Autophagy in Prostate Cancer and Androgen Suppression Therapy

 , and
, and
Abstract
:1. Prostate Cancer and Therapy
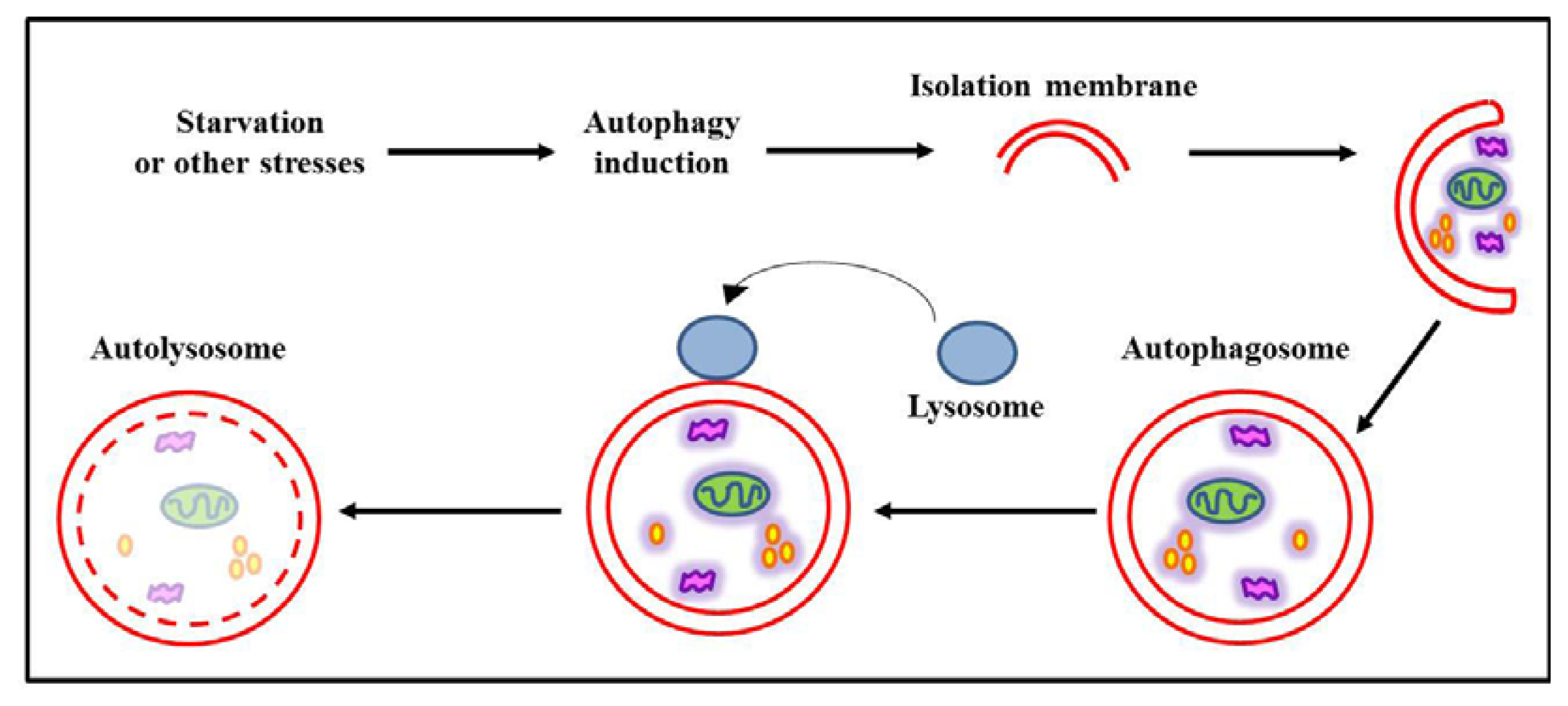
2. Autophagy
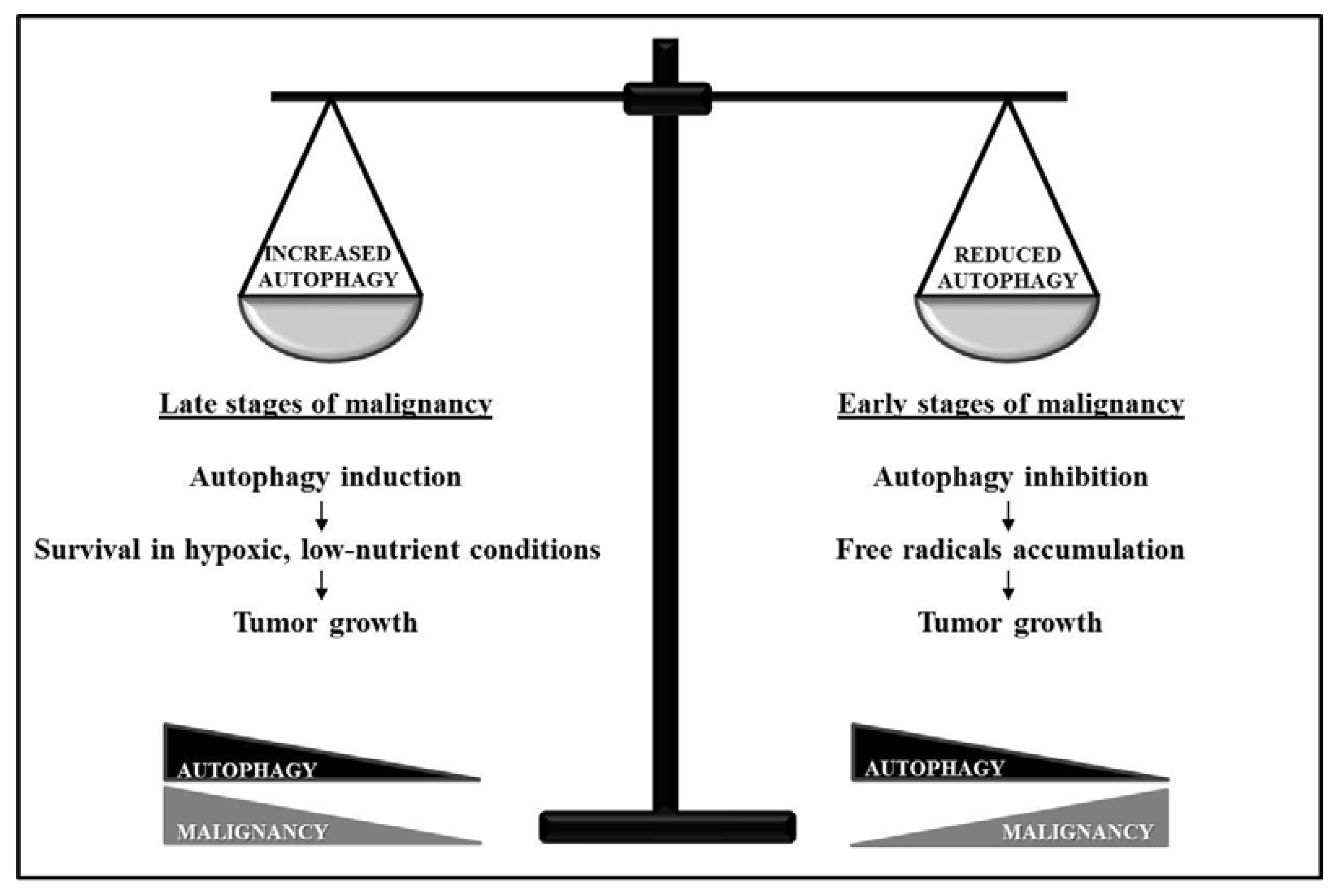
3. “Dual-Faced” Role of Autophagy in Cancer
4. Autophagy in Prostate Cancer
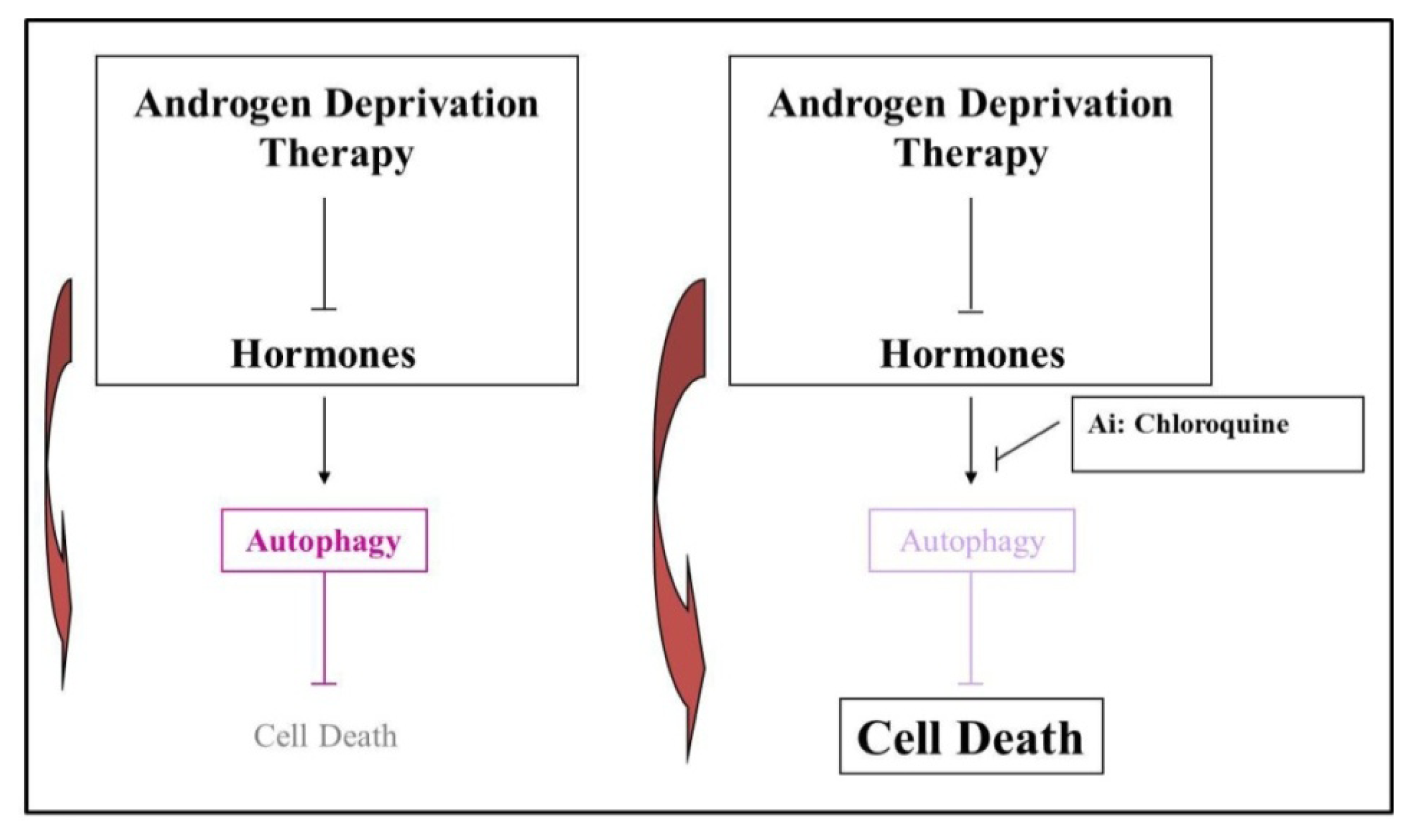
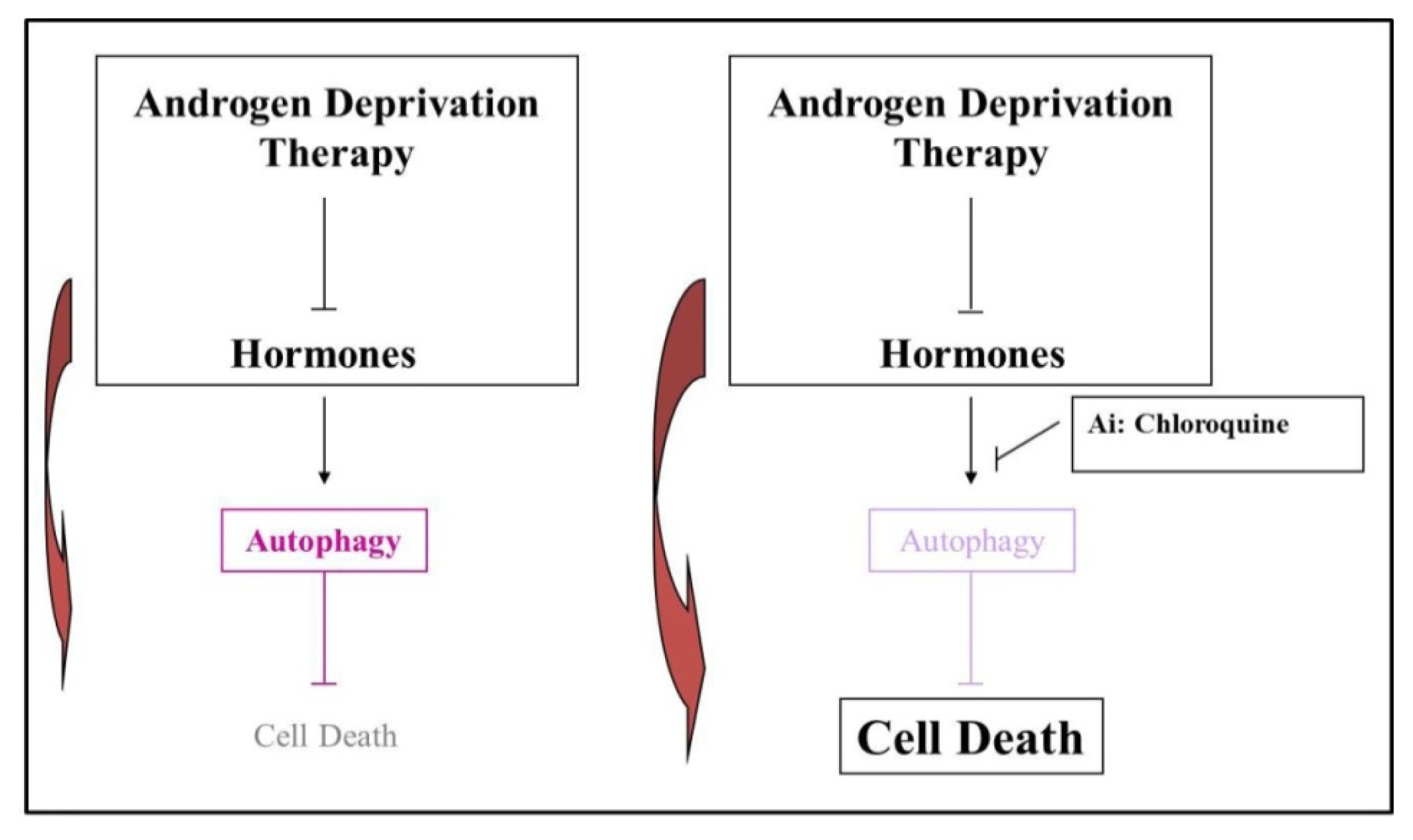
5. Androgen Deprivation Therapy (ADT) Stimulates an Autophagic Response
6. A Possible Therapeutic Role for Autophagy in Prostate Cancer
7. Autophagy Involvement in Modulating the Immune Response
8. Perspectives
9. Conclusions
Acknowledgments
Conflict of Interest
References
- Borley, N.; Feneley, M.R. Prostate cancer: Diagnosis and staging. Asian J. Androl 2009, 11, 74–80. [Google Scholar]
- Tadros, N.N.; Garzotto, M. Androgen deprivation therapy for prostate cancer: Not so simple. Asian J. Androl 2011, 13, 187–188. [Google Scholar]
- Boccon-Gibod, L.; van der Meulen, E.; Persson, B.E. An update on the use of gonadotropin-releasing hormone antagonists in prostate cancer. Ther. Adv. Urol 2011, 3, 127–140. [Google Scholar]
- Oudard, S. Progress in emerging therapies for advanced prostate cancer. Cancer Treat. Rev 2013, 39, 275–289. [Google Scholar]
- Mostaghel, E.A.; Page, S.T.; Lin, D.W.; Fazli, L.; Coleman, I.M.; True, L.D.; Knudsen, B.; Hess, D.L.; Nelson, C.C.; Matsumoto, A.M.; et al. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: Therapeutic implications for castration-resistant prostate cancer. Cancer Res 2007, 67, 5033–5041. [Google Scholar]
- Saraon, P.; Jarvi, K.; Diamandis, E.P. Molecular alterations during progression of prostate cancer to androgen independence. Clin. Chem 2011, 57, 1366–1375. [Google Scholar]
- Knudsen, K.E.; Scher, H.I. Starving the addiction: New opportunities for durable suppression of AR signaling in prostate cancer. Clin. Cancer Res 2009, 15, 4792–4798. [Google Scholar]
- Miyake, H.; Nelson, C.; Rennie, P.S.; Gleave, M.E. Overexpression of insulin-like growth factor binding protein-5 helps accelerate progression to androgen-independence in the human prostate LNCaP tumor model through activation of phosphatidylinositol 3′-kinase pathway. Endocrinology 2000, 141, 2257–2265. [Google Scholar]
- Rocchi, P.; So, A.; Kojima, S.; Signaevsky, M.; Beraldi, E.; Fazli, L.; Hurtado-Coll, A.; Yamanaka, K.; Gleave, M. Heat shock protein 27 increases after androgen ablation and plays a cytoprotective role in hormone-refractory prostate cancer. Cancer Res 2004, 64, 6595–6602. [Google Scholar]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol 2010, 221, 3–12. [Google Scholar]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar]
- Horbinski, C.; Mojesky, C.; Kyprianou, N. Live free or die: Tales of homeless (cells) in cancer. Am. J. Pathol 2010, 177, 1044–1052. [Google Scholar]
- Corcelle, E.A.; Puustinen, P.; Jaattela, M. Apoptosis and autophagy: Targeting autophagy signalling in cancer cells—’Trick or treats’? FEBS J 2009, 276, 6084–6096. [Google Scholar]
- Hippert, M.M.; O’Toole, P.S.; Thorburn, A. Autophagy in cancer: Good, bad, or both? Cancer Res 2006, 66, 9349–9351. [Google Scholar]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar]
- Arico, S.; Petiot, A.; Bauvy, C.; Dubbelhuis, P.F.; Meijer, A.J.; Codogno, P.; Ogier-Denis, E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem 2001, 276, 35243–35246. [Google Scholar]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar]
- Li, L.; Ishdorj, G.; Gibson, S.B. Reactive oxygen species regulation of autophagy in cancer: Implications for cancer treatment. Free Radic. Biol. Med 2012, 53, 1399–1410. [Google Scholar]
- Edinger, A.L.; Thompson, C.B. Defective autophagy leads to cancer. Cancer Cell 2003, 4, 422–424. [Google Scholar]
- Aita, V.M.; Liang, X.H.; Murty, V.V.; Pincus, D.L.; Yu, W.; Cayanis, E.; Kalachikov, S.; Gilliam, T.C.; Levine, B. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 1999, 59, 59–65. [Google Scholar]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest 2003, 112, 1809–1820. [Google Scholar]
- Dalby, K.N.; Tekedereli, I.; Lopez-Berestein, G.; Ozpolat, B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy 2010, 6, 322–329. [Google Scholar]
- Kung, H.J. Targeting tyrosine kinases and autophagy in prostate cancer. Horm. Cancer 2011, 2, 38–46. [Google Scholar]
- Guo, Z.; Dai, B.; Jiang, T.; Xu, K.; Xie, Y.; Kim, O.; Nesheiwat, I.; Kong, X.; Melamed, J.; Handratta, V.D.; et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 2006, 10, 309–319. [Google Scholar]
- Wu, Z.; Chang, P.C.; Yang, J.C.; Chu, C.Y.; Wang, L.Y.; Chen, N.T.; Ma, A.H.; Desai, S.J.; Lo, S.H.; Evans, C.P.; et al. Autophagy blockade sensitizes prostate cancer cells towards Src family kinase inhibitors. Genes Cancer 2010, 1, 40–49. [Google Scholar]
- Bristol, M.L.; Emery, S.M.; Maycotte, P.; Thorburn, A.; Chakradeo, S.; Gewirtz, D.A. Autophagy inhibition for chemosensitization and radiosensitization in cancer: Do the preclinical data support this therapeutic strategy? J. Pharmacol. Exp. Ther 2013, 344, 544–552. [Google Scholar]
- Kumano, M.; Furukawa, J.; Shiota, M.; Zardan, A.; Zhang, F.; Beraldi, E.; Wiedmann, R.M.; Fazli, L.; Zoubeidi, A.; Gleave, M.E. Cotargeting stress-activated Hsp27 and autophagy as a combinatorial strategy to amplify endoplasmic reticular stress in prostate cancer. Mol. Cancer Ther 2012, 11, 1661–1671. [Google Scholar]
- Lamoureux, F.; Thomas, C.; Crafter, C.; Kumano, M.; Zhang, F.; Davies, B.R.; Gleave, M.E.; Zoubeidi, A. Blocked autophagy using lysosomotropic agents sensitizes resistant prostate tumor cells to the novel Akt inhibitor AZD5363. Clin. Cancer Res 2013, 19, 833–844. [Google Scholar]
- Yu, L.; Tumati, V.; Tseng, S.F.; Hsu, F.M.; Kim, D.N.; Hong, D.; Hsieh, J.T.; Jacobs, C.; Kapur, P.; Saha, D. DAB2IP regulates autophagy in prostate cancer in response to combined treatment of radiation and a DNA-PKcs inhibitor. Neoplasia 2012, 14, 1203–1212. [Google Scholar]
- Chen, R.J.; Hung, C.M.; Chen, Y.L.; Wu, M.D.; Yuan, G.F.; Wang, Y.J. Monascuspiloin induces apoptosis and autophagic cell death in human prostate cancer cells via the Akt and AMPK signaling pathways. J. Agric. Food Chem 2012, 60, 7185–7193. [Google Scholar]
- Tai, S.; Sun, Y.; Liu, N.; Ding, B.; Hsia, E.; Bhuta, S.; Thor, R.K.; Damoiseaux, R.; Liang, C.; Huang, J. Combination of Rad001 (everolimus) and propachlor synergistically induces apoptosis through enhanced autophagy in prostate cancer cells. Mol. Cancer Ther 2012, 11, 1320–1331. [Google Scholar]
- He, Z.; Mangala, L.S.; Theriot, C.A.; Rohde, L.H.; Wu, H.; Zhang, Y. Cell killing and radiosensitizing effects of atorvastatin in PC3 prostate cancer cells. J. Radiat. Res 2012, 53, 225–233. [Google Scholar]
- Jain, R.K.; Safabakhsh, N.; Sckell, A.; Chen, Y.; Jiang, P.; Benjamin, L.; Yuan, F.; Keshet, E. Endothelial cell death, angiogenesis, and microvascular function after castration in an androgen-dependent tumor: Role of vascular endothelial growth factor. Proc. Natl. Acad. Sci. USA 1998, 95, 10820–10825. [Google Scholar]
- Chhipa, R.R.; Wu, Y.; Ip, C. AMPK-mediated autophagy is a survival mechanism in androgen-dependent prostate cancer cells subjected to androgen deprivation and hypoxia. Cell Signal 2011, 23, 1466–1472. [Google Scholar]
- Kaini, R.R.; Hu, C.A. Synergistic killing effect of chloroquine and androgen deprivation in LNCaP cells. Biochem. Biophys. Res. Commun 2012, 425, 150–156. [Google Scholar]
- Chhipa, R.R.; Wu, Y.; Mohler, J.L.; Ip, C. Survival advantage of AMPK activation to androgen-independent prostate cancer cells during energy stress. Cell Signal 2010, 22, 1554–1561. [Google Scholar]
- Meijer, A.J.; Codogno, P. AMP-activated protein kinase and autophagy. Autophagy 2007, 3, 238–240. [Google Scholar]
- Park, H.U.; Suy, S.; Danner, M.; Dailey, V.; Zhang, Y.; Li, H.; Hyduke, D.R.; Collins, B.T.; Gagnon, G.; Kallakury, B.; et al. AMP-activated protein kinase promotes human prostate cancer cell growth and survival. Mol. Cancer Ther 2009, 8, 733–741. [Google Scholar]
- Schoenlein, P.V.; Periyasamy-Thandavan, S.; Samaddar, J.S.; Jackson, W.H.; Barrett, J.T. Autophagy facilitates the progression of ERalpha-positive breast cancer cells to antiestrogen resistance. Autophagy 2009, 5, 400–403. [Google Scholar]
- Bennett, H.L.; Stockley, J.; Fleming, J.T.; Mandal, R.; O’Prey, J.; Ryan, K.M.; Robson, C.N.; Leung, H.Y. Does androgen-ablation therapy (AAT) associated autophagy have a pro-survival effect in LNCaP human prostate cancer cells? BJU Int 2012, 111, 672–682. [Google Scholar]
- Homewood, C.A.; Warhurst, D.C.; Peters, W.; Baggaley, V.C. Lysosomes, pH and the anti-malarial action of chloroquine. Nature 1972, 235, 50–52. [Google Scholar]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.M.; Weiss, W.A.; Takebe, N.; Timmer, W.; Dipaola, R.S.; Lotze, M.T.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res 2011, 17, 654–666. [Google Scholar]
- Sotelo, J.; Briceno, E.; Lopez-Gonzalez, M.A. Adding chloroquine to conventional treatment for glioblastoma multiforme: A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med 2006, 144, 337–343. [Google Scholar]
- Cheong, H.; Lu, C.; Lindsten, T.; Thompson, C.B. Therapeutic targets in cancer cell metabolism and autophagy. Nat. Biotechnol 2012, 30, 671–678. [Google Scholar]
- Tang, D.G.; Porter, A.T. Target to apoptosis: A hopeful weapon for prostate cancer. Prostate 1997, 32, 284–293. [Google Scholar]
- Hesry, V.; Piquet-Pellorce, C.; Travert, M.; Donaghy, L.; Jegou, B.; Patard, J.J.; Guillaudeux, T. Sensitivity of prostate cells to TRAIL-induced apoptosis increases with tumor progression: DR5 and caspase 8 are key players. Prostate 2006, 66, 987–995. [Google Scholar]
- Nakajima, Y.; DelliPizzi, A.M.; Mallouh, C.; Ferreri, N.R. TNF-mediated cytotoxicity and resistance in human prostate cancer cell lines. Prostate 1996, 29, 296–302. [Google Scholar]
- D’Arcangelo, D.; Giampietri, C.; Facchiano, F.; Facchiano, A. BAMM: A preliminary Bibliometric Analysis on Melanoma Manuscripts. Pigment Cell Melanoma Res 2013, 26, 415–417. [Google Scholar]
- Giampietri, C.; Petrungaro, S.; Facchiano, A.; Filippini, A.; Ziparo, E. Therapeutic implications of autophagy modulation in prostate cancer. J. Endocrinol. Invest 2012, 35, 945. [Google Scholar]
- Giampietri, C.; Petrungaro, S.; Padula, F.; D’Alessio, A.; Marini, E.S.; Facchiano, A.; Filippini, A.; Ziparo, E. Autophagy modulators sensitize prostate epithelial cancer cell lines to TNF-alpha-dependent apoptosis. Apoptosis 2012, 17, 1210–1222. [Google Scholar]
- He, W.; Wang, Q.; Xu, J.; Xu, X.; Padilla, M.T.; Ren, G.; Gou, X.; Lin, Y. Attenuation of TNFSF10/TRAIL-induced apoptosis by an autophagic survival pathway involving T. Autophagy 2012, 8, 1811–1821. [Google Scholar]
- Zhu, K.; Dunner, K., Jr; McConkey, D.J. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010, 29, 451–462. [Google Scholar]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.X.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.R.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J 2010, 29, 969–980. [Google Scholar]
- True, O.; Matthias, P. Interplay between histone deacetylases and autophagy—From cancer therapy to neurodegeneration. Immunol. Cell Biol 2012, 90, 78–84. [Google Scholar]
- Lu, J.V.; Walsh, C.M. Programmed necrosis and autophagy in immune function. Immunol. Rev 2012, 249, 205–217. [Google Scholar]
- Muzes, G.; Sipos, F. Anti-tumor immunity, autophagy and chemotherapy. World J. Gastroenterol 2012, 18, 6537–6540. [Google Scholar]
- Muranski, P.; Boni, A.; Antony, P.A.; Cassard, L.; Irvine, K.R.; Kaiser, A.; Paulos, C.M.; Palmer, D.C.; Touloukian, C.E.; Ptak, K.; et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 2008, 112, 362–373. [Google Scholar]
- Ahn, G.O.; Tseng, D.; Liao, C.H.; Dorie, M.J.; Czechowicz, A.; Brown, J.M. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc. Natl. Acad. Sci. USA 2010, 107, 8363–8368. [Google Scholar]
- Wildenberg, M.E.; Vos, A.C.; Wolfkamp, S.C.; Duijvestein, M.; Verhaar, A.P.; te Velde, A.A.; van den Brink, G.R.; Hommes, D.W. Autophagy attenuates the adaptive immune response by destabilizing the immunologic synapse. Gastroenterology 2012, 142, 1493–1503. [Google Scholar]
- Barron, D.A.; Rowley, D.R. The reactive stroma microenvironment and prostate cancer progression. Endocr. Relat. Cancer 2012, 19, R187–R204. [Google Scholar]
- Gerdes, M.J.; Dang, T.D.; Larsen, M.; Rowley, D.R. Transforming growth factor-beta1 induces nuclear to cytoplasmic distribution of androgen receptor and inhibits androgen response in prostate smooth muscle cells. Endocrinology 1998, 139, 3569–3577. [Google Scholar]
- Jones, E.; Pu, H.; Kyprianou, N. Targeting TGF-beta in prostate cancer: Therapeutic possibilities during tumor progression. Expert. Opin. Ther. Targets 2009, 13, 227–234. [Google Scholar]
- Salm, S.N.; Burger, P.E.; Coetzee, S.; Goto, K.; Moscatelli, D.; Wilson, E.L. TGF-{beta} maintains dormancy of prostatic stem cells in the proximal region of ducts. J. Cell Biol 2005, 170, 81–90. [Google Scholar]
- Stover, D.G.; Bierie, B.; Moses, H.L. A delicate balance: TGF-beta and the tumor microenvironment. J. Cell Biochem 2007, 101, 851–861. [Google Scholar]
- Zhu, B.; Kyprianou, N. Transforming growth factor beta and prostate cancer. Cancer Treat. Res 2005, 126, 157–173. [Google Scholar]
- Alonso-Magdalena, P.; Brossner, C.; Reiner, A.; Cheng, G.; Sugiyama, N.; Warner, M.; Gustafsson, J.A. A role for epithelial-mesenchymal transition in the etiology of benign prostatic hyperplasia. Proc. Natl. Acad. Sci. USA 2009, 106, 2859–2863. [Google Scholar]
- Ao, M.; Franco, O.E.; Park, D.; Raman, D.; Williams, K.; Hayward, S.W. Cross-talk between paracrine-acting cytokine and chemokine pathways promotes malignancy in benign human prostatic epithelium. Cancer Res 2007, 67, 4244–4253. [Google Scholar]
- Gann, P.H.; Klein, K.G.; Chatterton, R.T.; Ellman, A.E.; Grayhack, J.T.; Nadler, R.B.; Lee, C. Growth factors in expressed prostatic fluid from men with prostate cancer, BPH, and clinically normal prostates. Prostate 1999, 40, 248–255. [Google Scholar]
- Shoskes, D.A.; Albakri, Q.; Thomas, K.; Cook, D. Cytokine polymorphisms in men with chronic prostatitis/chronic pelvic pain syndrome: Association with diagnosis and treatment response. J. Urol 2002, 168, 331–335. [Google Scholar]
- Kiyono, K.; Suzuki, H.I.; Matsuyama, H.; Morishita, Y.; Komuro, A.; Kano, M.R.; Sugimoto, K.; Miyazono, K. Autophagy is activated by TGF-beta and potentiates TGF-beta-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res 2009, 69, 8844–8852. [Google Scholar]
- Lee, C.; Sintich, S.M.; Mathews, E.P.; Shah, A.H.; Kundu, S.D.; Perry, K.T.; Cho, J.S.; Ilio, K.Y.; Cronauer, M.V.; Janulis, L.; et al. Transforming growth factor-beta in benign and malignant prostate. Prostate 1999, 39, 285–290. [Google Scholar]
- Reynolds, A.R.; Kyprianou, N. Growth factor signalling in prostatic growth: Significance in tumor development and therapeutic targeting. Br. J. Pharmacol 2006, 147, S144–S152. [Google Scholar]
- Fuzio, P.; Ditonno, P.; Rutigliano, M.; Battaglia, M.; Bettocchi, C.; Loverre, A.; Grandaliano, G.; Perlino, E. Regulation of TGF-beta1 expression by androgen deprivation therapy of prostate cancer. Cancer Lett 2012, 318, 135–144. [Google Scholar]
- Calone, I.; Souchelnytskyi, S. Inhibition of TGFbeta signaling and its implications in anticancer treatments. Exp. Oncol 2012, 34, 9–16. [Google Scholar]
- Mirochnik, Y.; Veliceasa, D.; Williams, L.; Maxwell, K.; Yemelyanov, A.; Budunova, I.; Volpert, O.V. Androgen receptor drives cellular senescence. PLoS One 2012, 7, e31052. [Google Scholar]
- White, E.; Lowe, S.W. Eating to exit: Autophagy-enabled senescence revealed. Genes Dev 2009, 23, 784–787. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| NIH funded clinical trials | Interventions | Relation with autophagy | Number of Pubmed abstracts |
|---|---|---|---|
| 1 | Visualase Thermal Therapy System | ||
| 2 | Ferumoxytol | ++ | 1 |
| 3 | Flutamide | ||
| 4 | Abiraterone acetate; prednisone; veliparib | ++ (prednisone; veliparib) | 2 + 1 |
| 5 | Tivantinib | ||
| 6 | Metformin hydrochloride | +++ (metformin) | 42 |
| 7 | Radiation: fluorine F 18 sodium fluoride | +++ (radiation) | 359 |
| 8 | AMG 386; Abiraterone; Prednisone | ||
| 9 | Sulforaphane glucosinolate capsules; capsules with cellulose and magnesium stearate | ++ (sulphorane) | 7 |
| 10 | Akt inhibitor MK2206; bicalutamide | ++ (Akt inhibitor) | 5 |
| 11 | Cabozantinib; Docetaxel; Prednisone | ++ (docetaxel) | 9 |
| 12 | Akt inhibitor MK2206; hydroxychloroquine; | +++ (MK2206, hydroxychloroquine) | 5 + 37 |
| 13 | Finasteride | ++ (finasteride) | 1 |
| 14 | Counseling intervention | ||
| 15 | Bicalutamide; buserelin; flutamide; goserelin acetate; leuprolide acetate; orteronel; triptorelin | ++ (bicalutamide) | 3 |
| 16 | Docetaxel; goserelin acetate; leuprolide acetate; surgery | ++ (docetaxel) | 9 |
| 17 | Bicalutamide; buserelin; flutamide; goserelin acetate; leuprolide acetate; triptorelin; 3-dimensional conformal radiation therapy | ++ (bicalutamide) | 3 |
| 18 | Radiation: radiation therapy; selective external radiation therapy | +++ (radiation) | 359 |
| 19 | Bicalutamide; goserelin acetate | ++ (bicalutamide) | 3 |
| 20 | Radiation: 3-dimensional conformal radiation therapy; intensity-modulated radiation therapy; samarium Sm 153 lexidronam pentasodium | +++ (radiation) | 359 |
| 21 | Antiandrogen therapy; docetaxel | ++ (docetaxel) | 9 |
| 22 | Abiraterone acetate; prednisone; | ++ (prednisone) | 2 |
| 23 | Bicalutamide; flutamide; radiation therapy | +++ (bicalutamide; radiation therapy) | 3 + 359 |
| 24 | MR Imaging of the prostate using Amide-Proton-Transfer | ||
| 25 | Genistein | ++ | 9 |
| 26 | Abiraterone acetate; degarelix; goserelin acetate; leuprolide acetate; orchiectomy | ||
| 27 | Biological: Ad5-CMV-NIS; liothyronine sodium; iodine I 131 | +++ (liothyronine, radiation) | 2 + 359 |
| 28 | Abiraterone acetate; dasatinib; prednisone | ++ (dasatinib) | 9 |
| 29 | Hypofractionated radiation therapy | +++ | 359 |
| 30 | Hydroxychloroquine | +++ | 37 |
| 31 | Ipilimumab | ||
| 32 | Cabazitaxel; prednisone; octreotide pamoate; octreotide acetate | ++ (prednisone ) | 2 |
| 33 | Axitinib; therapeutic conventional surgery | ||
| 34 | Radiation: radiation therapy | +++ (radiation) | 359 |
| 35 | Oral L-arginine; | +++ | 35 |
| 36 | Laser interstitial thermal therapy | ||
| 37 | Oral microencapsulated diindolylmethane | ++ (diindolylmethane) | 3 |
| 38 | Radiation: stereotactic body radiation therapy | +++ (radiation) | 359 |
| 39 | Lenalidomide; cyclophosphamide | ++ (cyclophosphamide) | 5 |
| 40 | Abiraterone acetate | ||
| 41 | Cinacalcet hydrochloride | ||
| 42 | Motexafin gadolinium | ||
| 43 | Radiation; Androgen Deprivation Therapy (ADT); L-BLP25 | +++ (radiation) | 359 |
| 44 | Atorvastatin calcium | ++ | 7 |
| 45 | Transrectal prostate biopsy | ||
| 46 | Docetaxel; pasireotide; prednisone | ++ (docetaxel, prednisone) | 9 + 2 |
| 47 | Proton Beam Therapy; Intensity Modulated Radiation Therapy | +++ (radiation) | 359 |
| 48 | Dietary intervention; nutritional support | ++ (dietary intervention) | 5 |
| 49 | Purified isoflavones; Methyl cellulose blend | ++ (isoflavones) | 21 |
| 50 | Therapeutic conventional surgery | ||
| 51 | Information Gathering; | ||
| 52 | Proteomic profiling comprising MALDI-TOF MS, | ||
| 53 | External beam radiation therapy; goserelin acetate | +++ (radiation) | 359 |
| 54 | Robot-assisted laparoscopic surgery | ||
| 55 | Docetaxel; prostate biopsy; phenelzine sulfate | ++ (docetaxel) | 9 |
| 56 | TNFerade™ | ||
| 57 | Radiation: brachytherapy; iodine I 125; palladium Pd 103 | +++ (radiation) | 359 |
| 58 | Dietary Suppl. Se-methyl-seleno-L-cysteine; selenomethionine | ++ (selenomethionine) | 2 |
| 59 | Behavioral: BF+GROUP; BF+PHONE | ||
| 60 | Survey administration | ||
| 61 | Gemcitabine; cisplatin; bevacizumab | +++ (gemcitabine; cisplatin) | 13 + 103 |
| 62 | Aerobic exercise | ++ | 2 |
| 63 | Memantine hydrochloride | ||
| 64 | Texotere (Docetaxel); Alimta (Pemetrexed) | ++ (Docetaxel, Pemetrexed) | 9 + 3 |
| 65 | NK cells +CliniMACs CD3 and CD56 systems | ++ (CD3) | 7 |
| 66 | Lapatinib; paclitaxel | ++ (Lapatinib; paclitaxel) | 10 + 40 |
| 67 | Radiation: radiation therapy; stereotactic radiosurgery | +++ (radiation) | 359 |
| 68 | Nicotine Replacement Patch | ++ (nicotine) | 3 |
| 69 | Hyperthermia; Radiation: HDR brachytherapy | +++ (hyperthermia) | 32 |
| 70 | Behavioral: BF+GROUP; BF+PHONE | ||
| 71 | Brachytherapy | +++ (radiation) | 359 |
| 72 | Veliparib | ++ | 1 |
| 73 | Behavioral: MR Therapy; Relaxing Music (RM) Therapy | ||
| 74 | Radiation: Bone marrow sparing IMRT radiation therapy | +++ (radiation) | 359 |
| 75 | Polyphenon E; | ||
| 76 | Selenium; vitamin E; selenium placebo | +++ (selenium; vitamin E) | 13 + 26 |
| 77 | Cabozantinib; FDG PET CT; NaF PET CT | ||
| 78 | Biological: Autologous Ad HER2 dendritic cell vaccine | ||
| 79 | Biological: recombinant albumin fusion protein sEphB4-HSA | ||
| 80 | Behavioral: Home environs-based lifestyle counseling |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ziparo, E.; Petrungaro, S.; Marini, E.S.; Starace, D.; Conti, S.; Facchiano, A.; Filippini, A.; Giampietri, C. Autophagy in Prostate Cancer and Androgen Suppression Therapy. Int. J. Mol. Sci. 2013, 14, 12090-12106. https://doi.org/10.3390/ijms140612090
Ziparo E, Petrungaro S, Marini ES, Starace D, Conti S, Facchiano A, Filippini A, Giampietri C. Autophagy in Prostate Cancer and Androgen Suppression Therapy. International Journal of Molecular Sciences. 2013; 14(6):12090-12106. https://doi.org/10.3390/ijms140612090
Chicago/Turabian StyleZiparo, Elio, Simonetta Petrungaro, Elettra Sara Marini, Donatella Starace, Silvia Conti, Antonio Facchiano, Antonio Filippini, and Claudia Giampietri. 2013. "Autophagy in Prostate Cancer and Androgen Suppression Therapy" International Journal of Molecular Sciences 14, no. 6: 12090-12106. https://doi.org/10.3390/ijms140612090