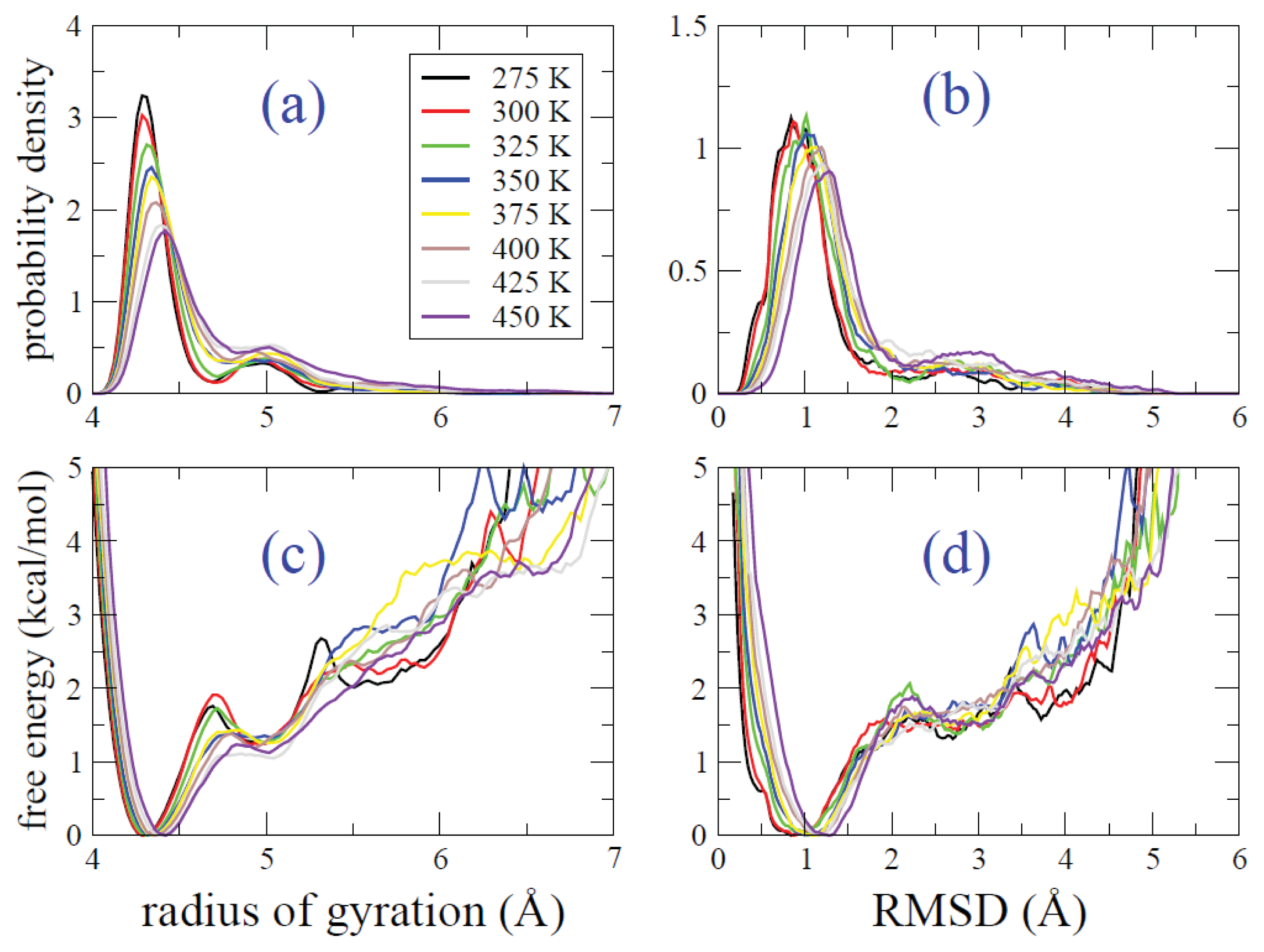
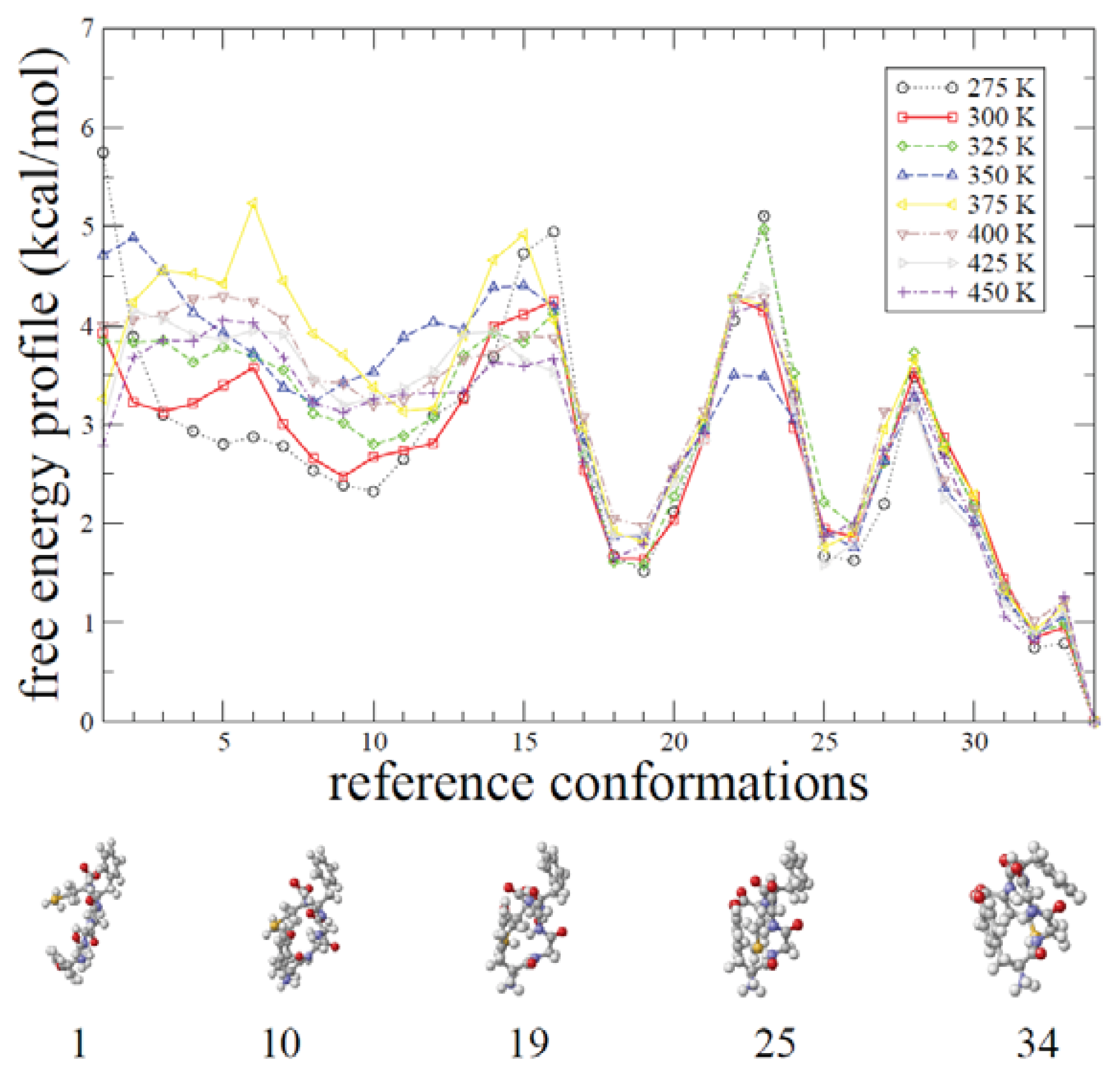
2.2. REMD Simulations: Thermodynamic Analysis
3. Theoretical Methods
3.1. ADMD Method: Background and Setup Parameters
3.2. REMD Simulations: Setup Details
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar]
- Fersht, A.R.; Daggett, V. Protein folding and unfolding at atomic resolution. Cell 2002, 108, 573–582. [Google Scholar]
- Wolynes, P.G. Latest folding game results: Protein A barely frustrates computationalists. Proc. Natl. Acad. Sci. USA 2004, 101, 6837–6838. [Google Scholar]
- Gouda, H.; Torigoe, H.; Saito, A.; Sato, M.; Arata, Y.; Shimada, I. Three-dimensional solution structure of the B domain of staphylococcal protein A: Comparisons of the solution and crystal structures. Biochemistry 1992, 31, 9665–9672. [Google Scholar]
- Witte, K.; Skolnick, J.; Wong, C. A synthetic retrotransition (Backward Reading) sequence of the right-handed three-helix bundle domain (10–53) of protein A shows similarity in conformation as predicted by computation. J. Am. Chem. Soc 1998, 120, 13042–13045. [Google Scholar]
- Shaw, D.E.; Maragakis, P.; Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Eastwood, M.P.; Bank, J.A.; Jumper, J.M.; Salmon, J.K.; Shan, Y.; et al. Atomic-level characterization of the structural dynamics of proteins. Science 2010, 330, 341–346. [Google Scholar]
- Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Shaw, D.E. How fast-folding proteins fold. Science 2011, 334, 517–520. [Google Scholar]
- Chodera, J.D.; Pande, V.S. Splitting probabilities as a test of reaction coordinate choice in single-molecule experiments. Phys. Rev. Lett 2011, 107, 098102. [Google Scholar]
- Passerone, D.; Parrinello, M. Action-derived molecular dynamics in the study of rare events. Phys. Rev. Lett 2001, 87, 108302. [Google Scholar]
- Lee, I.-H.; Lee, J.; Lee, S. Kinetic energy control in action-derived molecular dynamics simulations. Phys. Rev. B 2003, 68, 064303. [Google Scholar]
- Lee, I.-H.; Kim, S.-Y.; Lee, J. Dynamic folding pathway models of α-helix and β-hairpin structures. Chem. Phys. Lett 2005, 412, 307–312. [Google Scholar]
- Lee, I.-H.; Kim, S.-Y.; Lee, J. Dynamic folding pathway models of the villin headpiece subdomain (HP-36) structure. J. Comput. Chem 2010, 31, 57–65. [Google Scholar]
- Lee, I.-H.; Kim, S.-Y.; Lee, J. Folding models of min-protein FSD-1. J. Phys. Chem. B 2012, 116, 6916–6922. [Google Scholar]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett 1999, 314, 141–151. [Google Scholar]
- Swendsen, R.H.; Wang, J.S. Replica Monte Carlo simulation of spin-glasses. Phys. Rev. Lett 1986, 57, 2607–2609. [Google Scholar]
- Lee, J.; Scheraga, H.A.; Rackovsky, S. New optimization method for conformational energy calculations on polypeptides: Conformational space annealing. J. Comput. Chem 1997, 18, 1222–1232. [Google Scholar]
- Perez, J.J.; Villar, H.O.; Loew, G.H. Characterization of low-energy conformational domains for met-enkephalin. J. Comput. Aided Mol. Des. 1992, 6, 175–190. [Google Scholar]
- Jin, A.Y.; Leung, F.Y.; Weaver, D.F. Three variations of genetic algorithm for searching biomolecular conformation space: Comparison of GAP 1.0, 2.0, and 3.0. J. Comput. Chem 1999, 20, 1329–1342. [Google Scholar]
- Isogai, Y.; Nemethy, G.; Scheraga, H.A. Enkephalin: Conformational analysis by means of empirical energy calculations. Proc. Natl. Acad. Sci. USA 1977, 74, 414–418. [Google Scholar]
- Montcalm, T.; Weili, C.; Hong, Z.; Guarnieri, F.; Wilson, S.R. Simulated annealing of met-enkephalin: low energy states and their relevance to membrane-bound, solution and solid-state conformations. J. Mol. Struc. Theochem 1994, 308, 37–51. [Google Scholar]
- Garbay-Jaureguiberry, C.; Roques, B.P.; Oberlin, R.; Anteunis, M.; Lala, A.K. Preferential conformation of the endogenous opiate-like pentapeptide met-enkephalin in DMSO-d6 solution determined by high field H NMR. Biochem. Biophys. Res. Commun 1976, 71, 558–565. [Google Scholar]
- Jones, C.R.; Gibbons, W.A.; Garsky, V. Proton magnetic resonance studies of conformation and flexibility of encephalin peptides. Nature 1976, 262, 779–782. [Google Scholar]
- Roques, B.P.; Garbay-Jaureguiberry, C.; Oberlin, R.; Anteunis, M.; Lala, A.K. Conformation of met-enkephalin determined by high field PMR spectroscopy. Nature 1976, 262, 778–779. [Google Scholar]
- Khaled, M.A.; Long, M.M.; Thompson, W.D.; Bradley, R.J.; Brown, G.B.; Urry, D.W. Conformational states of enkephalins in solution. Biochem. Biophys. Res. Commun 1977, 76, 224–231. [Google Scholar]
- Graham, W.H.; Carter, E.S., II; Hickes, R.P. Conformational analysis of met-enkephalin in both aqueous solution and in the presence of sodium dodecyl sulfate micelles using multidimensional NMR and molecular modeling. Biopolymers 1992, 32, 1755–1764. [Google Scholar]
- Renugopalakrishnan, V.; Rapaka, R.S.; Collette, T.W.; Carreira, L.A.; Bhatnagar, R.S. Conformational states of Leu5- and Met5-enkephalins in solution. Biochem. Biophys. Res. Commun 1985, 126, 1029–1035. [Google Scholar]
- Schiller, P.W. Fluorescence study on the conformation of a cyclic enkephalin analog in aqueous solution. Biochem. Biophys. Res. Commun 1983, 114, 268–274. [Google Scholar]
- Spirtes, M.A.; Schwartz, R.W.; Mattice, W.L.; Coy, D.H. Circular dichroism and absorption study of the structure of methionine-enkephalin in solution. Biochem. Biophys. Res. Commun 1978, 81, 602–609. [Google Scholar]
- D’Alagni, M.; Delfini, M.; di Nola, A.; Eisenberg, M.; Paci, M.; Roda, L.G.; Veglia, G. Conformational study of Met-enkephalin-Arg-Phe in the presence of phosphatidylserine vesicles. Eur. J. Biochem 1996, 240, 540–549. [Google Scholar]
- Smith, G.D.; Griffin, J.F. Conformation of Leu5 enkephalin from X-ray diffraction: Features important for recognition at opiate receptor. Science 1978, 199, 1214–1216. [Google Scholar]
- Deschamps, J.R.; George, C.; Flippen-Anderson, J.L. Structural studies of opioid peptides: A review of recent progress in X-ray diffraction studies. Biopolymers 1996, 40, 121–139. [Google Scholar]
- Lee, I.-H.; Kim, S.-Y. Dynamics of the neuropeptide met-enkephalin by using action-derived molecular dynamics. J. Korean Phys. Soc 2008, 53, 1764–1769. [Google Scholar]
- Kearsley, S.K. An algorithm for the simultaneous superposition of a structural series. J. Comput. Chem 1990, 11, 1187–1192. [Google Scholar]
- Coutsias, E.A.; Seok, C.; Dill, K.A. Using quaternions to calculate RMSD. J. Comput. Chem 2004, 25, 1849–1857. [Google Scholar]
- Kinoshita, M.; Okamoto, Y.; Hirata, F. Solvation structure and stability of peptides in aqueous solutions analyzed by the reference interaction site model theory. J. Chem. Phys 1997, 107, 1586–1599. [Google Scholar]
- Mitsutake, A.; Kinoshita, M.; Okamoto, Y.; Hirata, F. Combination of the replica-exchange Monte Carlo method and the reference interaction site model theory for simulating a peptide molecule in aqueous solution. J. Phys. Chem. B 2004, 108, 19002–19012. [Google Scholar]
- Li, Z.; Scheraga, H.A. Structure and free energy of complex thermodynamic systems. J. Mol. Struct. (Theochem. ) 1988, 179, 333–352. [Google Scholar]
- Qiu, D.; Shenkin, P.S.; Hollinger, F.P.; Still, W.C. The GB/SA continuum model for solvation. A fast analytical method for the calculation of approximate Born radii. J. Phys. Chem. A 1997, 101, 3005–3014. [Google Scholar]
- Ponder, J.W.; Richard, F.M. An efficient Newton-like method for molecular mechanics energy minimization of large molecules. J. Comput. Chem 1987, 8, 1016–1024. [Google Scholar]
- Eisenmenger, F.; Hansmann, U.H.E. Variation of the energy landscape of a small peptide under a change from the ECEPP/2 force field to ECEPP/3. J. Phys. Chem. B 1997, 101, 3304–3310. [Google Scholar]
- Hayryan, S.; Hu, C.-K.; Hu, S.-Y.; Shang, R.-J. Multicanonical parallel simulations of proteins with continuous potentials. J. Comput. Chem 2001, 22, 1287–1296. [Google Scholar]
- Vengadesan, K.; Gautham, N. Conformational studies on enkephalins using the MOLS technique. Biopolymers 2004, 74, 476–494. [Google Scholar]
- Zhan, L.; Chen, J.Z.Y.; Liu, W.-K. Conformational study of met-enkephalin based on the ECEPP force fields. Biophys. J 2006, 91, 2399–2404. [Google Scholar]
- Elber, R.; Cárdenas, A.; Ghosh, A.; Stern, H.A. Bridging the gap between reaction pathways, long time dynamics and calculation of rates. Adv. Chem. Phys 2003, 126, 93–129. [Google Scholar]
- Onsager, L.; Machlup, S. Fluctuations and irreversible processes. Phys. Rev 1953, 91, 1505–1512. [Google Scholar]
- Verlet, L. Computer “Experiments” on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev 1967, 159, 98–103. [Google Scholar]
- Lee, I.-H.; Kim, S.-Y.; Lee, J. Dynamics of conformational isomerization in alanine dipeptide and valine dipeptide. J. Korean Phys. Soc 2005, 46, 601–605. [Google Scholar]
- Kim, Y.-H.; Lee, I.-H.; Chang, K.J.; Lee, S. Dynamics of fullerene coalescence. Phys. Rev. Lett 2003, 90, 065501. [Google Scholar]
- Lee, I.-H.; Kim, H.; Lee, J. Dynamic pathway model for the formation of C60. J. Chem. Phys 2004, 120, 4672–4676. [Google Scholar]
- Lee, I.-H.; Jun, S.; Kim, H.; Kim, S.Y.; Lee, Y. Adatom-assisted structural transformations of fullerenes. Appl. Phys. Lett 2006, 88, 011913. [Google Scholar]
- Pendurti, S.; Jun, S.; Lee, I.-H.; Prasad, V. Cooperative atomic motions and core rearrangement in dislocation cross slip. Appl. Phys. Lett 2006, 88, 201908. [Google Scholar]
- Kim, S.Y.; Lee, I.-H.; Jun, S.; Lee, Y.; Im, S. Coalescence and T-junction formation of carbon nanotubes: Action-derived molecular dynamics simulations. Phys. Rev. B 2006, 74, 195409. [Google Scholar]
- Kim, S.Y.; Lee, I.-H.; Jun, S. Transition-pathway models of atomic diffusion on fcc metal surfaces. I. Flat surfaces. Phys. Rev. B 2007, 76, 245407. [Google Scholar]
- Kim, S.Y.; Lee, I.-H.; Jun, S. Transition-pathway models of atomic diffusion on fcc metal surfaces. II. Stepped surfaces. Phys. Rev. B 2007, 76, 245408. [Google Scholar]
- Lee, Y.; Han, J.; Lee, I.-H.; Im, S. Mobility of a 5|7 defect in carbon nanotubes. Nanotechnology 2011, 22, 105707. [Google Scholar]
- Lee, I.-H.; Kim, S.Y.; Jun, S. An introductory overview of action-derived molecular dynamics for multiple time-scale simulations. Comput. Methods Appl. Mech. Eng 2004, 193, 1633–1644. [Google Scholar]
- Zagrovic, B.; Sorin, E.J.; Pande, V. β-hairpin folding simulations in atomistic detail using an implicit solvent model. J. Mol. Biol 2001, 313, 151–169. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, I.-H.; Kim, S.-Y.; Lee, J. Transition Pathway and Its Free-Energy Profile: A Protocol for Protein Folding Simulations. Int. J. Mol. Sci. 2013, 14, 16058-16075. https://doi.org/10.3390/ijms140816058
Lee I-H, Kim S-Y, Lee J. Transition Pathway and Its Free-Energy Profile: A Protocol for Protein Folding Simulations. International Journal of Molecular Sciences. 2013; 14(8):16058-16075. https://doi.org/10.3390/ijms140816058
Chicago/Turabian StyleLee, In-Ho, Seung-Yeon Kim, and Jooyoung Lee. 2013. "Transition Pathway and Its Free-Energy Profile: A Protocol for Protein Folding Simulations" International Journal of Molecular Sciences 14, no. 8: 16058-16075. https://doi.org/10.3390/ijms140816058