Mechanisms of Radiation Toxicity in Transformed and Non-Transformed Cells


{kind=link}
Abstract
:1. Introduction
2. Ionizing Radiation-Induced Damage to Biological Molecules
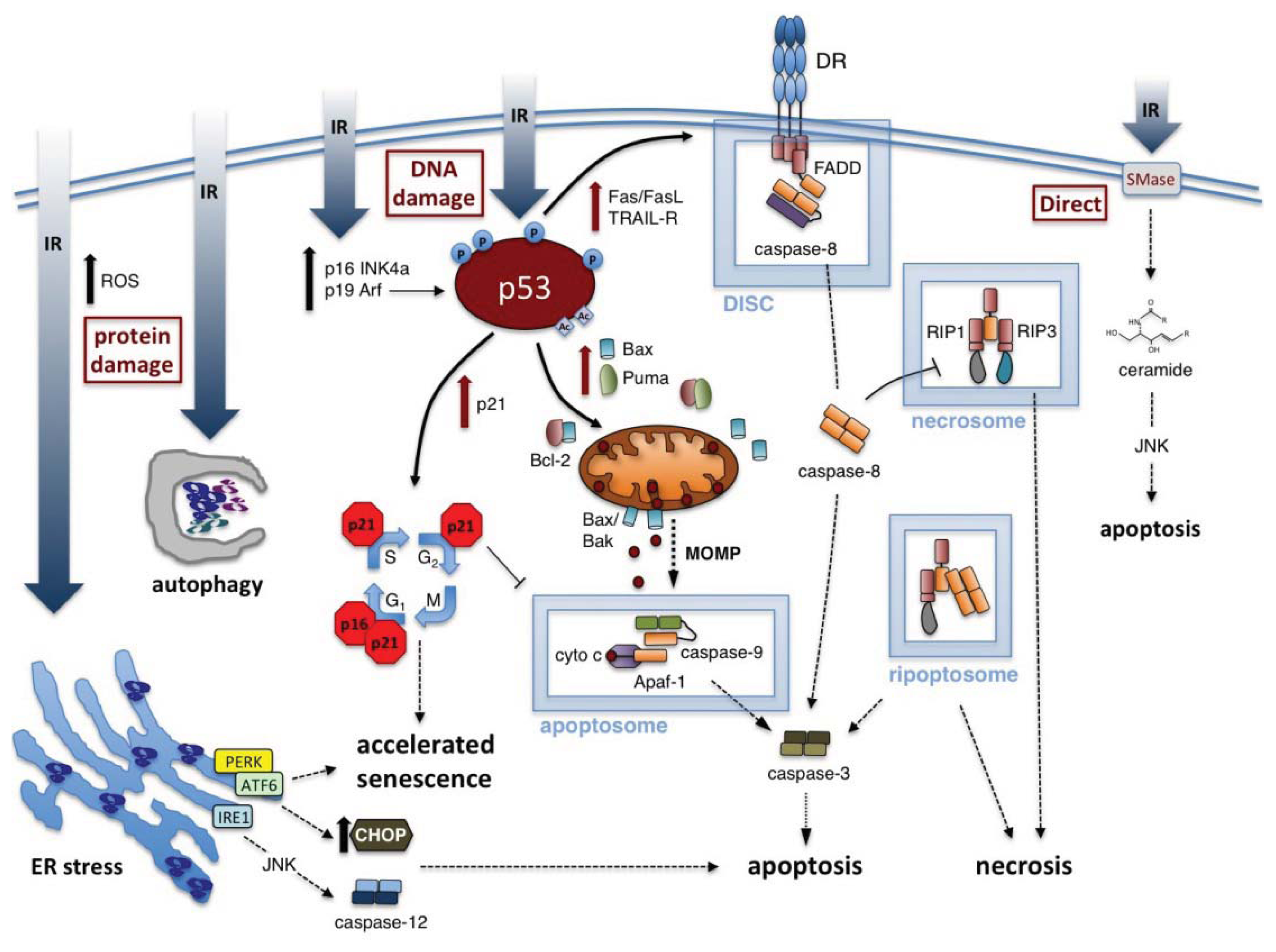
3. Ionizing Radiation-Induced Cell Toxicities
3.1. Radiation-Induced Apoptosis
3.1.1. Intrinsic Apoptosis Induced by IR
3.1.2. Extrinsic Apoptosis Induced by IR
3.1.3. ER Stress and Activation of Apoptosis by IR
3.2. Radiation-Induced Necrosis
3.3. Radiation-Induced Autophagy
3.4. Radiation-Induced Accelerated Senescence
3.5. Radiation Effects in Bystander Cells
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Posner, E. Reception of Rontgen’s discovery in Britain and U.S.A. Br. Med. J 1970, 4, 357–360. [Google Scholar]
- Rontgen, W. Eine neue art von strahlen. Nature 1895, 53, 274. [Google Scholar]
- Becquerel, A. On the invisible rays emitted by phosphorescent bodies. Comptes Rendus 1896, 122, 501–503. [Google Scholar]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci 2012, 9, 193–199. [Google Scholar]
- Waselenko, J.K.; MacVittie, T.J.; Blakely, W.F.; Pesik, N.; Wiley, A.L.; Dickerson, W.E.; Tsu, H.; Confer, D.L.; Coleman, C.N.; Seed, T.; et al. Medical management of the acute radiation syndrome: Recommendations of the strategic national stockpile radiation working group. Ann. Intern. Med 2004, 140, 1037–1051. [Google Scholar]
- Xiao, M.; Whitnall, M.H. Pharmacological countermeasures for the acute radiation syndrome. Curr. Mol. Pharmacol 2009, 2, 122–133. [Google Scholar]
- Coleman, C.N.; Blakely, W.F.; Fike, J.R.; MacVittie, T.J.; Metting, N.F.; Mitchell, J.B.; Moulder, J.E.; Preston, R.J.; Seed, T.M.; Stone, H.B.; et al. Molecular and cellular biology of moderate-dose (1–10 Gy) radiation and potential mechanisms of radiation protection: Report of a workshop at Bethesda, Maryland, December 17–18, 2001. Radiat. Res 2003, 159, 812–834. [Google Scholar]
- Friedman, E.J. Immune modulation by ionizing radiation and its implications for cancer immunotherapy. Curr. Pharm. Des 2002, 8, 1765–1780. [Google Scholar]
- Zhao, W.; Robbins, M.E. Inflammation and chronic oxidative stress in radiation-induced late normal tissue injury: Therapeutic implications. Curr. Med. Chem 2009, 16, 130–143. [Google Scholar]
- Calveley, V.L.; Khan, M.A.; Yeung, I.W.; Vandyk, J.; Hill, R.P. Partial volume rat lung irradiation: Temporal fluctuations of in-field and out-of-field DNA damage and inflammatory cytokines following irradiation. Int. J. Radiat. Biol 2005, 81, 887–899. [Google Scholar]
- Gobbel, G.T.; Chan, P.H. Neuronal death is an active, caspase-dependent process after moderate but not severe DNA damage. J. Neurochem 2001, 76, 520–531. [Google Scholar]
- Iglesias-Bartolome, R.; Patel, V.; Cotrim, A.; Leelahavanichkul, K.; Molinolo, A.A.; Mitchell, J.B.; Gutkind, J.S. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell 2012, 11, 401–414. [Google Scholar]
- Weiss, J.F.; Landauer, M.R. History and development of radiation-protective agents. Int. J. Radiat. Biol 2009, 85, 539–573. [Google Scholar]
- Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist, 6th ed; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2006. [Google Scholar]
- Naumov, S.; von Sonntag, C. The energetics of rearrangement and water elimination reactions in the radiolysis of the DNA bases in aqueous solution (eaq- and *OH Attack): DFT calculations. Radiat. Res 2008, 169, 355–363. [Google Scholar]
- Rhee, S.G. Redox signaling: Hydrogen peroxide as intracellular messenger. Exp. Mol. Med 1999, 31, 53–59. [Google Scholar]
- Rhee, S.G.; Chang, T.S.; Bae, Y.S.; Lee, S.R.; Kang, S.W. Cellular regulation by hydrogen peroxide. J. Am. Soc. Nephrol 2003, 14, S211–S215. [Google Scholar]
- Bubici, C.; Papa, S.; Dean, K.; Franzoso, G. Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: Molecular basis and biological significance. Oncogene 2006, 25, 6731–6748. [Google Scholar]
- Hutchinson, F. The molecular basis for radiation effects on cells. Cancer Res 1966, 26, 2045–2052. [Google Scholar]
- Pollycove, M.; Feinendegen, L.E. Radiation-induced versus endogenous DNA damage: Possible effect of inducible protective responses in mitigating endogenous damage. Hum. Exp. Toxicol 2003, 22, 290–306, , discussion 307, 315–317, 319–323.. [Google Scholar]
- Jonathan, E.C.; Bernhard, E.J.; McKenna, W.G. How does radiation kill cells? Curr. Opin. Chem. Biol 1999, 3, 77–83. [Google Scholar]
- Shuryak, I.; Brenner, D.J. Mechanistic analysis of the contributions of DNA and protein damage to radiation-induced cell death. Radiat. Res 2012, 178, 17–24. [Google Scholar]
- Qiu, W.; Leibowitz, B.; Zhang, L.; Yu, J. Growth factors protect intestinal stem cells from radiation-induced apoptosis by suppressing puma through the PI3K/Akt/p53 axis. Oncogene 2010, 29, 1622–1632. [Google Scholar]
- Daly, M.J.; Gaidamakova, E.K.; Matrosova, V.Y.; Vasilenko, A.; Zhai, M.; Leapman, R.D.; Lai, B.; Ravel, B.; Li, S.M.; Kemner, K.M.; et al. Protein oxidation implicated as the primary determinant of bacterial radioresistance. PLoS Biol 2007, 5, e92. [Google Scholar]
- Krisko, A.; Leroy, M.; Radman, M.; Meselson, M. Extreme anti-oxidant protection against ionizing radiation in bdelloid rotifers. Proc. Natl. Acad. Sci. USA 2012, 109, 2354–2357. [Google Scholar]
- Du, J.; Gebicki, J.M. Proteins are major initial cell targets of hydroxyl free radicals. Int. J. Biochem. Cell. Biol 2004, 36, 2334–2343. [Google Scholar]
- Panganiban, R.A.; Mungunsukh, O.; Day, R.M. X-irradiation induces er stress, apoptosis, and senescence in pulmonary artery endothelial cells. Int. J. Radiat. Biol. 2012. [Google Scholar] [CrossRef]
- Tamulevicius, P.; Wang, M.; Iliakis, G. Homology-directed repair is required for the development of radioresistance during S phase: Interplay between double-strand break repair and checkpoint response. Radiat. Res 2007, 167, 1–11. [Google Scholar]
- Guerci, A.M.; Dulout, F.N.; Grillo, C.A.; Seoane, A.I. differential response of two cell lines sequentially irradiated with low X-ray doses. Int. J. Radiat. Biol 2005, 81, 367–372. [Google Scholar]
- Vogin, G.; Foray, N. The law of bergonie and tribondeau: A nice formula for a first approximation. Int. J. Radiat. Biol 2013, 89, 2–8. [Google Scholar]
- Surova, O.; Zhivotovsky, B. Various modes of cell death induced by DNA damage. Oncogene 2012. [Google Scholar] [CrossRef]
- Rothkamm, K.; Lobrich, M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low X-ray doses. Proc. Natl. Acad. Sci. USA 2003, 100, 5057–5062. [Google Scholar]
- Escribano-Diaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.; Tkac, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtiP controls DNA repair pathway choice. Mol. Cell 2013, 49, 872–883. [Google Scholar]
- Kaliberov, S.A.; Buchsbaum, D.J. Chapter Seven—Cancer treatment with gene therapy and radiation therapy. Adv. Cancer Res 2012, 115, 221–263. [Google Scholar]
- Verheij, M.; Bartelink, H. Radiation-induced apoptosis. Cell Tissue Res 2000, 301, 133–142. [Google Scholar]
- Muller, M. Cellular senescence: Molecular mechanisms, in vivo significance, and redox considerations. Antioxid. Redox Signal 2009, 11, 59–98. [Google Scholar]
- Xiao, M.; Inal, C.E.; Parekh, V.I.; Chang, C.M.; Whitnall, M.H. 5-androstenediol promotes survival of gamma-irradiated human hematopoietic progenitors through induction of nuclear factor-kappa B activation and granulocyte colony-stimulating factor expression. Mol. Pharmacol 2007, 72, 370–379. [Google Scholar]
- Kapty, J.; Murray, D.; Mercer, J. Radiotracers for noninvasive molecular imaging of tumor cell death. Cancer Biother. Radiopharm 2010, 25, 615–628. [Google Scholar]
- Lindsay, K.J.; Coates, P.J.; Lorimore, S.A.; Wright, E.G. The genetic basis of tissue responses to ionizing radiation. Br. J. Radiol 2007, 80, S2–S6. [Google Scholar]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol 2013, 87, 1157–1180. [Google Scholar]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar]
- Yoshida, K.; Kubo, Y.; Kusunoki, Y.; Morishita, Y.; Nagamura, H.; Hayashi, I.; Kyoizumi, S.; Seyama, T.; Nakachi, K.; Hayashi, T. Caspase-independent cell death without generation of reactive oxygen species in irradiated MOLT-4 human leukemia cells. Cell. Immunol 2009, 255, 61–68. [Google Scholar]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell. Biol 2007, 8, 405–413. [Google Scholar]
- Assefa, Z.; Van Laethem, A.; Garmyn, M.; Agostinis, P. Ultraviolet radiation-induced apoptosis in keratinocytes: On the role of cytosolic factors. Biochim. Biophys. Acta 2005, 1755, 90–106. [Google Scholar]
- Earnshaw, W.C.; Martins, L.M.; Kaufmann, S.H. Mammalian caspases: Structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem 1999, 68, 383–424. [Google Scholar]
- Han, Y.; Wang, Y.; Xu, H.T.; Yang, L.H.; Wei, Q.; Liu, Y.; Zhang, Y.; Zhao, Y.; Dai, S.D.; Miao, Y.; et al. X-radiation induces non-small-cell lung cancer apoptosis by upregulation of axin expression. Int. J. Radiat. Oncol. Biol. Phys 2009, 75, 518–526. [Google Scholar]
- Rodel, C.; Haas, J.; Groth, A.; Grabenbauer, G.G.; Sauer, R.; Rodel, F. Spontaneous and radiation-induced apoptosis in colorectal carcinoma cells with different intrinsic radiosensitivities: Survivin as a radioresistance factor. Int. J. Radiat. Oncol. Biol. Phys 2003, 55, 1341–1347. [Google Scholar]
- Kyprianou, N.; Rock, S. Radiation-induced apoptosis of human prostate cancer cells is independent of mutant P53 overexpression. Anticancer Res 1998, 18, 897–905. [Google Scholar]
- Petit-Frere, C.; Capulas, E.; Lyon, D.A.; Norbury, C.J.; Lowe, J.E.; Clingen, P.H.; Riballo, E.; Green, M.H.; Arlett, C.F. Apoptosis and cytokine release induced by ionizing or ultraviolet b radiation in primary and immortalized human keratinocytes. Carcinogenesis 2000, 21, 1087–1095. [Google Scholar]
- Waters, K.M.; Stenoien, D.L.; Sowa, M.B.; von Neubeck, C.; Chrisler, W.B.; Tan, R.; Sontag, R.L.; Weber, T.J. Annexin A2 modulates radiation-sensitive transcriptional programming and cell fate. Radiat. Res 2013, 179, 53–61. [Google Scholar]
- Claro, S.; Kanashiro, C.A.; Oshiro, M.E.; Ferreira, A.T.; Khalil, R.A. Alpha- and epsilon-protein kinase C activity during smooth muscle cell apoptosis in response to gamma-radiation. J. Pharmacol. Exp. Ther 2007, 322, 964–972. [Google Scholar]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Beclin 1 interactome controls the crosstalk between apoptosis, autophagy and inflammasome activation: Impact on the aging process. Ageing Res. Rev 2013, 12, 520–534. [Google Scholar]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med 2011, 17, 860–866. [Google Scholar]
- Weber, G.F.; Menko, A.S. The canonical intrinsic mitochondrial death pathway has a non-apoptotic role in signaling lens cell differentiation. J. Biol. Chem 2005, 280, 22135–22145. [Google Scholar]
- Jurgensmeier, J.M.; Xie, Z.; Deveraux, Q.; Ellerby, L.; Bredesen, D.; Reed, J.C. Bax directly induces release of cytochrome C from isolated mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 4997–5002. [Google Scholar]
- Galluzzi, L.; Joza, N.; Tasdemir, E.; Maiuri, M.C.; Hengartner, M.; Abrams, J.M.; Tavernarakis, N.; Penninger, J.; Madeo, F.; Kroemer, G. No death without life: Vital functions of apoptotic effectors. Cell Death Differ 2008, 15, 1113–1123. [Google Scholar]
- Srinivasula, S.M.; Ahmad, M.; Fernandes-Alnemri, T.; Alnemri, E.S. Autoactivation of procaspase-9 by apaf-1-mediated oligomerization. Mol. Cell 1998, 1, 949–957. [Google Scholar]
- Kwon, Y.W.; Ueda, S.; Ueno, M.; Yodoi, J.; Masutani, H. Mechanism of p53-dependent apoptosis induced by 3-methylcholanthrene: Involvement ofp p3 phosphorylation and p38 mapk. J. Biol. Chem 2002, 277, 1837–1844. [Google Scholar]
- Lippens, S.; Hoste, E.; Vandenabeele, P.; Agostinis, P.; Declercq, W. Cell death in the skin. Apoptosis 2009, 14, 549–569. [Google Scholar]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of EIF2alpha dephosphorylation protects cells from Er stress. Science 2005, 307, 935–939. [Google Scholar]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the atm kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar]
- Chow, J.; Tron, V.A. Molecular aspects of ultraviolet radiation-induced apoptosis in the skin. J. Cutan. Med. Surg 2005, 9, 289–295. [Google Scholar]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar]
- Linares, L.K.; Kiernan, R.; Triboulet, R.; Chable-Bessia, C.; Latreille, D.; Cuvier, O.; Lacroix, M.; Le Cam, L.; Coux, O.; Benkirane, M. Intrinsic ubiquitination activity of pcaf controls the stability of the oncoprotein Hdm2. Nat. Cell Biol 2007, 9, 331–338. [Google Scholar]
- Chernov, M.V.; Ramana, C.V.; Adler, V.V.; Stark, G.R. Stabilization and activation of p53 are regulated independently by different phosphorylation events. Proc. Natl. Acad. Sci. USA 1998, 95, 2284–2289. [Google Scholar]
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. Biophys. Acta 2009, 1787, 414–420. [Google Scholar]
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar]
- Kang, M.H.; Reynolds, C.P. Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin. Cancer Res 2009, 15, 1126–1132. [Google Scholar]
- Susnow, N.; Zeng, L.; Margineantu, D.; Hockenbery, D.M. Bcl-2 family proteins as regulators of oxidative stress. Semin. Cancer Biol 2009, 19, 42–49. [Google Scholar]
- Jeffers, J.R.; Parganas, E.; Lee, Y.; Yang, C.; Wang, J.; Brennan, J.; MacLean, K.H.; Han, J.; Chittenden, T.; Ihle, J.N.; et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 2003, 4, 321–328. [Google Scholar]
- Villunger, A.; Michalak, E.M.; Coultas, L.; Mullauer, F.; Bock, G.; Ausserlechner, M.J.; Adams, J.M.; Strasser, A. p53- and drug-induced apoptotic responses mediated by bh3-only proteins puma and noxa. Science 2003, 302, 1036–1038. [Google Scholar]
- Mancini, F.; Moretti, F. Mitochondrial MDM4 (MDMX): An unpredicted role in the p53-mediated intrinsic apoptotic pathway. Cell Cycle 2009, 8, 3854–3859. [Google Scholar]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar]
- Zhu, H.B.; Yang, K.; Xie, Y.Q.; Lin, Y.W.; Mao, Q.Q.; Xie, L.P. Silencing of mutant p53 by sirna induces cell cycle arrest and apoptosis in human bladder cancer cells. World J. Surg. Oncol 2013, 11, 22. [Google Scholar]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar]
- Clarke, A.R.; Purdie, C.A.; Harrison, D.J.; Morris, R.G.; Bird, C.C.; Hooper, M.L.; Wyllie, A.H. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature 1993, 362, 849–852. [Google Scholar]
- Lowe, S.W.; Schmitt, E.M.; Smith, S.W.; Osborne, B.A.; Jacks, T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 1993, 362, 847–849. [Google Scholar]
- Shaw, P.; Bovey, R.; Tardy, S.; Sahli, R.; Sordat, B.; Costa, J. Induction of apoptosis by wild-type p53 in a human colon tumor-derived cell line. Proc. Natl. Acad. Sci. USA 1992, 89, 4495–4499. [Google Scholar]
- Yonish-Rouach, E.; Resnitzky, D.; Lotem, J.; Sachs, L.; Kimchi, A.; Oren, M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 1991, 352, 345–347. [Google Scholar]
- Kolesnick, R.; Fuks, Z. Radiation and ceramide-induced apoptosis. Oncogene 2003, 22, 5897–5906. [Google Scholar]
- Lozano, J.; Menendez, S.; Morales, A.; Ehleiter, D.; Liao, W.C.; Wagman, R.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Cell autonomous apoptosis defects in acid sphingomyelinase knockout fibroblasts. J. Biol. Chem 2001, 276, 442–448. [Google Scholar]
- Lee, H.; Rotolo, J.A.; Mesicek, J.; Penate-Medina, T.; Rimner, A.; Liao, W.C.; Yin, X.; Ragupathi, G.; Ehleiter, D.; Gulbins, E.; et al. Mitochondrial ceramide-rich macrodomains functionalize bax upon irradiation. PLoS One 2011, 6, e19783. [Google Scholar]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell. Biol 2010, 11, 621–632. [Google Scholar]
- Finnberg, N.; Gruber, J.J.; Fei, P.; Rudolph, D.; Bric, A.; Kim, S.H.; Burns, T.F.; Ajuha, H.; Page, R.; Wu, G.S.; et al. DR5 knockout mice are compromised in radiation-induced apoptosis. Mol. Cell. Biol 2005, 25, 2000–2013. [Google Scholar]
- Kuribayashi, K.; Finnberg, N.; Jeffers, J.R.; Zambetti, G.P.; El-Deiry, W.S. The relative contribution of pro-apoptotic p53-target genes in the triggering of apoptosis following DNA damage in vitro and in vivo. Cell Cycle 2011, 10, 2380–2389. [Google Scholar]
- Luce, A.; Courtin, A.; Levalois, C.; Altmeyer-Morel, S.; Romeo, P.H.; Chevillard, S.; Lebeau, J. Death receptor pathways mediate targeted and non-targeted effects of ionizing radiations in breast cancer cells. Carcinogenesis 2009, 30, 432–439. [Google Scholar]
- Belka, C.; Heinrich, V.; Marini, P.; Faltin, H.; Schulze-Osthoff, K.; Bamberg, M.; Budach, W. Ionizing radiation and the activation of caspase-8 in highly apoptosis-sensitive lymphoma Cells. Int. J. Radiat. Biol 1999, 75, 1257–1264. [Google Scholar]
- Belkacemi, Y.; Piel, G.; Rat, P.; Julia, F.; Touboul, E.; Housset, M.; Warnet, J.M. Ionizing radiation-induced death in bovine lens epithelial cells: Mechanisms and influence of irradiation dose rate. Int. J. Cancer 2000, 90, 138–144. [Google Scholar]
- Afshar, G.; Jelluma, N.; Yang, X.; Basila, D.; Arvold, N.D.; Karlsson, A.; Yount, G.L.; Dansen, T.B.; Koller, E.; Haas-Kogan, D.A. Radiation-induced caspase-8 mediates p53-independent apoptosis in glioma cells. Cancer Res 2006, 66, 4223–4232. [Google Scholar]
- Eriksson, D.; Lofroth, P.O.; Johansson, L.; Riklund, K.; Stigbrand, T. Apoptotic signalling in HeLa Hep2 cells following 5 Gy of cobalt-60 gamma radiation. Anticancer Res 2009, 29, 4361–4366. [Google Scholar]
- Kuribayashi, K.; Mayes, P.A.; El-Deiry, W.S. What are caspases 3 and 7 doing upstream of the mitochondria? Cancer Biol. Ther 2006, 5, 763–765. [Google Scholar]
- Hock, A.K.; Vousden, K.H. Tumor suppression by p53: Fall of the triumvirate? Cell 2012, 149, 1183–1185. [Google Scholar]
- Burns, T.F.; Bernhard, E.J.; El-Deiry, W.S. Tissue specific expression of p53 target genes suggests a key role for killer/Dr5 in p53-dependent apoptosis in vivo. Oncogene 2001, 20, 4601–4612. [Google Scholar]
- Fei, P.; Bernhard, E.J.; El-Deiry, W.S. Tissue-specific induction of p53 targets in vivo. Cancer Res 2002, 62, 7316–7327. [Google Scholar]
- Kataoka, T. The caspase-8 modulator C-FLIP. Crit. Rev. Immunol 2005, 25, 31–58. [Google Scholar]
- Safa, A.R.; Day, T.W.; Wu, C.H. Cellular flice-like inhibitory protein (C-FLIP): A novel target for cancer therapy. Curr. Cancer Drug Targets 2008, 8, 37–46. [Google Scholar]
- de Almagro, M.C.; Vucic, D. The inhibitor of apoptosis (IAP) proteins are critical regulators of signaling pathways and targets for anti-cancer therapy. Exp. Oncol 2012, 34, 200–211. [Google Scholar]
- Tan, W.Q.; Wang, J.X.; Lin, Z.Q.; Li, Y.R.; Lin, Y.; Li, P.F. Novel cardiac apoptotic pathway: The dephosphorylation of apoptosis repressor with caspase recruitment domain by calcineurin. Circulation 2008, 118, 2268–2276. [Google Scholar]
- Ludwig-Galezowska, A.H.; Flanagan, L.; Rehm, M. Apoptosis repressor with caspase recruitment domain, a multifunctional modulator of cell death. J. Cell. Mol. Med 2011, 15, 1044–1053. [Google Scholar]
- Zhang, N.; Wang, X.; Huo, Q.; Li, X.; Wang, H.; Schneider, P.; Hu, G.; Yang, Q. The oncogene metadherin modulates the apoptotic pathway based on the tumor necrosis factor superfamily member trail (tumor necrosis factor-related apoptosis-inducing ligand) in breast cancer. J. Biol. Chem 2013, 288, 9396–9407. [Google Scholar]
- Ola, M.S.; Nawaz, M.; Ahsan, H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol. Cell. Biochem 2011, 351, 41–58. [Google Scholar]
- Mott, J.L.; Kobayashi, S.; Bronk, S.F.; Gores, G.J. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene 2007, 26, 6133–6140. [Google Scholar]
- Huerta, S.; Heinzerling, J.H.; Anguiano-Hernandez, Y.M.; Huerta-Yepez, S.; Lin, J.; Chen, D.; Bonavida, B.; Livingston, E.H. Modification of gene products involved in resistance to apoptosis in metastatic colon cancer cells: Roles of Fas, Apaf-1, NFkappaB, IAPs, Smac/Diablo, and AIF. J. Surg. Res 2007, 142, 184–194. [Google Scholar]
- Fiandalo, M.V.; Schwarze, S.R.; Kyprianou, N. Proteasomal regulation of caspase-8 in cancer cell apoptosis. Apoptosis 2013, 18, 766–776. [Google Scholar]
- Gaud, G.; Guillemot, D.; Jacob, Y.; Favre, M.; Vuillier, F. EVER2 protein binds TRADD to promote TNF-α-induced apoptosis. Cell Death Dis 2013, 4, e499. [Google Scholar]
- Hensley, P.; Mishra, M.; Kyprianou, N. Targeting caspases in cancer therapeutics. Biol. Chem 2013, 394, 831–843. [Google Scholar]
- Koutsounas, I.; Giaginis, C.; Patsouris, E.; Theocharis, S. Current evidence for histone deacetylase inhibitors in pancreatic cancer. World J. Gastroenterol 2013, 19, 813–828. [Google Scholar]
- Tomek, M.; Akiyama, T.; Dass, C.R. Role of Bcl-2 in tumour cell survival and implications for pharmacotherapy. J. Pharm. Pharmacol 2012, 64, 1695–1702. [Google Scholar]
- Lai, E.; Teodoro, T.; Volchuk, A. Endoplasmic reticulum stress: Signaling the unfolded protein response. Physiology 2007, 22, 193–201. [Google Scholar]
- Kim, I.; Shu, C.W.; Xu, W.; Shiau, C.W.; Grant, D.; Vasile, S.; Cosford, N.D.; Reed, J.C. Chemical biology investigation of cell death pathways activated by endoplasmic reticulum stress reveals cytoprotective modulators of Ask1. J. Biol. Chem 2009, 284, 1593–1603. [Google Scholar]
- Tsang, K.Y.; Chan, D.; Bateman, J.F.; Cheah, K.S. In vivo cellular adaptation to ER stress: Survival strategies with double-edged consequences. J. Cell Sci 2010, 123, 2145–2154. [Google Scholar]
- Min, S.K.; Lee, S.K.; Park, J.S.; Lee, J.; Paeng, J.Y.; Lee, S.I.; Lee, H.J.; Kim, Y.; Pae, H.O.; Kim, E.C. Endoplasmic reticulum stress is involved in hydrogen peroxide induced apoptosis in immortalized and malignant human oral keratinocytes. J. Oral Pathol. Med 2008, 37, 490–498. [Google Scholar]
- Zhang, B.; Wang, Y.; Pang, X.; Su, Y.; Ai, G.; Wang, T. ER stress induced by ionising radiation in IEC-6 cells. Int. J. Radiat. Biol 2010, 86, 429–435. [Google Scholar]
- Baumeister, P.; Luo, S.; Skarnes, W.C.; Sui, G.; Seto, E.; Shi, Y.; Lee, A.S. Endoplasmic reticulum stress induction of the Grp78/BiP promoter: Activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol. Cell. Biol 2005, 25, 4529–4540. [Google Scholar]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar]
- Boyce, M.; Yuan, J. Cellular response to endoplasmic reticulum stress: A matter of life or death. Cell Death Differ 2006, 13, 363–373. [Google Scholar]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol 2000, 20, 6755–6767. [Google Scholar]
- Gupta, S.; Deepti, A.; Deegan, S.; Lisbona, F.; Hetz, C.; Samali, A. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1alpha-XBP1 signaling through a physical interaction. PLoS Biol 2010, 8, e1000410. [Google Scholar]
- Brewer, J.W.; Diehl, J.A. PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc. Natl. Acad. Sci. USA 2000, 97, 12625–12630. [Google Scholar]
- Yoneda, T.; Imaizumi, K.; Oono, K.; Yui, D.; Gomi, F.; Katayama, T.; Tohyama, M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem 2001, 276, 13935–13940. [Google Scholar]
- Park, M.T.; Song, M.J.; Lee, H.; Oh, E.T.; Choi, B.H.; Jeong, S.Y.; Choi, E.K.; Park, H.J. β-lapachone significantly increases the effect of ionizing radiation to cause mitochondrial apoptosis via JNK activation in cancer cells. PLoS One 2011, 6, e25976. [Google Scholar]
- England, K.; Driscoll, C.O.; Cotter, T.G. ROS and protein oxidation in early stages of cytotoxic drug induced apoptosis. Free Radic. Res 2006, 40, 1124–1137. [Google Scholar]
- Yokouchi, M.; Hiramatsu, N.; Hayakawa, K.; Okamura, M.; Du, S.; Kasai, A.; Takano, Y.; Shitamura, A.; Shimada, T.; Yao, J.; et al. Involvement of selective reactive oxygen species upstream of proapoptotic branches of unfolded protein response. J. Biol. Chem 2008, 283, 4252–4260. [Google Scholar]
- Guan, L.; Han, B.; Li, Z.; Hua, F.; Huang, F.; Wei, W.; Yang, Y.; Xu, C. Sodium selenite induces apoptosis by ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction in human acute promyelocytic leukemia NB4 cells. Apoptosis 2009, 14, 218–225. [Google Scholar]
- Tagawa, Y.; Hiramatsu, N.; Kasai, A.; Hayakawa, K.; Okamura, M.; Yao, J.; Kitamura, M. Induction of apoptosis by cigarette smoke via ROS-dependent endoplasmic reticulum stress and CCAAT/enhancer-binding protein-homologous protein (CHOP). Free Radic. Biol. Med 2008, 45, 50–59. [Google Scholar]
- He, L.; Kim, S.O.; Kwon, O.; Jeong, S.J.; Kim, M.S.; Lee, H.G.; Osada, H.; Jung, M.; Ahn, J.S.; Kim, B.Y. ATM blocks tunicamycin-induced endoplasmic reticulum stress. FEBS Lett 2009, 583, 903–908. [Google Scholar]
- Lim, M.J.; Ahn, J.Y.; Han, Y.; Yu, C.H.; Kim, M.H.; Lee, S.L.; Lim, D.S.; Song, J.Y. Acriflavine enhances radiosensitivity of colon cancer cells through endoplasmic reticulum stress-mediated apoptosis. Int. J. Biochem. Cell Biol 2012, 44, 1214–1222. [Google Scholar]
- Pang, X.L.; He, G.; Liu, Y.B.; Wang, Y.; Zhang, B. Endoplasmic reticulum stress sensitizes human esophageal cancer cell to radiation. World J. Gastroenterol 2013, 19, 1736–1748. [Google Scholar]
- Chiu, H.W.; Fang, W.H.; Chen, Y.L.; Wu, M.D.; Yuan, G.F.; Ho, S.Y.; Wang, Y.J. Monascuspiloin enhances the radiation sensitivity of human prostate cancer cells by stimulating endoplasmic reticulum stress and inducing autophagy. PLoS One 2012, 7, e40462. [Google Scholar]
- Golstein, P.; Kroemer, G. Cell death by necrosis: Towards a molecular definition. Trends Biochem. Sci 2007, 32, 37–43. [Google Scholar]
- Denecker, G.; Vercammen, D.; Steemans, M.; Vanden Berghe, T.; Brouckaert, G.; Van Loo, G.; Zhivotovsky, B.; Fiers, W.; Grooten, J.; Declercq, W.; et al. Death receptor-induced apoptotic and necrotic cell death: Differential role of caspases and mitochondria. Cell Death Differ 2001, 8, 829–840. [Google Scholar]
- Soti, C.; Sreedhar, A.S.; Csermely, P. Apoptosis, necrosis and cellular senescence: Chaperone occupancy as a potential switch. Aging Cell 2003, 2, 39–45. [Google Scholar]
- Cronin, T.D.; Brauer, R.O. Radiodermatitis and necrosis. Surgery 1949, 26, 665–672. [Google Scholar]
- Akagi, Y.; Ito, K.; Sawada, S. Radiation-induced apoptosis and necrosis in MOLT-4 cells: A study of dose-effect relationships and their modification. Int. J. Radiat. Biol 1993, 64, 47–56. [Google Scholar]
- Maccomb, W.S. Necrosis in treatment of intraoral cancer by radiation therapy. Am. J. Roentgenol. Radium. Ther. Nucl. Med 1962, 87, 431–440. [Google Scholar]
- Jella, K.K.; Garcia, A.; McClean, B.; Byrne, H.J.; Lyng, F.M. Cell death pathways in directly irradiated cells and cells exposed to medium from irradiated cells. Int. J. Radiat. Biol 2012, 89, 182–190. [Google Scholar]
- Vavrova, J.; Marekova, M.; Vokurkova, D. Radiation-induced apoptosis and cell cycle progression in Tp53-deficient human leukemia cell line HL-60. Neoplasma 2001, 48, 26–33. [Google Scholar]
- Mullins, M.E.; Barest, G.D.; Schaefer, P.W.; Hochberg, F.H.; Gonzalez, R.G.; Lev, M.H. Radiation necrosis versus glioma recurrence: Conventional MR imaging clues to diagnosis. Am. J. Neuroradiol 2005, 26, 1967–1972. [Google Scholar]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 2013, 38, 209–223. [Google Scholar]
- Golden, E.B.; Pellicciotta, I.; Demaria, S.; Barcellos-Hoff, M.H.; Formenti, S.C. The convergence of radiation and immunogenic cell death signaling pathways. Front. Oncol 2012, 2, 88. [Google Scholar]
- Vanden Berghe, T.; Vanlangenakker, N.; Parthoens, E.; Deckers, W.; Devos, M.; Festjens, N.; Guerin, C.J.; Brunk, U.T.; Declercq, W.; Vandenabeele, P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ 2010, 17, 922–930. [Google Scholar]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol 2008, 4, 313–321. [Google Scholar]
- Nehs, M.A.; Lin, C.I.; Kozono, D.E.; Whang, E.E.; Cho, N.L.; Zhu, K.; Moalem, J.; Moore, F.D., Jr; Ruan, D.T. Necroptosis is a novel mechanism of radiation-induced cell death in anaplastic thyroid and adrenocortical cancers. Surgery 2011, 150, 1032–1039. [Google Scholar]
- Dodson, M.; Darley-Usmar, V.; Zhang, J. Cellular metabolic and autophagic pathways: Traffic control by redox signaling. Free Radic. Biol. Med 2013, 63, 207–221. [Google Scholar]
- Denton, D.; Nicolson, S.; Kumar, S. Cell death by autophagy: Facts and apparent artefacts. Cell Death Differ 2012, 19, 87–95. [Google Scholar]
- Yi, H.; Liang, B.; Jia, J.; Liang, N.; Xu, H.; Ju, G.; Ma, S.; Liu, X. Differential roles of mir-199a-5p in radiation-induced autophagy in breast cancer cells. FEBS Lett 2013, 587, 436–443. [Google Scholar]
- Palumbo, S.; Comincini, S. Autophagy and ionizing radiation in tumors: The “survive or not survive” dilemma. J. Cell. Physiol 2013, 228, 1–8. [Google Scholar]
- Kim, K.W.; Speirs, C.K.; Jung, D.K.; Lu, B. The zinc ionophore PCI-5002 radiosensitizes non-small cell lung cancer cells by enhancing autophagic cell death. J. Thorac. Oncol 2011, 6, 1542–1552. [Google Scholar]
- Yu, L.; Tumati, V.; Tseng, S.F.; Hsu, F.M.; Kim, D.N.; Hong, D.; Hsieh, J.T.; Jacobs, C.; Kapur, P.; Saha, D. DAB2IP regulates autophagy in prostate cancer in response to combined treatment of radiation and a DNA-PKcs inhibitor. Neoplasia 2012, 14, 1203–1212. [Google Scholar]
- Zois, C.E.; Giatromanolaki, A.; Sivridis, E.; Papaiakovou, M.; Kainulainen, H.; Koukourakis, M.I. “Autophagic Flux” in normal mouse tissues: Focus on endogenous LC3A processing”. Autophagy 2011, 7, 1371–1378. [Google Scholar]
- Firat, E.; Weyerbrock, A.; Gaedicke, S.; Grosu, A.L.; Niedermann, G. Chloroquine or chloroquine-PI3K/Akt pathway inhibitor combinations strongly promote γ-irradiation-induced cell death in primary stem-like glioma cells. PLoS One 2012, 7, e47357. [Google Scholar]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int. J. Cancer 2011, 129, 2720–2731. [Google Scholar]
- Chang, S.H.; Minai-Tehrani, A.; Shin, J.Y.; Park, S.; Kim, J.E.; Yu, K.N.; Hong, S.H.; Hong, C.M.; Lee, K.H.; Beck, G.R., Jr; et al. Beclin1-induced autophagy abrogates radioresistance of lung cancer cells by suppressing osteopontin. J. Radiat. Res. 2012, 53, 422–432. [Google Scholar]
- Gewirtz, D.A. Autophagy and senescence in cancer therapy. J. Cell. Physiol. 2013. [Google Scholar] [CrossRef]
- Kim, E.J.; Jeong, J.H.; Bae, S.; Kang, S.; Kim, C.H.; Lim, Y.B. mTOR inhibitors radiosensitize PTEN-deficient non-small-cell lung cancer cells harboring an EGFR activating mutation by inducing autophagy. J. Cell. Biochem 2013, 114, 1248–1256. [Google Scholar]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res 1965, 37, 614–636. [Google Scholar]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res 1961, 25, 585–621. [Google Scholar]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell. Biol 2007, 8, 729–740. [Google Scholar]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol 2013, 75, 685–705. [Google Scholar]
- Rodier, F.; Campisi, J.; Bhaumik, D. Two faces of p53: Aging and tumor suppression. Nucleic Acids Res 2007, 35, 7475–7484. [Google Scholar]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Invest 2013, 123, 966–972. [Google Scholar]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar]
- Tominaga, K.; Pereira-Smith, O.M. The role of chromatin reorganization in the process of cellular senescence. Curr. Drug Targets 2012, 13, 1593–1602. [Google Scholar]
- Hill, R.; Bodzak, E.; Blough, M.D.; Lee, P.W. p53 binding to the p21 promoter is dependent on the nature of DNA damage. Cell Cycle 2008, 7, 2535–2543. [Google Scholar]
- Solomon, J.M.; Pasupuleti, R.; Xu, L.; McDonagh, T.; Curtis, R.; DiStefano, P.S.; Huber, L.J. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell. Biol 2006, 26, 28–38. [Google Scholar]
- Sperka, T.; Wang, J.; Rudolph, K.L. DNA damage checkpoints in stem cells, ageing and cancer. Nat. Rev. Mol. Cell. Biol 2012, 13, 579–590. [Google Scholar]
- el-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar]
- el-Deiry, W.S.; Harper, J.W.; O’Connor, P.M.; Velculescu, V.E.; Canman, C.E.; Jackman, J.; Pietenpol, J.A.; Burrell, M.; Hill, D.E.; Wang, Y.; et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res 1994, 54, 1169–1174. [Google Scholar]
- Satyanarayana, A.; Hilton, M.B.; Kaldis, P. p21 inhibits Cdk1 in the absence of Cdk2 to maintain the G1/S phase DNA damage checkpoint. Mol. Biol. Cell 2008, 19, 65–77. [Google Scholar]
- Niculescu, A.B., III; Chen, X.; Smeets, M.; Hengst, L.; Prives, C.; Reed, S.I. Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol. Cell. Biol. 1998, 18, 629–643. [Google Scholar]
- Andorfer, P.; Rotheneder, H. EAPP: Gatekeeper at the crossroad of apoptosis and p21-mediated cell-cycle arrest. Oncogene 2011, 30, 2679–2690. [Google Scholar]
- Roninson, I.B. Oncogenic functions of tumour suppressor P21(Waf1/Cip1/Sdi1): Association with cell senescence and tumour-promoting activities of stromal fibroblasts. Cancer Lett 2002, 179, 1–14. [Google Scholar]
- Asada, M.; Yamada, T.; Ichijo, H.; Delia, D.; Miyazono, K.; Fukumuro, K.; Mizutani, S. Apoptosis inhibitory activity of cytoplasmic p21(Cip1/Waf1) in monocytic differentiation. EMBO J 1999, 18, 1223–1234. [Google Scholar]
- Baptiste-Okoh, N.; Barsotti, A.M.; Prives, C. Caspase 2 is both required for p53-mediated apoptosis and downregulated by p53 in a p21-dependent manner. Cell Cycle 2008, 7, 1133–1138. [Google Scholar]
- Suzuki, A.; Tsutomi, Y.; Miura, M.; Akahane, K. Caspase 3 inactivation to suppress fas-mediated apoptosis: Identification of binding domain with p21 and ILP and inactivation machinery by p21. Oncogene 1999, 18, 1239–1244. [Google Scholar]
- Meng, A.; Wang, Y.; Van Zant, G.; Zhou, D. Ionizing radiation and busulfan induce premature senescence in murine bone marrow hematopoietic cells. Cancer Res 2003, 63, 5414–5419. [Google Scholar]
- Wang, Y.; Schulte, B.A.; LaRue, A.C.; Ogawa, M.; Zhou, D. Total body irradiation selectively induces murine hematopoietic stem cell senescence. Blood 2006, 107, 358–366. [Google Scholar]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar]
- Sherr, C.J. The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell. Biol 2001, 2, 731–737. [Google Scholar]
- Bertwistle, D.; Sugimoto, M.; Sherr, C.J. Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Mol. Cell. Biol 2004, 24, 985–996. [Google Scholar]
- Blagosklonny, M.V. Cell cycle arrest is not senescence. Aging 2011, 3, 94–101. [Google Scholar]
- Blagosklonny, M.V. Progeria, rapamycin and normal aging: Recent breakthrough. Aging 2011, 3, 685–691. [Google Scholar]
- Anisimov, V.N.; Zabezhinski, M.A.; Popovich, I.G.; Piskunova, T.S.; Semenchenko, A.V.; Tyndyk, M.L.; Yurova, M.N.; Rosenfeld, S.V.; Blagosklonny, M.V. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle 2011, 10, 4230–4236. [Google Scholar]
- Demidenko, Z.N.; Blagosklonny, M.V. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle 2008, 7, 3355–3361. [Google Scholar]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar]
- Haferkamp, S.; Tran, S.L.; Becker, T.M.; Scurr, L.L.; Kefford, R.F.; Rizos, H. The relative contributions of the p53 and pRb pathways in oncogene-induced melanocyte senescence. Aging 2009, 1, 542–556. [Google Scholar]
- Mirzayans, R.; Andrais, B.; Hansen, G.; Murray, D. Role of p16(Ink4a) in replicative senescence and DNA damage-induced premature senescence in p53-deficient human cells. Biochem. Res. Int. 2012, 2012. [Google Scholar] [CrossRef]
- Hei, T.K.; Zhou, H.; Chai, Y.; Ponnaiya, B.; Ivanov, V.N. Radiation induced non-targeted response: Mechanism and potential clinical implications. Curr. Mol. Pharmacol 2011, 4, 96–105. [Google Scholar]
- Waldren, C.A. Classical radiation biology dogma, bystander effects and paradigm shifts. Hum. Exp. Toxicol 2004, 23, 95–100. [Google Scholar]
- Blyth, B.J.; Sykes, P.J. Radiation-induced bystander effects: What are they, and how relevant are they to human radiation exposures? Radiat. Res 2011, 176, 139–157. [Google Scholar]
- Baskar, R. Emerging role of radiation induced bystander effects: Cell communications and carcinogenesis. Genome Integr. 2010, 1. [Google Scholar] [CrossRef]
- Mothersill, C.; Seymour, C. Medium from irradiated human epithelial cells but not human fibroblasts reduces the clonogenic survival of unirradiated cells. Int. J. Radiat. Biol 1997, 71, 421–427. [Google Scholar]
- Sedelnikova, O.A.; Nakamura, A.; Kovalchuk, O.; Koturbash, I.; Mitchell, S.A.; Marino, S.A.; Brenner, D.J.; Bonner, W.M. DNA double-strand breaks form in bystander cells after microbeam irradiation of three-dimensional human tissue models. Cancer Res 2007, 67, 4295–4302. [Google Scholar]
- Khan, M.A.; Van Dyk, J.; Yeung, I.W.; Hill, R.P. Partial volume rat lung irradiation; assessment of early DNA damage in different lung regions and effect of radical scavengers. Radiother. Oncol 2003, 66, 95–102. [Google Scholar]
- Sokolov, M.V.; Smilenov, L.B.; Hall, E.J.; Panyutin, I.G.; Bonner, W.M.; Sedelnikova, O.A. Ionizing radiation induces DNA double-strand breaks in bystander primary human fibroblasts. Oncogene 2005, 24, 7257–7265. [Google Scholar]
- Yang, H.; Asaad, N.; Held, K.D. Medium-mediated intercellular communication is involved in bystander responses of X-ray-irradiated normal human fibroblasts. Oncogene 2005, 24, 2096–2103. [Google Scholar]
- Zhou, H.; Ivanov, V.N.; Gillespie, J.; Geard, C.R.; Amundson, S.A.; Brenner, D.J.; Yu, Z.; Lieberman, H.B.; Hei, T.K. Mechanism of radiation-induced bystander effect: Role of the cyclooxygenase-2 signaling pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 14641–14646. [Google Scholar]
- Nagasawa, H.; Little, J.B. Induction of sister chromatid exchanges by extremely low doses of alpha-particles. Cancer Res 1992, 52, 6394–6396. [Google Scholar]
- Iyer, R.; Lehnert, B.E.; Svensson, R. Factors underlying the cell growth-related bystander responses to alpha particles. Cancer Res 2000, 60, 1290–1298. [Google Scholar]
- Baskar, R.; Balajee, A.S.; Geard, C.R. Effects of low and high let radiations on bystander human lung fibroblast cell survival. Int. J. Radiat. Biol 2007, 83, 551–559. [Google Scholar]
- Han, W.; Chen, S.; Yu, K.N.; Wu, L. Nitric oxide mediated DNA double strand breaks induced in proliferating bystander cells after alpha-particle irradiation. Mutat. Res 2010, 684, 81–89. [Google Scholar]
- Khan, M.A.; Hill, R.P.; Van Dyk, J. Partial volume rat lung irradiation: An evaluation of early DNA damage. Int. J. Radiat. Oncol. Biol. Phys 1998, 40, 467–476. [Google Scholar]
- Koturbash, I.; Rugo, R.E.; Hendricks, C.A.; Loree, J.; Thibault, B.; Kutanzi, K.; Pogribny, I.; Yanch, J.C.; Engelward, B.P.; Kovalchuk, O. Irradiation induces DNA damage and modulates epigenetic effectors in distant bystander tissue in vivo. Oncogene 2006, 25, 4267–4275. [Google Scholar]
- Muller, K.; Meineke, V. Radiation-induced mast cell mediators differentially modulate chemokine release from dermal fibroblasts. J. Dermatol. Sci 2011, 61, 199–205. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Panganiban, R.-A.M.; Snow, A.L.; Day, R.M. Mechanisms of Radiation Toxicity in Transformed and Non-Transformed Cells. Int. J. Mol. Sci. 2013, 14, 15931-15958. https://doi.org/10.3390/ijms140815931
Panganiban R-AM, Snow AL, Day RM. Mechanisms of Radiation Toxicity in Transformed and Non-Transformed Cells. International Journal of Molecular Sciences. 2013; 14(8):15931-15958. https://doi.org/10.3390/ijms140815931
Chicago/Turabian StylePanganiban, Ronald-Allan M., Andrew L. Snow, and Regina M. Day. 2013. "Mechanisms of Radiation Toxicity in Transformed and Non-Transformed Cells" International Journal of Molecular Sciences 14, no. 8: 15931-15958. https://doi.org/10.3390/ijms140815931