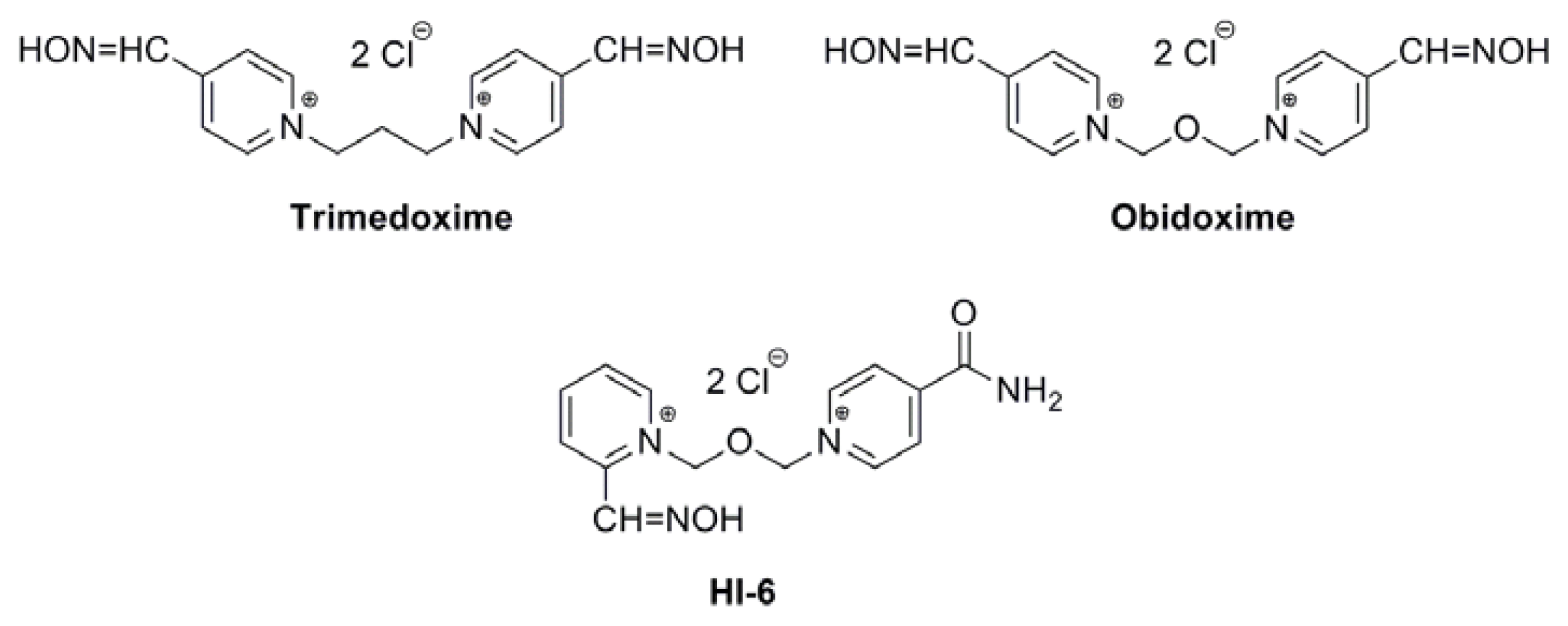
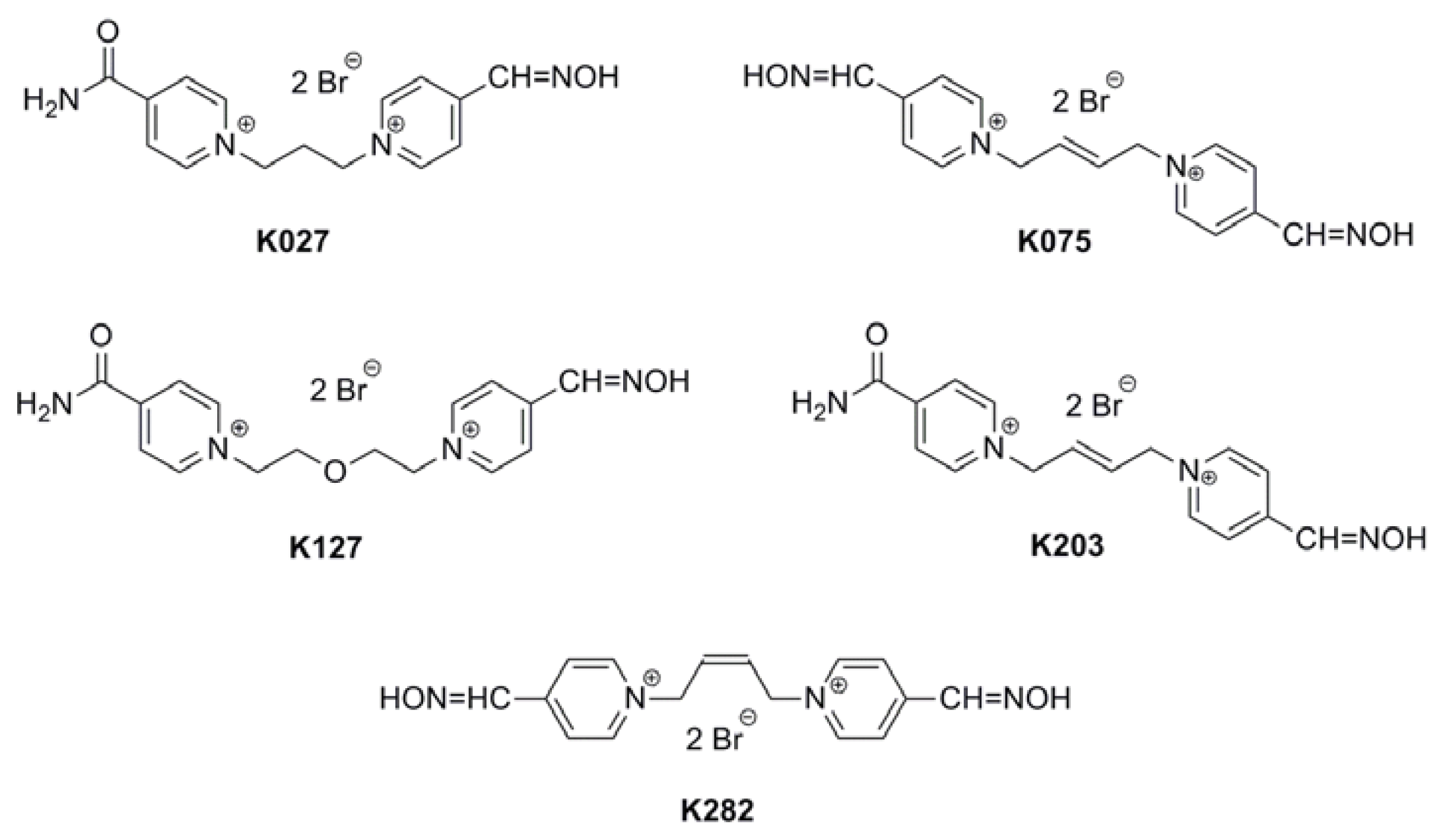
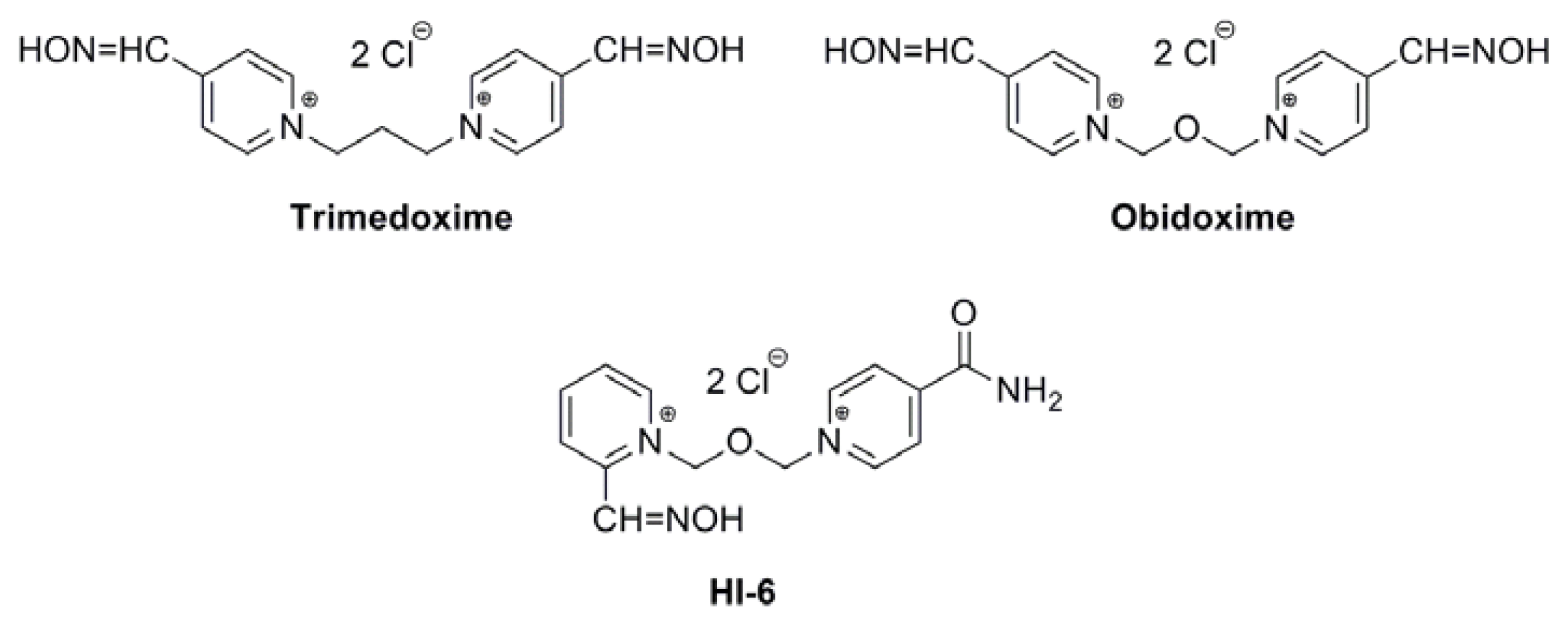
Acetylcholinesterase Reactivators (HI-6, Obidoxime, Trimedoxime, K027, K075, K127, K203, K282): Structural Evaluation of Human Serum Albumin Binding and Absorption Kinetics



Abstract
:1. Introduction
2. Results and Discussion
2.1. Tolerability
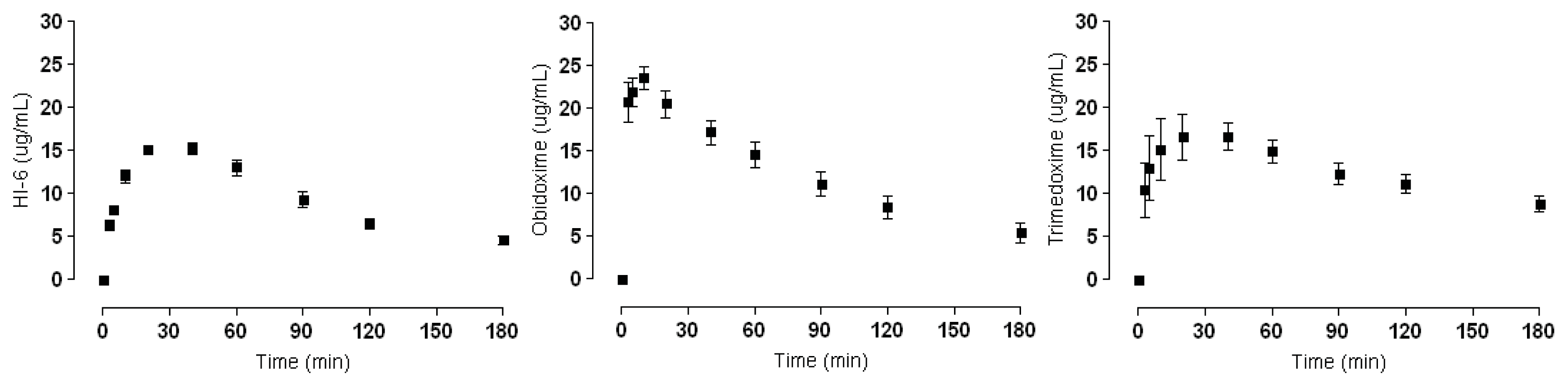
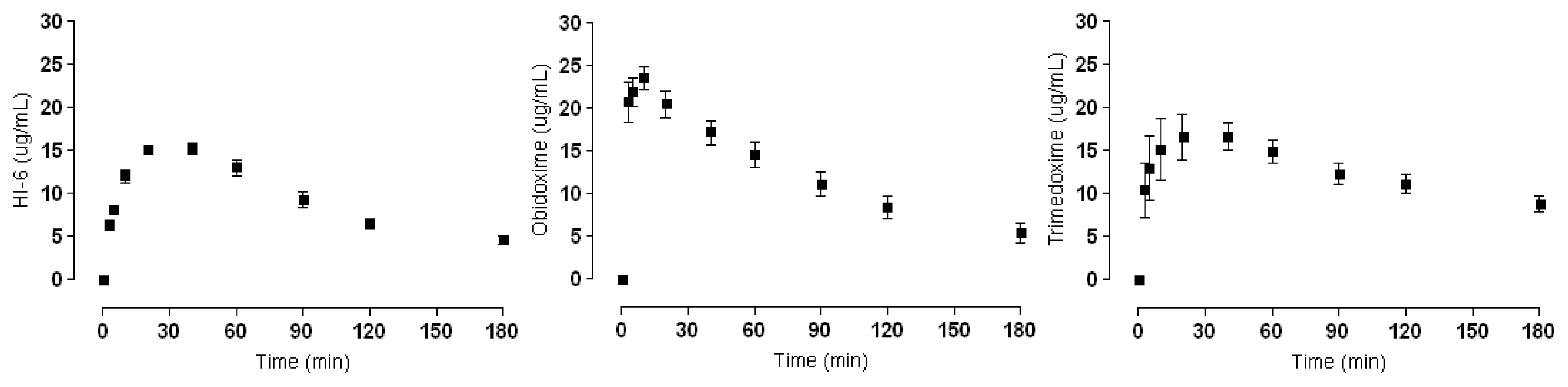
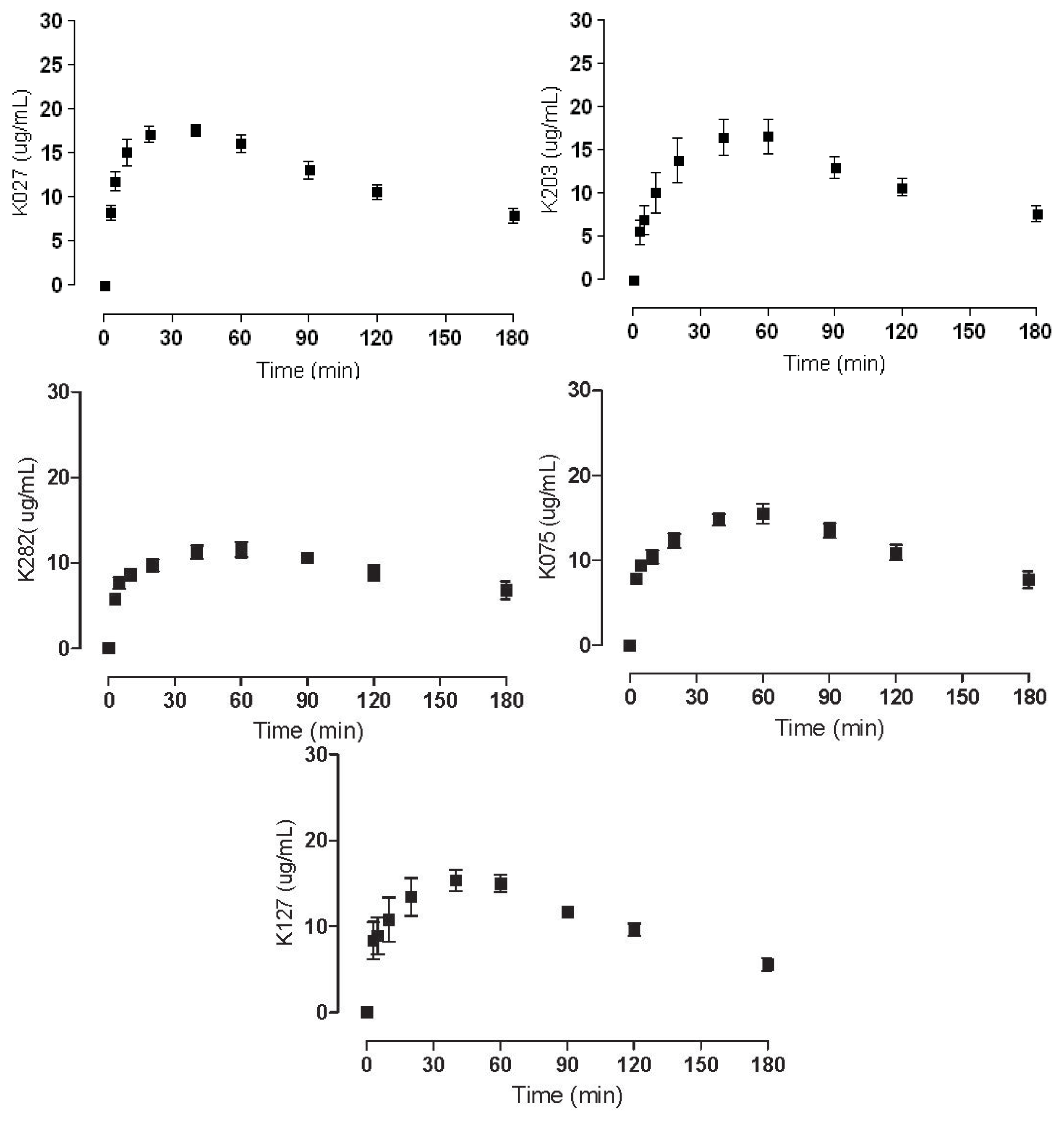
2.2. Plasma Concentrations
2.3. HSA Binding
2.4. Discussion
3. Experimental Section
3.1. Chemicals
3.2. Apparatus
3.3. Sample Preparation
3.4. Calibration
3.5. Pharmacokinetic Studies
3.6. HPLC Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Singh, S.S. Preclinical pharmacokinetics: An approach towards safer and efficacious drugs. Curr. Drug Metab 2006, 7, 165–182. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, S.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery. Adv. Drug Deliv. Rev 1997, 23, 3–25. [Google Scholar]
- Walters, W.P. Going further than Lipinski’s rule in drug design. Expert Opin. Drug Discov 2012, 7, 99–107. [Google Scholar]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol 1975, 11, 824–832. [Google Scholar]
- Hage, D.S.; Austin, J. High-performance affinity chromatography and immobilized serum albumin as probes for drug and hormone binding. J. Chromatogr. B Biomed. Sci. Appl 2000, 739, 39–54. [Google Scholar]
- Kim, H.S.; Wainer, I.W. Rapid analysis of the interactions between drugs and human serum albumin (HSA) using high-performance affinity chromatography (HPAC). J. Chromatogr. B Biomed. Sci. Appl 2008, 870, 22–26. [Google Scholar]
- Kragh-Hansen, U. Relations between high-affinity binding sites for l-tryptophan, diazepam, salicylate and Phenol Red on human serum albumin. Biochem. J 1983, 209, 135–142. [Google Scholar]
- Kragh-Hansen, U. Relations between high-affinity binding sites of markers for binding regions on human serum albumin. Biochem. J 1985, 225, 629–638. [Google Scholar]
- Kragh-Hansen, U. Evidence for a large and flexible region of human serum albumin possessing high affinity binding sites for salicylate, warfarin, and other ligands. Mol. Pharmacol 1988, 34, 160–171. [Google Scholar]
- Joseph, K.S.; Moser, A.C.; Basiaga, S.B.G.; Schiel, J.E.S.; Hage, D.S. Evaluation of alternatives to warfarin as probes for Sudlow site I of human serum albumin: Characterization by high-performance affinity chromatography. J. Chromatogr. A 2009, 1216, 3492–3500. [Google Scholar]
- Petersen, C.E.; Ha, C.E.; Harohalli, K.; Feix, J.B.; Bhagavan, N.V. A dynamic model for bilirubin binding to human serum albumin. J. Biol. Chem 2000, 275, 20985–20995. [Google Scholar]
- Ghuman, J.; Zunszain, P.A.; Peptitpas, I.; Bhattacharay, A.A.; Otagari, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol 2005, 353, 38–52. [Google Scholar]
- Varshney, A.; Ahmad, B.; Khan, R.H. Comparative studies of unfolding and binding of ligands to human serum albumin in the presence of fatty acid: Spectroscopic approach. Int. J. Biol. Macromol 2008, 42, 483–490. [Google Scholar]
- Dill, K.A.; Shortie, D. Denatured states of proteins. Ann. Rev. Biochem 1991, 60, 795–825. [Google Scholar]
- Petitpas, I.; Petersen, C.E.; Ha, C.E.; Bhattacharay, A.A.; Zunszain, P.A.; Ghuman, J. Structural basis of albumin-thyroxine interactions and familial dysalbuminemic hyperthyroxinemia. Proc. Natl. Acad. Sci. USA 2003, 100, 6440–6445. [Google Scholar]
- Petitpas, I.; Grune, T.; Bhattacharya, A.A.; Curry, S. Crystal structures of human serum albumin complexed with monounsaturated and polyunsaturated fatty acids. J. Mol. Biol 2001, 314, 955–960. [Google Scholar]
- Zemek, F.; Korabecny, J.; Sepsova, V.; Karasova, J.Z.; Musilek, K.; Kuca, K. Albumin and α1-acid glycoprotein: Old acquaintances. Expert Opin. Drug Metab. Toxicol 2013, 9, 943–954. [Google Scholar]
- Bajgar, J. Optimal choice of acetylcholinesterase reactivators for antidotal treatment of nerve agent intoxication. Acta Medica 2010, 53, 207–211. [Google Scholar]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol 2013, 11, 315–335. [Google Scholar]
- Litchfield, M.H. Estimates of acute pesticide poisoning in agricultural workers in less developed countries. Toxicol. Rev 2005, 24, 271–278. [Google Scholar]
- Karasova, J.Z.; Chladek, J.; Hroch, M.; Josef, F.; Hnidkova, D.; Kuca, K. Pharmacokinetic study of two acetylcholinesterase reactivators, trimedoxime and newly synthesized oxime K027, in rat plasma. J. Appl. Toxicol 2013, 33, 18–23. [Google Scholar]
- Kalasz, H.; Szegi, P.; Janoki, G.; Balogh, L.; Postenyi, Z.; Musilek, K.; Petroianu, G.A.; Siddiq, A.; Tekes, K. Study on medicinal chemistry of K203 in wistar rats and beagle dogs. Curr. Med. Chem 2013, 20, 2137–2144. [Google Scholar]
- Karasova, J.Z.; Novotny, L.; Antos, K.; Zivna, H.; Kuca, K. Time-dependent changes in concentration of two clinically used acetylcholinesterase reactivators (HI-6 and obidoxime) in rat plasma determined by HPLC techniques after in vivo administration. Anal. Sci 2010, 26, 63–67. [Google Scholar]
- Kassa, J.; Misik, J.; Karasova, J.Z. A comparison of the potency of a novel bispyridinium oxime K203 and currently available oximes (obidoxime, HI-6) to counteract the acute neurotoxicity of sarin in rats. Basic Clin. Pharmacol. Toxicol 2012, 111, 333–338. [Google Scholar]
- Karasova, J.Z.; Pohanka, M.; Pavlik, M.; Kassa, J.; Musilek, K.; Zemek, F.; Kuca, K. Distribution study of acetylcholinesterase reactivators after application of therapeutic doses in guinea pigs. Toxicol. Lett 2011, 205, S116. [Google Scholar]
- Kuca, K.; Musilek, K.; Jun, D.; Pohanka, M.; Ghosh, K.K.; Hrabinova, M. Oxime K027: Novel low-toxic candidate for the universal reactivator of nerve agent-and pesticide-inhibited acetylcholinesterase. J. Enzyme Inhib. Med. Chem 2010, 25, 509–512. [Google Scholar]
- Matos, K.S.; da Cunha, E.F.F.; Goncalves, A.S.; Wilter, A.; Kuca, K.; Franca, T.C.C.; Ramalho, T.C. First principles calculations of thermodynamics and kinetic parameters and molecular dynamics simulations of acetylcholinesterase reactivators: Can mouse data provide new insights into humans? J. Biomol. Struct. Dyn 2012, 30, 546–558. [Google Scholar]
- Worek, F.; Thiermann, H. Reactivation of organophosphate-inhibited human acetylcholinesterase by isontrosoacetone (MINA): A kinetic analysis. Chem. Biol. Interact 2011, 194, 91–96. [Google Scholar]
- Skovira, J.W.; O’Donnell, J.C.; Koplovitz, I.; Kan, R.K.; McDonough, J.H.; Snih, T.M. Reactivation of brain acetylcholinesterase by monoisonitrosoacetone increases the therapeutic efficacy against nerve agents in guinea pigs. Chem. Biol. Interact 2010, 187, 318–324. [Google Scholar]
- Kovarik, Z.; Macek, N.; Sit, R.K.; Radic, Z.; Fokin, V.V.; Baarry Sharpless, K.; Taylor, P. Centrally acting oximes in reactivation of tabun-phosphoramidated AChE. Chem. Biol. Interact 2013, 203, 77–80. [Google Scholar]
- Karasova, J.Z.; Kassa, J.; Jung, Y.S.; Musilek, K.; Pohanka, M.; Kuca, K. Effect of several new and currently available oxime cholinesterase reactivators on tabun-intoxicated rats. Int. J. Mol. Sci 2008, 9, 2243–2252. [Google Scholar]
- Kassa, J.; Karasova, J.Z.; Tesarova, S.; Kuca, K.; Musilek, K. A comparison of the ability of newly-developed bispyridinium oxime K203 and currently available oximes (trimedoxime, obidoxime, HI-6) to counteract the acute neurotoxicity of soman in rats. Toxicol. Mech. Methods 2010, 20, 445–451. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AChE Reactivators | Plasma Concentration (Cmax μg/mL) |
|---|---|
| HI-6 | 15.26 ± 1.71 |
| Obidoxime | 22.76 ± 4.28 |
| Trimedoxime | 16.64 ± 4.25 |
| K027 | 17.61 ± 1.60 |
| K075 | 15.50 ± 2.82 |
| K127 | 15.35 ± 3.28 |
| K203 | 16.63 ± 5.29 |
| K282 | 11.56 ± 2.31 |
| AChE Reactivators | Binding (%) a |
|---|---|
| HI-6 | 1 |
| Obidoxime | 7 |
| Trimedoxime | 6 |
| K027 | 5 |
| K075 | 10 |
| K127 | 4 |
| K203 | 15 |
| K282 | 12 |
| Tested Substance | Administered Dose i.m. (mg/kg) |
|---|---|
| HI-6 | 22.23 |
| Obidoxime | 22.23 |
| Trimedoxime | 22.07 |
| K027 | 22.07 |
| K075 | 23.00 |
| K127 | 24.39 |
| K203 | 23.00 |
| K282 | 23.00 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zemek, F.; Zdarova, J.K.; Sepsova, V.; Kuca, K. Acetylcholinesterase Reactivators (HI-6, Obidoxime, Trimedoxime, K027, K075, K127, K203, K282): Structural Evaluation of Human Serum Albumin Binding and Absorption Kinetics. Int. J. Mol. Sci. 2013, 14, 16076-16086. https://doi.org/10.3390/ijms140816076
Zemek F, Zdarova JK, Sepsova V, Kuca K. Acetylcholinesterase Reactivators (HI-6, Obidoxime, Trimedoxime, K027, K075, K127, K203, K282): Structural Evaluation of Human Serum Albumin Binding and Absorption Kinetics. International Journal of Molecular Sciences. 2013; 14(8):16076-16086. https://doi.org/10.3390/ijms140816076
Chicago/Turabian StyleZemek, Filip, Jana Karasova Zdarova, Vendula Sepsova, and Kamil Kuca. 2013. "Acetylcholinesterase Reactivators (HI-6, Obidoxime, Trimedoxime, K027, K075, K127, K203, K282): Structural Evaluation of Human Serum Albumin Binding and Absorption Kinetics" International Journal of Molecular Sciences 14, no. 8: 16076-16086. https://doi.org/10.3390/ijms140816076
APA StyleZemek, F., Zdarova, J. K., Sepsova, V., & Kuca, K. (2013). Acetylcholinesterase Reactivators (HI-6, Obidoxime, Trimedoxime, K027, K075, K127, K203, K282): Structural Evaluation of Human Serum Albumin Binding and Absorption Kinetics. International Journal of Molecular Sciences, 14(8), 16076-16086. https://doi.org/10.3390/ijms140816076