Oxidative Stress Mechanisms Underlying Parkinson’s Disease-Associated Neurodegeneration in C. elegans


{kind=link}
{kind=link}
Abstract
:1. Introduction
2 . C. elegans as a Model for Neurodegeneration
3. Assessing Oxidative Stress in C. elegans
3.1. Neurotoxins: PD-Mimetics
3.2. Measuring ROS Production in Vivo
3.3. Using Pharmacological Agents to Assess the Role of ROS in Dopamine Neuron Degeneration
3.4. Methods to Quantify Oxidative Damage
4. Oxidative Stress and PD-Associated Neurodegeneration in C. elegans
4.1. DJ-1
4.2. Parkin and PINK1
4.3. Nrf2
5. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar]
- Dawson, T.M.; Ko, H.S.; Dawson, V.L. Genetic animal models of Parkinson’s disease. Neuron 2010, 66, 646–661. [Google Scholar]
- Varcin, M.; Bentea, E.; Michotte, Y.; Sarre, S. Oxidative stress in genetic mouse models of Parkinson’s disease. Oxid. Med. Cell. Longev 2012, 2012. [Google Scholar] [CrossRef]
- Boada, J.; Cutillas, B.; Roig, T.; Bermudez, J.; Ambrosio, S. Mpp+-induced mitochondrial dysfunction is potentiated by dopamine. Biochem. Biophys. Res. Commun 2000, 268, 916–920. [Google Scholar]
- Keane, P.C.; Kurzawa, M.; Blain, P.G.; Morris, C.M. Mitochondrial dysfunction in Parkinson’s disease. Parkinson’s Dis 2011, 2011. [Google Scholar] [CrossRef]
- Cacciatore, I.; Baldassarre, L.; Fornasari, E.; Mollica, A.; Pinnen, F. Recent advances in the treatment of neurodegenerative diseases based on GSH delivery systems. Oxid. Med. Cell. Longev 2012, 2012. [Google Scholar] [CrossRef]
- Sulston, J.; Dew, M.; Brenner, S. Dopaminergic neurons in the nematode Caenorhabditis elegans. J. Comp. Neurol 1975, 163, 215–226. [Google Scholar]
- Jayanthi, L.D.; Apparsundaram, S.; Malone, M.D.; Ward, E.; Miller, D.M.; Eppler, M.; Blakely, R.D. The Caenorhabditis elegans gene T23G5.5 encodes an antidepressant- and cocaine-sensitive dopamine transporter. Mol. Pharmacol 1998, 54, 601–609. [Google Scholar]
- Duerr, J.S.; Frisby, D.L.; Gaskin, J.; Duke, A.; Asermely, K.; Huddleston, D.; Eiden, L.E.; Rand, J.B. The cat-1 gene of Caenorhabditis elegans encodes a vesicular monoamine transporter required for specific monoamine-dependent behaviors. J. Neurosci 1999, 19, 72–84. [Google Scholar]
- Sawin, E.R.; Ranganathan, R.; Horvitz, H.R. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 2000, 26, 619–631. [Google Scholar]
- Chase, D.L.; Koelle, M.R. Biogenic amine neurotransmitters in C. elegans. WormBook 2007. [Google Scholar] [CrossRef]
- Nass, R.; Hall, D.H.; Miller, D.M., 3rd; Blakely, R.D. Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 3264–3269. [Google Scholar]
- Consortium, C.E.S. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science 1998, 282, 2012–2018. [Google Scholar]
- Martinez-Finley, E.J.; Avila, D.S.; Chakraborty, S.; Aschner, M. Insights from Caenorhabditis elegans on the role of metals in neurodegenerative diseases. Metallomics Integr. Biomet. Sci 2011, 3, 271–279. [Google Scholar]
- Ahringer, J. Reverse Genetics. In Wormbook; The C elegans Research Community: Cambridge, UK, 2006. [Google Scholar]
- Isik, M.; Berezikov, E. Biolistic transformation of Caenorhabditis elegans. Methods Mol. Biol 2013, 940, 77–86. [Google Scholar]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar]
- Simmer, F.; Tijsterman, M.; Parrish, S.; Koushika, S.P.; Nonet, M.L.; Fire, A.; Ahringer, J.; Plasterk, R.H. Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr. Biol 2002, 12, 1317–1319. [Google Scholar]
- Timmons, L.; Court, D.L.; Fire, A. Ingestion of bacterially expressed dsrnas can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 2001, 263, 103–112. [Google Scholar]
- Tabara, H.; Grishok, A.; Mello, C.C. RNAi in C. elegans: Soaking in the genome sequence. Science 1998, 282, 430–431. [Google Scholar]
- Timmons, L. Endogenous inhibitors of RNA interference in Caenorhabditis elegans. BioEssays 2004, 26, 715–718. [Google Scholar]
- Frokjaer-Jensen, C.; Davis, M.W.; Hollopeter, G.; Taylor, J.; Harris, T.W.; Nix, P.; Lofgren, R.; Prestgard-Duke, M.; Bastiani, M.; Moerman, D.G.; et al. Targeted gene deletions in C. elegans using transposon excision. Nat. Methods 2010, 7, 451–453. [Google Scholar]
- Antoshechkin, I.; Sternberg, P.W. The versatile worm: Genetic and genomic resources for Caenorhabditis elegans research. Nat. Rev. Genet 2007, 8, 518–532. [Google Scholar]
- Blandini, F.; Armentero, M.T. Animal models of Parkinson’s disease. FEBS J 2012, 279, 1156–1166. [Google Scholar]
- Blesa, J.; Phani, S.; Jackson-Lewis, V.; Przedborski, S. Classic and new animal models of Parkinson’s disease. J. Biomed. Biotechnol 2012, 2012. [Google Scholar] [CrossRef]
- Hoban, D.B.; Connaughton, E.; Connaughton, C.; Hogan, G.; Thornton, C.; Mulcahy, P.; Moloney, T.C.; Dowd, E. Further characterisation of the LPS model of parkinson’s disease: A comparison of intra-nigral and intra-striatal lipopolysaccharide administration on motor function, microgliosis and nigrostriatal neurodegeneration in the rat. Brain Behav. Immun 2013, 27, 91–100. [Google Scholar]
- Tucci, M.L.; Harrington, A.J.; Caldwell, G.A.; Caldwell, K.A. Modeling dopamine neuron degeneration in Caenorhabditis elegans. Methods Mol. Biol 2011, 793, 129–148. [Google Scholar]
- Ali, S.J.; Rajini, P.S. Elicitation of dopaminergic features of Parkinson’s disease in C. elegans by monocrotophos, an organophosphorous insecticide. CNS Neurol. Disord. Drug Targets 2012, 11, 993–1000. [Google Scholar]
- Wang, Y.M.; Pu, P.; Le, W.D. ATP depletion is the major cause of MPP+ induced dopamine neuronal death and worm lethality in α-synuclein transgenic C. elegans. Neurosci. Bull 2007, 23, 329–335. [Google Scholar]
- Benedetto, A.; Au, C.; Avila, D.S.; Milatovic, D.; Aschner, M. Extracellular dopamine potentiates Mn-induced oxidative stress, lifespan reduction, and dopaminergic neurodegeneration in a BLI-3-dependent manner in Caenorhabditis elegans. PLoS Genet 2010, 6, e1001084. [Google Scholar]
- Avila, D.S.; Benedetto, A.; Au, C.; Manarin, F.; Erikson, K.; Soares, F.A.; Rocha, J.B.; Aschner, M. Organotellurium and organoselenium compounds attenuate mn-induced toxicity in Caenorhabditis elegans by preventing oxidative stress. Free Radic. Biol. Med 2012, 52, 1903–1910. [Google Scholar]
- Jellinger, K.; Linert, L.; Kienzl, E.; Herlinger, E.; Youdim, M.B. Chemical evidence for 6-hydroxydopamine to be an endogenous toxic factor in the pathogenesis of Parkinson’s disease. J. Neural Transm. Suppl 1995, 46, 297–314. [Google Scholar]
- Slivka, A.; Cohen, G. Hydroxyl radical attack on dopamine. J. Biol. Chem 1985, 260, 15466–15472. [Google Scholar]
- Cohen, G.; Heikkila, R.E. The generation of hydrogen peroxide, superoxide radical, and hydroxyl radical by 6-hydroxydopamine, dialuric acid, and related cytotoxic agents. J. Biol. Chem 1974, 249, 2447–2452. [Google Scholar]
- Graham, D.G.; Tiffany, S.M.; Bell, W.R., Jr.; Gutknecht, W.F. Autoxidation versus covalent binding of quinones as the mechanism of toxicity of dopamine, 6-hydroxydopamine, and related compounds toward C1300 neuroblastoma cells in vitro. Mol. Pharmacol. 1978, 14, 644–653. [Google Scholar]
- Glinka, Y.Y.; Youdim, M.B. Inhibition of mitochondrial complexes I and II by 6-hydroxydopamine. Eur. J. Pharmacol 1995, 292, 329–332. [Google Scholar]
- Glinka, Y.; Gassen, M.; Youdim, M.B. Mechanism of 6-hydroxydopamine neurotoxicity. J. Neural Transm. Suppl 1997, 50, 55–66. [Google Scholar]
- Jenner, P. Oxidative mechanisms in nigral cell death in Parkinson’s disease. Mov. Disord 1998, 13, 24–34. [Google Scholar]
- Kitayama, S.; Shimada, S.; Uhl, G.R. Parkinsonism-inducing neurotoxin MPP+: Uptake and toxicity in nonneuronal cos cells expressing dopamine transporter cdna. Ann. Neurol 1992, 32, 109–111. [Google Scholar]
- Dingley, S.; Polyak, E.; Lightfoot, R.; Ostrovsky, J.; Rao, M.; Greco, T.; Ischiropoulos, H.; Falk, M.J. Mitochondrial respiratory chain dysfunction variably increases oxidant stress in Caenorhabditis elegans. Mitochondrion 2010, 10, 125–136. [Google Scholar]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab 2007, 6, 280–293. [Google Scholar]
- Vanfleteren, J.R.; de Vreese, A. Rate of aerobic metabolism and superoxide production rate potential in the nematode Caenorhabditis elegans. J. Exp. Zool 1996, 274, 93–100. [Google Scholar]
- Burkitt, M.J.; Wardman, P. Cytochrome C is a potent catalyst of dichlorofluorescin oxidation: Implications for the role of reactive oxygen species in apoptosis. Biochem. Biophys. Res. Commun 2001, 282, 329–333. [Google Scholar]
- Bonini, M.G.; Rota, C.; Tomasi, A.; Mason, R.P. The oxidation of 2′,7′-dichlorofluorescin to reactive oxygen species: A self-fulfilling prophesy? Free Radic. Biol. Med 2006, 40, 968–975. [Google Scholar]
- Hempel, S.L.; Buettner, G.R.; O’Malley, Y.Q.; Wessels, D.A.; Flaherty, D.M. Dihydrofluorescein diacetate is superior for detecting intracellular oxidants: Comparison with 2′,7′-dichlorodihydrofluorescein diacetate, 5(and 6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate, and dihydrorhodamine 123. Free Radic. Biol. Med 1999, 27, 146–159. [Google Scholar]
- Folkes, L.K.; Patel, K.B.; Wardman, P.; Wrona, M. Kinetics of reaction of nitrogen dioxide with dihydrorhodamine and the reaction of the dihydrorhodamine radical with oxygen: Implications for quantifying peroxynitrite formation in cells. Arch. Biochem. Biophys 2009, 484, 122–126. [Google Scholar]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J., 2nd; Ischiropoulos, H. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radic. Biol. Med 2012, 52, 1–6. [Google Scholar]
- Back, P.; de Vos, W.H.; Depuydt, G.G.; Matthijssens, F.; Vanfleteren, J.R.; Braeckman, B.P. Exploring real-time in vivo redox biology of developing and aging Caenorhabditis elegans. Free Radic. Biol. Med 2012, 52, 850–859. [Google Scholar]
- Knoefler, D.; Thamsen, M.; Koniczek, M.; Niemuth, N.J.; Diederich, A.K.; Jakob, U. Quantitative in vivo redox sensors uncover oxidative stress as an early event in life. Mol. Cell 2012, 47, 767–776. [Google Scholar]
- Belousov, V.V.; Fradkov, A.F.; Lukyanov, K.A.; Staroverov, D.B.; Shakhbazov, K.S.; Terskikh, A.V.; Lukyanov, S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat. Methods 2006, 3, 281–286. [Google Scholar]
- Aslund, F.; Zheng, M.; Beckwith, J.; Storz, G. Regulation of the OxyR transcription factor by hydrogen peroxide and the cellular thiol-disulfide status. Proc. Natl. Acad. Sci. USA 1999, 96, 6161–6165. [Google Scholar]
- De Castro, E.; Hegi de Castro, S.; Johnson, T.E. Isolation of long-lived mutants in Caenorhabditis elegans using selection for resistance to juglone. Free Radic. Biol. Med 2004, 37, 139–145. [Google Scholar]
- Inbaraj, J.J.; Chignell, C.F. Cytotoxic action of juglone and plumbagin: A mechanistic study using HaCaT keratinocytes. Chem. Res. Toxicol 2004, 17, 55–62. [Google Scholar]
- Hartwig, K.; Heidler, T.; Moch, J.; Daniel, H.; Wenzel, U. Feeding a ROS-generator to Caenorhabditis elegans leads to increased expression of small heat shock protein HSP-16.2 and hormesis. Genes Nutr 2009, 4, 59–67. [Google Scholar]
- Przybysz, A.J.; Choe, K.P.; Roberts, L.J.; Strange, K. Increased age reduces DAF-16 and SKN-1 signaling and the hormetic response of Caenorhabditis elegans to the xenobiotic juglone. Mechan. Ageing Dev 2009, 130, 357–369. [Google Scholar]
- Ishii, N.; Takahashi, K.; Tomita, S.; Keino, T.; Honda, S.; Yoshino, K.; Suzuki, K. A methyl viologen-sensitive mutant of the nematode Caenorhabditis elegans. Mutat. Res 1990, 237, 165–171. [Google Scholar]
- Khare, S.; Gomez, T.; Linster, C.L.; Clarke, S.G. Defective responses to oxidative stress in protein l-isoaspartyl repair-deficient Caenorhabditis elegans. Mech. Ageing Dev 2009, 130, 670–680. [Google Scholar]
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 2006, 23, 607–618. [Google Scholar]
- Vanfleteren, J.R. Oxidative stress and ageing in Caenorhabditis elegans. Biochem. J 1993, 292, 605–608. [Google Scholar]
- Blum, J.; Fridovich, I. Superoxide, hydrogen peroxide, and oxygen toxicity in two free-living nematode species. Arch. Biochem. Biophys 1983, 222, 35–43. [Google Scholar]
- Lee, S.S.; Lee, R.Y.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Ruvkun, G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet 2003, 33, 40–48. [Google Scholar]
- Ventura, N.; Rea, S.; Henderson, S.T.; Condo, I.; Johnson, T.E.; Testi, R. Reduced expression of frataxin extends the lifespan of Caenorhabditis elegans. Aging Cell 2005, 4, 109–112. [Google Scholar]
- Yamamoto, K.; Honda, S.; Ishii, N. Properties of an oxygen-sensitive mutant mev-3 of the nematode Caenorhabditis elegans. Mutat. Res 1996, 358, 1–6. [Google Scholar]
- Feng, J.; Bussiere, F.; Hekimi, S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev. Cell 2001, 1, 633–644. [Google Scholar]
- Montine, K.S.; Quinn, J.F.; Zhang, J.; Fessel, J.P.; Roberts, L.J., 2nd; Morrow, J.D.; Montine, T.J. Isoprostanes and related products of lipid peroxidation in neurodegenerative diseases. Chem. Phys. Lipids 2004, 128, 117–124. [Google Scholar]
- Milne, G.L.; Yin, H.; Hardy, K.D.; Davies, S.S.; Roberts, L.J., 2nd. Isoprostane generation and function. Chem. Rev 2011, 111, 5973–5996. [Google Scholar]
- Gao, L.; Yin, H.; Milne, G.L.; Porter, N.A.; Morrow, J.D. Formation of F-ring isoprostane-like compounds (F3-isoprostanes) in vivo from eicosapentaenoic acid. J. Biol. Chem 2006, 281, 14092–14099. [Google Scholar]
- Labuschagne, C.F.; Stigter, E.C.; Hendriks, M.M.; Berger, R.; Rokach, J.; Korswagen, H.C.; Brenkman, A.B. Quantification of in vivo oxidative damage in Caenorhabditis elegans during aging by endogenous F3-isoprostane measurement. Aging Cell 2013, 12, 214–223. [Google Scholar]
- Adachi, H.; Fujiwara, Y.; Ishii, N. Effects of oxygen on protein carbonyl and aging in Caenorhabditis elegans mutants with long (age-1) and short (mev-1) life spans. J. Gerontol. A Biol. Sci. Med. Sci 1998, 53, B240–B244. [Google Scholar]
- Petriv, O.I.; Rachubinski, R.A. Lack of peroxisomal catalase causes a progeric phenotype in Caenorhabditis elegans. J. Biol. Chem 2004, 279, 19996–20001. [Google Scholar]
- Yanase, S.; Yasuda, K.; Ishii, N. Adaptive responses to oxidative damage in three mutants of Caenorhabditis elegans (age-1, mev-1 and daf-16) that affect life span. Mech. Ageing Dev 2002, 123, 1579–1587. [Google Scholar]
- Kayser, E.B.; Sedensky, M.M.; Morgan, P.G. The effects of complex I function and oxidative damage on lifespan and anesthetic sensitivity in Caenorhabditis elegans. Mech. Ageing Dev 2004, 125, 455–464. [Google Scholar]
- Yang, Y.Y.; Gangoiti, J.A.; Sedensky, M.M.; Morgan, P.G. The effect of different ubiquinones on lifespan in Caenorhabditis elegans. Mech. Ageing Dev 2009, 130, 370–376. [Google Scholar]
- Davies, S.S.; Talati, M.; Wang, X.; Mernaugh, R.L.; Amarnath, V.; Fessel, J.; Meyrick, B.O.; Sheller, J.; Roberts, L.J., 2nd. Localization of isoketal adducts in vivo using a single-chain antibody. Free Rad. Biol. Med 2004, 36, 1163–1174. [Google Scholar]
- Choe, K.P.; Przybysz, A.J.; Strange, K. The WD40 repeat protein WDR-23 functions with the CUL4/DDB1 ubiquitin ligase to regulate nuclear abundance and activity of SKN-1 in Caenorhabditis elegans. Mol. Cell. Biol 2009, 29, 2704–2715. [Google Scholar]
- Leung, C.K.; Deonarine, A.; Strange, K.; Choe, K.P. High-throughput screening and biosensing with fluorescent C. elegans strains. J. Vis. Exp 2011. [Google Scholar] [CrossRef]
- Leung, C.K.; Empinado, H.; Choe, K.P. Depletion of a nucleolar protein activates xenobiotic detoxification genes in Caenorhabditis elegans via Nrf/SKN-1 and p53/CEP-1. Free Radic. Biol. Med 2012, 52, 937–950. [Google Scholar]
- Bandyopadhyay, S.; Cookson, M.R. Evolutionary and functional relationships within the DJ1 superfamily. BMC Evol. Biol 2004, 4, 6. [Google Scholar]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the dj-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar]
- Miller, D.W.; Ahmad, R.; Hague, S.; Baptista, M.J.; Canet-Aviles, R.; McLendon, C.; Carter, D.M.; Zhu, P.P.; Stadler, J.; Chandran, J.; et al. L166P mutant DJ-1, causative for recessive Parkinson’s disease, is degraded through the ubiquitin-proteasome system. J. Biol. Chem 2003, 278, 36588–36595. [Google Scholar]
- Macedo, M.G.; Anar, B.; Bronner, I.F.; Cannella, M.; Squitieri, F.; Bonifati, V.; Hoogeveen, A.; Heutink, P.; Rizzu, P. The DJ-1l166P mutant protein associated with early onset Parkinson’s disease is unstable and forms higher-order protein complexes. Hum. Mol. Genet 2003, 12, 2807–2816. [Google Scholar]
- Shendelman, S.; Jonason, A.; Martinat, C.; Leete, T.; Abeliovich, A. DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol 2004, 2, e362. [Google Scholar]
- Mitsumoto, A.; Nakagawa, Y. DJ-1 is an indicator for endogenous reactive oxygen species elicited by endotoxin. Free Radic. Res 2001, 35, 885–893. [Google Scholar]
- Mitsumoto, A.; Nakagawa, Y.; Takeuchi, A.; Okawa, K.; Iwamatsu, A.; Takanezawa, Y. Oxidized forms of peroxiredoxins and DJ-1 on two-dimensional gels increased in response to sublethal levels of paraquat. Free Radic. Res 2001, 35, 301–310. [Google Scholar]
- Wang, X.; Petrie, T.G.; Liu, Y.; Liu, J.; Fujioka, H.; Zhu, X. Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J. Neurochem 2012, 121, 830–839. [Google Scholar]
- Lev, N.; Ickowicz, D.; Barhum, Y.; Lev, S.; Melamed, E.; Offen, D. DJ-1 protects against dopamine toxicity. J. Neural Transm 2009, 116, 151–160. [Google Scholar]
- Lev, N.; Barhum, Y.; Pilosof, N.S.; Ickowicz, D.; Cohen, H.Y.; Melamed, E.; Offen, D. DJ-1 protects against dopamine toxicity: Implications for Parkinson’s disease and aging. J. Gerontol. A Biol. Sci. Med. Sci 2013, 68, 215–225. [Google Scholar]
- Andres-Mateos, E.; Perier, C.; Zhang, L.; Blanchard-Fillion, B.; Greco, T.M.; Thomas, B.; Ko, H.S.; Sasaki, M.; Ischiropoulos, H.; Przedborski, S.; et al. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc. Natl. Acad. Sci. USA 2007, 104, 14807–14812. [Google Scholar]
- Canet-Aviles, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar]
- Choi, J.; Sullards, M.C.; Olzmann, J.A.; Rees, H.D.; Weintraub, S.T.; Bostwick, D.E.; Gearing, M.; Levey, A.I.; Chin, L.S.; Li, L. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J. Biol. Chem 2006, 281, 10816–10824. [Google Scholar]
- Taira, T.; Saito, Y.; Niki, T.; Iguchi-Ariga, S.M.; Takahashi, K.; Ariga, H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep 2004, 5, 213–218. [Google Scholar]
- Trempe, J.F.; Fon, E.A. Structure and function of parkin, PINK1, and DJ-1, the three musketeers of neuroprotection. Front Neurol 2013, 4, 38. [Google Scholar]
- Girotto, S.; Sturlese, M.; Bellanda, M.; Tessari, I.; Cappellini, R.; Bisaglia, M.; Bubacco, L.; Mammi, S. Dopamine-derived quinones affect the structure of the redox sensor DJ-1 through modifications at Cys-106 and Cys-53. J. Biol. Chem 2012, 287, 18738–18749. [Google Scholar]
- Mullett, S.J.; di Maio, R.; Greenamyre, J.T.; Hinkle, D.A. DJ-1 expression modulates astrocyte-mediated protection against neuronal oxidative stress. J. Mol. Neurosci 2013, 49, 507–511. [Google Scholar]
- Meulener, M.; Whitworth, A.J.; Armstrong-Gold, C.E.; Rizzu, P.; Heutink, P.; Wes, P.D.; Pallanck, L.J.; Bonini, N.M. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson’s disease. Curr. Biol 2005, 15, 1572–1577. [Google Scholar]
- Shim, J.H.; Yoon, S.H.; Kim, K.H.; Han, J.Y.; Ha, J.Y.; Hyun, D.H.; Paek, S.H.; Kang, U.J.; Zhuang, X.; Son, J.H. The antioxidant trolox helps recovery from the familial Parkinson’s disease-specific mitochondrial deficits caused by PINK1- and DJ-1-deficiency in dopaminergic neuronal cells. Mitochondrion 2011, 11, 707–715. [Google Scholar]
- Wilhelmus, M.M.; Nijland, P.G.; Drukarch, B.; de Vries, H.E.; van Horssen, J. Involvement and interplay of parkin, PINK1, and DJ1 in neurodegenerative and neuroinflammatory disorders. Free Radic. Biol. Med 2012, 53, 983–992. [Google Scholar]
- Kim, R.H.; Smith, P.D.; Aleyasin, H.; Hayley, S.; Mount, M.P.; Pownall, S.; Wakeham, A.; You-Ten, A.J.; Kalia, S.K.; Horne, P.; et al. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. USA 2005, 102, 5215–5220. [Google Scholar]
- Hao, L.Y.; Giasson, B.I.; Bonini, N.M. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc. Natl. Acad. Sci. USA 2010, 107, 9747–9752. [Google Scholar]
- Joselin, A.P.; Hewitt, S.J.; Callaghan, S.M.; Kim, R.H.; Chung, Y.H.; Mak, T.W.; Shen, J.; Slack, R.S.; Park, D.S. ROS-dependent regulation of parkin and DJ-1 localization during oxidative stress in neurons. Hum. Mol. Genet 2012, 21, 4888–4903. [Google Scholar]
- Xiong, H.; Wang, D.; Chen, L.; Choo, Y.S.; Ma, H.; Tang, C.; Xia, K.; Jiang, W.; Ronai, Z.; Zhuang, X.; et al. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J. Clin. Invest 2009, 119, 650–660. [Google Scholar]
- Thomas, K.J.; McCoy, M.K.; Blackinton, J.; Beilina, A.; van der Brug, M.; Sandebring, A.; Miller, D.; Maric, D.; Cedazo-Minguez, A.; Cookson, M.R. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet 2011, 20, 40–50. [Google Scholar]
- Zhang, L.; Shimoji, M.; Thomas, B.; Moore, D.J.; Yu, S.W.; Marupudi, N.I.; Torp, R.; Torgner, I.A.; Ottersen, O.P.; Dawson, T.M.; et al. Mitochondrial localization of the Parkinson’s disease related protein DJ-1: Implications for pathogenesis. Hum. Mol. Genet 2005, 14, 2063–2073. [Google Scholar]
- Lucas, J.I.; Marin, I. A new evolutionary paradigm for the parkinson disease geneDJ-1. Mol. Biol. Evol 2007, 24, 551–561. [Google Scholar]
- Lee, J.Y.; Song, J.; Kwon, K.; Jang, S.; Kim, C.; Baek, K.; Kim, J.; Park, C. Human DJ-1 and its homologs are novel glyoxalases. Hum. Mol. Genet 2012, 21, 3215–3225. [Google Scholar]
- Ved, R.; Saha, S.; Westlund, B.; Perier, C.; Burnam, L.; Sluder, A.; Hoener, M.; Rodrigues, C.M.; Alfonso, A.; Steer, C.; et al. Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of alpha-synuclein, parkin, and DJ-1 inCaenorhabditis elegans. J. Biol. Chem 2005, 280, 42655–42668. [Google Scholar]
- Lee, J.Y.; Kim, C.; Kim, J.; Park, C. DJR-1.2 of Caenorhabditis elegans is induced by DAF-16 in the dauer state. Gene 2013, 524, 373–376. [Google Scholar]
- Zhang, Y.; Gao, J.; Chung, K.K.; Huang, H.; Dawson, V.L.; Dawson, T.M. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc. Natl. Acad. Sci. USA 2000, 97, 13354–13359. [Google Scholar]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar]
- Horowitz, J.M.; Myers, J.; Stachowiak, M.K.; Torres, G. Identification and distribution of parkin in rat brain. Neuroreport 1999, 10, 3393–3397. [Google Scholar]
- Shimura, H.; Schlossmacher, M.G.; Hattori, N.; Frosch, M.P.; Trockenbacher, A.; Schneider, R.; Mizuno, Y.; Kosik, K.S.; Selkoe, D.J. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: Implications for Parkinson’s disease. Science 2001, 293, 263–269. [Google Scholar]
- Chung, K.K.; Zhang, Y.; Lim, K.L.; Tanaka, Y.; Huang, H.; Gao, J.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: Implications for lewy-body formation in Parkinson disease. Nat. Med 2001, 7, 1144–1150. [Google Scholar]
- Imai, Y.; Soda, M.; Inoue, H.; Hattori, N.; Mizuno, Y.; Takahashi, R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of parkin. Cell 2001, 105, 891–902. [Google Scholar]
- Sriram, S.R.; Li, X.; Ko, H.S.; Chung, K.K.; Wong, E.; Lim, K.L.; Dawson, V.L.; Dawson, T.M. Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Hum. Mol. Genet 2005, 14, 2571–2586. [Google Scholar]
- Goldberg, M.S.; Fleming, S.M.; Palacino, J.J.; Cepeda, C.; Lam, H.A.; Bhatnagar, A.; Meloni, E.G.; Wu, N.; Ackerson, L.C.; Klapstein, G.J.; et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem 2003, 278, 43628–43635. [Google Scholar]
- Jiang, H.; Jiang, Q.; Feng, J. Parkin increases dopamine uptake by enhancing the cell surface expression of dopamine transporter. J. Biol. Chem 2004, 279, 54380–54386. [Google Scholar]
- Kitada, T.; Pisani, A.; Karouani, M.; Haburcak, M.; Martella, G.; Tscherter, A.; Platania, P.; Wu, B.; Pothos, E.N.; Shen, J. Impaired dopamine release and synaptic plasticity in the striatum of parkin−/− mice. J. Neurochem 2009, 110, 613–621. [Google Scholar]
- Helton, T.D.; Otsuka, T.; Lee, M.C.; Mu, Y.; Ehlers, M.D. Pruning and loss of excitatory synapses by the parkin ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2008, 105, 19492–19497. [Google Scholar]
- Sang, T.K.; Chang, H.Y.; Lawless, G.M.; Ratnaparkhi, A.; Mee, L.; Ackerson, L.C.; Maidment, N.T.; Krantz, D.E.; Jackson, G.R. A drosophila model of mutant human parkin-induced toxicity demonstrates selective loss of dopaminergic neurons and dependence on cellular dopamine. J. Neurosci 2007, 27, 981–992. [Google Scholar]
- Lu, X.H.; Fleming, S.M.; Meurers, B.; Ackerson, L.C.; Mortazavi, F.; Lo, V.; Hernandez, D.; Sulzer, D.; Jackson, G.R.; Maidment, N.T.; et al. Bacterial artificial chromosome transgenic mice expressing a truncated mutant parkin exhibit age-dependent hypokinetic motor deficits, dopaminergic neuron degeneration, and accumulation of proteinase K-resistant alpha-synuclein. J. Neurosci 2009, 29, 1962–1976. [Google Scholar]
- Higashi, Y.; Asanuma, M.; Miyazaki, I.; Hattori, N.; Mizuno, Y.; Ogawa, N. Parkin attenuates manganese-induced dopaminergic cell death. J. Neurochem 2004, 89, 1490–1497. [Google Scholar]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar]
- Kessler, K.R.; Hamscho, N.; Morales, B.; Menzel, C.; Barrero, F.; Vives, F.; Gispert, S.; Auburger, G. Dopaminergic function in a family with the PARK6 form of autosomal recessive Parkinson’s syndrome. J. Neural Transm 2005, 112, 1345–1353. [Google Scholar]
- Kitada, T.; Pisani, A.; Porter, D.R.; Yamaguchi, H.; Tscherter, A.; Martella, G.; Bonsi, P.; Zhang, C.; Pothos, E.N.; Shen, J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. USA 2007, 104, 11441–11446. [Google Scholar]
- Gandhi, S.; Muqit, M.M.; Stanyer, L.; Healy, D.G.; Abou-Sleiman, P.M.; Hargreaves, I.; Heales, S.; Ganguly, M.; Parsons, L.; Lees, A.J.; et al. PINK1 protein in normal human brain and Parkinson’s disease. Brain J. Neurol 2006, 129, 1720–1731. [Google Scholar]
- Sim, C.H.; Lio, D.S.; Mok, S.S.; Masters, C.L.; Hill, A.F.; Culvenor, J.G.; Cheng, H.C. C-terminal truncation and Parkinson’s disease-associated mutations down-regulate the protein serine/threonine kinase activity of PTEN-induced kinase-1. Hum. Mol. Genet 2006, 15, 3251–3262. [Google Scholar]
- Haque, M.E.; Thomas, K.J.; D’Souza, C.; Callaghan, S.; Kitada, T.; Slack, R.S.; Fraser, P.; Cookson, M.R.; Tandon, A.; Park, D.S. Cytoplasmic PINK1 activity protects neurons from dopaminergic neurotoxin MPTP. Proc. Natl. Acad. Sci. USA 2008, 105, 1716–1721. [Google Scholar]
- Petit, A.; Kawarai, T.; Paitel, E.; Sanjo, N.; Maj, M.; Scheid, M.; Chen, F.; Gu, Y.; Hasegawa, H.; Salehi-Rad, S.; et al. Wild-type PINK1 prevents basal and induced neuronal apoptosis, a protective effect abrogated by Parkinson disease-related mutations. J. Biol. Chem 2005, 280, 34025–34032. [Google Scholar]
- Hoepken, H.H.; Gispert, S.; Morales, B.; Wingerter, O.; del Turco, D.; Mulsch, A.; Nussbaum, R.L.; Muller, K.; Drose, S.; Brandt, U.; et al. Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol. Dis 2007, 25, 401–411. [Google Scholar]
- Kim, Y.; Park, J.; Kim, S.; Song, S.; Kwon, S.K.; Lee, S.H.; Kitada, T.; Kim, J.M.; Chung, J. PINK1 controls mitochondrial localization of parkin through direct phosphorylation. Biochem. Biophys. Res. Commun 2008, 377, 975–980. [Google Scholar]
- Iguchi, M.; Kujuro, Y.; Okatsu, K.; Koyano, F.; Kosako, H.; Kimura, M.; Suzuki, N.; Uchiyama, S.; Tanaka, K.; Matsuda, N. Parkin catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J. Biol. Chem 2013, 288, 22019–22032. [Google Scholar]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.M.; et al. Mitochondrial dysfunction in drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar]
- Deng, H.; Dodson, M.W.; Huang, H.; Guo, M. The Parkinson’s disease genes PINK1 and parkin promote mitochondrial fission and/or inhibit fusion in drosophila. Proc. Natl. Acad. Sci. USA 2008, 105, 14503–14508. [Google Scholar]
- Okatsu, K.; Oka, T.; Iguchi, M.; Imamura, K.; Kosako, H.; Tani, N.; Kimura, M.; Go, E.; Koyano, F.; Funayama, M.; et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for parkin recruitment to damaged mitochondria. Nat. Commun 2012, 3, 1016. [Google Scholar]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol 2008, 183, 795–803. [Google Scholar]
- Kawajiri, S.; Saiki, S.; Sato, S.; Sato, F.; Hatano, T.; Eguchi, H.; Hattori, N. PINK1 is recruited to mitochondria with parkin and associates with LC3 in mitophagy. FEBS Lett 2010, 584, 1073–1079. [Google Scholar]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar]
- Lee, S.; Sterky, F.H.; Mourier, A.; Terzioglu, M.; Cullheim, S.; Olson, L.; Larsson, N.G. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum. Mol. Genet 2012, 21, 4827–4835. [Google Scholar]
- Sun, Y.; Vashisht, A.A.; Tchieu, J.; Wohlschlegel, J.A.; Dreier, L. Voltage-dependent anion channels (VDACS) recruit parkin to defective mitochondria to promote mitochondrial autophagy. J. Biol. Chem 2012, 287, 40652–40660. [Google Scholar]
- Springer, W.; Hoppe, T.; Schmidt, E.; Baumeister, R. A Caenorhabditis elegans parkin mutant with altered solubility couples alpha-synuclein aggregation to proteotoxic stress. Hum. Mol. Genet 2005, 14, 3407–3423. [Google Scholar]
- Samann, J.; Hegermann, J.; von Gromoff, E.; Eimer, S.; Baumeister, R.; Schmidt, E. Caenorhabditits elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J. Biol. Chem 2009, 284, 16482–16491. [Google Scholar]
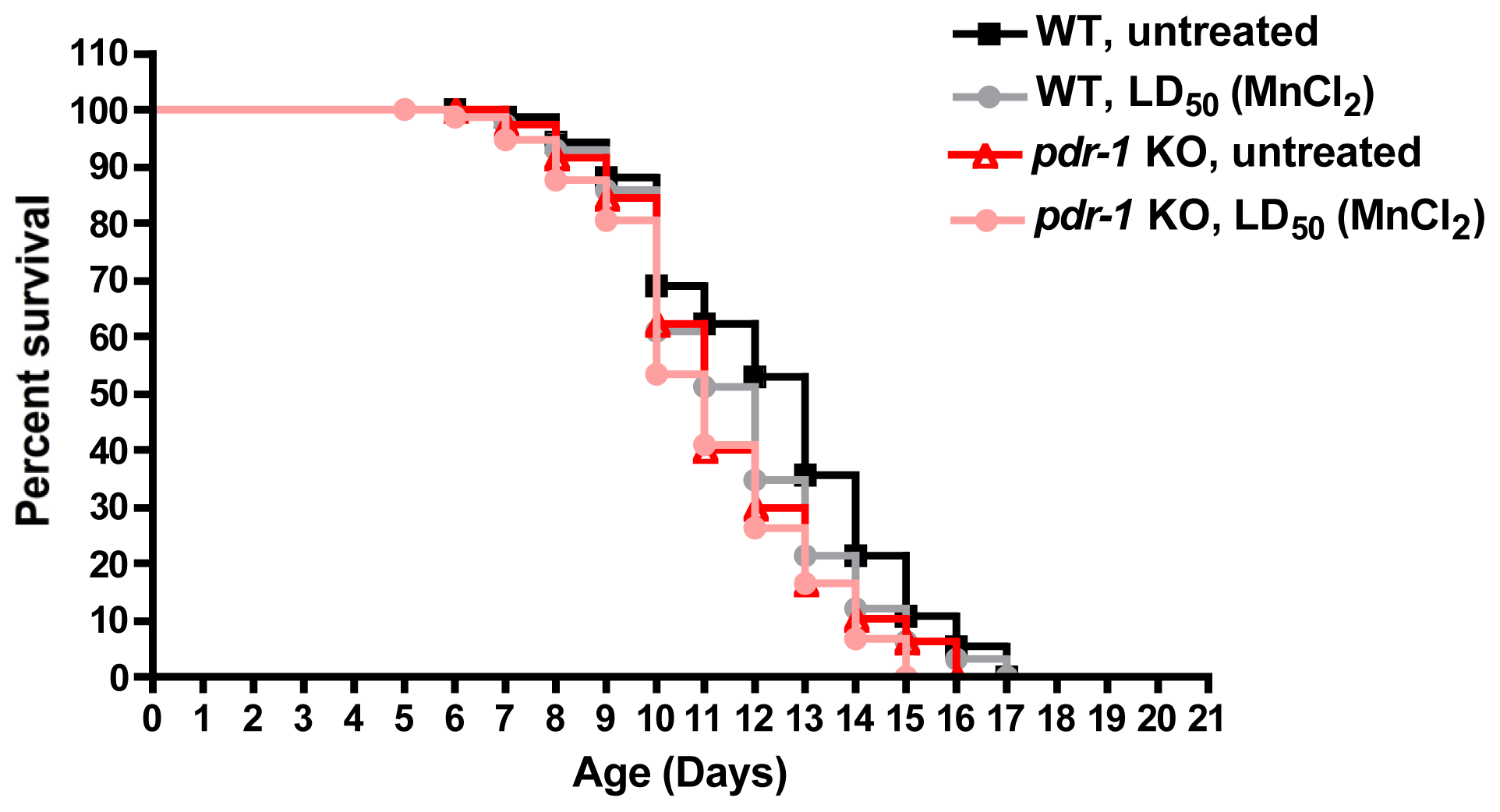
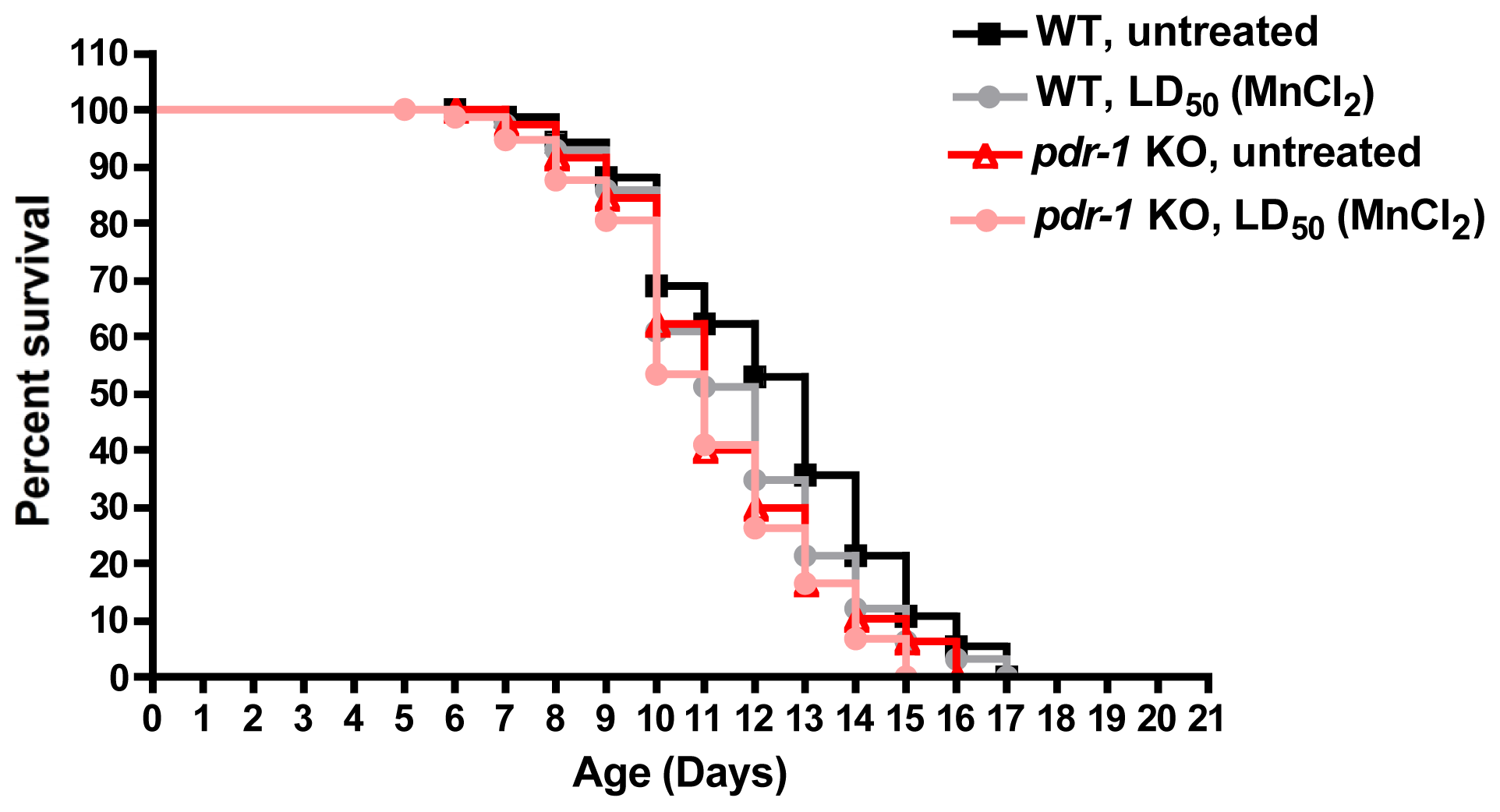
- Martinez-Finley, E.J.; Chakraborty, S.; Slaughter, J.C.; Aschner, M. Early-life exposure to methylmercury in wildtype and PDR-1/parkin knockout C. elegans. Neurochem. Res 2013, 38, 1543–1552. [Google Scholar]
- Chakraborty, S.; Aschner, M.; Vanderbilt, University; Nashville, M.I. Personal communication, 2013.
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar]
- Funayama, M.; Hasegawa, K.; Kowa, H.; Saito, M.; Tsuji, S.; Obata, F. A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann. Neurol 2002, 51, 296–301. [Google Scholar]
- West, A.B.; Moore, D.J.; Biskup, S.; Bugayenko, A.; Smith, W.W.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 16842–16847. [Google Scholar]
- Mortiboys, H.; Johansen, K.K.; Aasly, J.O.; Bandmann, O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 2010, 75, 2017–2020. [Google Scholar]
- Smith, W.W.; Pei, Z.; Jiang, H.; Moore, D.J.; Liang, Y.; West, A.B.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 18676–18681. [Google Scholar]
- Cherra, S.J., 3rd; Steer, E.; Gusdon, A.M.; Kiselyov, K.; Chu, C.T. Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am. J. Pathol 2013, 182, 474–484. [Google Scholar]
- Venderova, K.; Kabbach, G.; Abdel-Messih, E.; Zhang, Y.; Parks, R.J.; Imai, Y.; Gehrke, S.; Ngsee, J.; Lavoie, M.J.; Slack, R.S.; et al. Leucine-rich repeat kinase 2 interacts with parkin, DJ-1 and PINK-1 in a drosophila melanogaster model of Parkinson’s disease. Hum. Mol. Genet 2009, 18, 4390–4404. [Google Scholar]
- Ng, C.H.; Mok, S.Z.; Koh, C.; Ouyang, X.; Fivaz, M.L.; Tan, E.K.; Dawson, V.L.; Dawson, T.M.; Yu, F.; Lim, K.L. Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in drosophila. J. Neurosci 2009, 29, 11257–11262. [Google Scholar]
- Sakaguchi-Nakashima, A.; Meir, J.Y.; Jin, Y.; Matsumoto, K.; Hisamoto, N. LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr. Biol 2007, 17, 592–598. [Google Scholar]
- Saha, S.; Guillily, M.D.; Ferree, A.; Lanceta, J.; Chan, D.; Ghosh, J.; Hsu, C.H.; Segal, L.; Raghavan, K.; Matsumoto, K.; et al. LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J. Neurosci 2009, 29, 9210–9218. [Google Scholar]
- Di Domenico, F.; Sultana, R.; Ferree, A.; Smith, K.; Barone, E.; Perluigi, M.; Coccia, R.; Pierce, W.; Cai, J.; Mancuso, C.; et al. Redox proteomics analyses of the influence of co-expression of wild-type or mutated LRRK2 and Tau on C. elegans protein expression and oxidative modification: Relevance to Parkinson disease. Antioxid. Redox Signal 2012, 17, 1490–1506. [Google Scholar]
- Yao, C.; Johnson, W.M.; Gao, Y.; Wang, W.; Zhang, J.; Deak, M.; Alessi, D.R.; Zhu, X.; Mieyal, J.J.; Roder, H.; et al. Kinase inhibitors arrest neurodegeneration in cell and C. elegans models of LRRK2 toxicity. Hum. Mol. Genet 2013, 22, 328–344. [Google Scholar]
- Kobayashi, M.; Yamamoto, M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv. Enzym. Regul 2006, 46, 113–140. [Google Scholar]
- Lee, J.M.; Li, J.; Johnson, D.A.; Stein, T.D.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Johnson, J.A. Nrf2, a multi-organ protector? FASEB J 2005, 19, 1061–1066. [Google Scholar]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar]
- Chan, K.; Han, X.D.; Kan, Y.W. An important function of Nrf2 in combating oxidative stress: Detoxification of acetaminophen. Proc. Natl. Acad. Sci. USA 2001, 98, 4611–4616. [Google Scholar]
- An, J.H.; Blackwell, T.K. SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev 2003, 17, 1882–1893. [Google Scholar]
- Bishop, N.A.; Guarente, L. Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature 2007, 447, 545–549. [Google Scholar]
- Tullet, J.M.; Hertweck, M.; An, J.H.; Baker, J.; Hwang, J.Y.; Liu, S.; Oliveira, R.P.; Baumeister, R.; Blackwell, T.K. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 2008, 132, 1025–1038. [Google Scholar]
- Pi, J.; Bai, Y.; Reece, J.M.; Williams, J.; Liu, D.; Freeman, M.L.; Fahl, W.E.; Shugar, D.; Liu, J.; Qu, W.; et al. Molecular mechanism of human Nrf2 activation and degradation: Role of sequential phosphorylation by protein kinase CK2. Free Radic. Biol. Med 2007, 42, 1797–1806. [Google Scholar]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med 2009, 47, 1304–1309. [Google Scholar]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem 2003, 278, 21592–21600. [Google Scholar]
- Park, S.K.; Tedesco, P.M.; Johnson, T.E. Oxidative stress and longevity in Caenorhabditis elegans as mediated by SKN-1. Aging Cell 2009, 8, 258–269. [Google Scholar]
- Oliveira, R.P.; Porter Abate, J.; Dilks, K.; Landis, J.; Ashraf, J.; Murphy, C.T.; Blackwell, T.K. Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging Cell 2009, 8, 524–541. [Google Scholar]
- Wang, J.; Robida-Stubbs, S.; Tullet, J.M.; Rual, J.F.; Vidal, M.; Blackwell, T.K. Rnai screening implicates a SKN-1-dependent transcriptional response in stress resistance and longevity deriving from translation inhibition. PLoS Genet 2010, 6, e1001048. [Google Scholar]
- Link, C.D.; Johnson, C.J. Reporter transgenes for study of oxidant stress in Caenorhabditis elegans. Methods Enzymol 2002, 353, 497–505. [Google Scholar]
- Papp, D.; Csermely, P.; Soti, C. A role for SKN-1/Nrf in pathogen resistance and immunosenescence in Caenorhabditis elegans. PLoS Pathog 2012, 8, e1002673. [Google Scholar]
- Hoeven, R.; McCallum, K.C.; Cruz, M.R.; Garsin, D.A. Ce-Duox1/BLI-3 generated reactive oxygen species trigger protective SKN-1 activity via p38 MAPK signaling during infection in C. elegans. PLoS Pathog 2011, 7, e1002453. [Google Scholar]
- An, J.H.; Vranas, K.; Lucke, M.; Inoue, H.; Hisamoto, N.; Matsumoto, K.; Blackwell, T.K. Regulation of the Caenorhabditis elegans oxidative stress defense protein SKN-1 by glycogen synthase kinase-3. Proc. Natl. Acad. Sci. USA 2005, 102, 16275–16280. [Google Scholar]
- Bathla, G.; Hegde, A.N. MRI and CT appearances in metabolic encephalopathies due to systemic diseases in adults. Clin. Radiol 2013, 68, 545–554. [Google Scholar]
- Miller, J.W.; Selhub, J.; Joseph, J.A. Oxidative damage caused by free radicals produced during catecholamine autoxidation: Protective effects of o-methylation and melatonin. Free Radic. Biol. Med 1996, 21, 241–249. [Google Scholar]
- Salazar, J.; Mena, N.; Hunot, S.; Prigent, A.; Alvarez-Fischer, D.; Arredondo, M.; Duyckaerts, C.; Sazdovitch, V.; Zhao, L.; Garrick, L.M.; et al. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2008, 105, 18578–18583. [Google Scholar]
- Worth, P.F. How to treat Parkinson’s disease in 2013. Clin. Med 2013, 13, 93–96. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chakraborty, S.; Bornhorst, J.; Nguyen, T.T.; Aschner, M. Oxidative Stress Mechanisms Underlying Parkinson’s Disease-Associated Neurodegeneration in C. elegans. Int. J. Mol. Sci. 2013, 14, 23103-23128. https://doi.org/10.3390/ijms141123103
Chakraborty S, Bornhorst J, Nguyen TT, Aschner M. Oxidative Stress Mechanisms Underlying Parkinson’s Disease-Associated Neurodegeneration in C. elegans. International Journal of Molecular Sciences. 2013; 14(11):23103-23128. https://doi.org/10.3390/ijms141123103
Chicago/Turabian StyleChakraborty, Sudipta, Julia Bornhorst, Thuy T. Nguyen, and Michael Aschner. 2013. "Oxidative Stress Mechanisms Underlying Parkinson’s Disease-Associated Neurodegeneration in C. elegans" International Journal of Molecular Sciences 14, no. 11: 23103-23128. https://doi.org/10.3390/ijms141123103