How Parkinsonian Toxins Dysregulate the Autophagy Machinery



Abstract
:
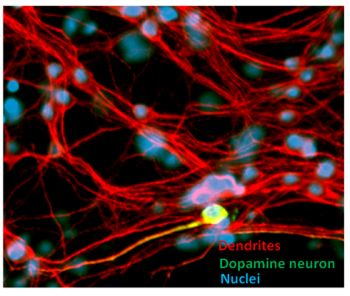{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Overview on the Selective Sensitivity of Dopamine Neurons to Oxidative Stress
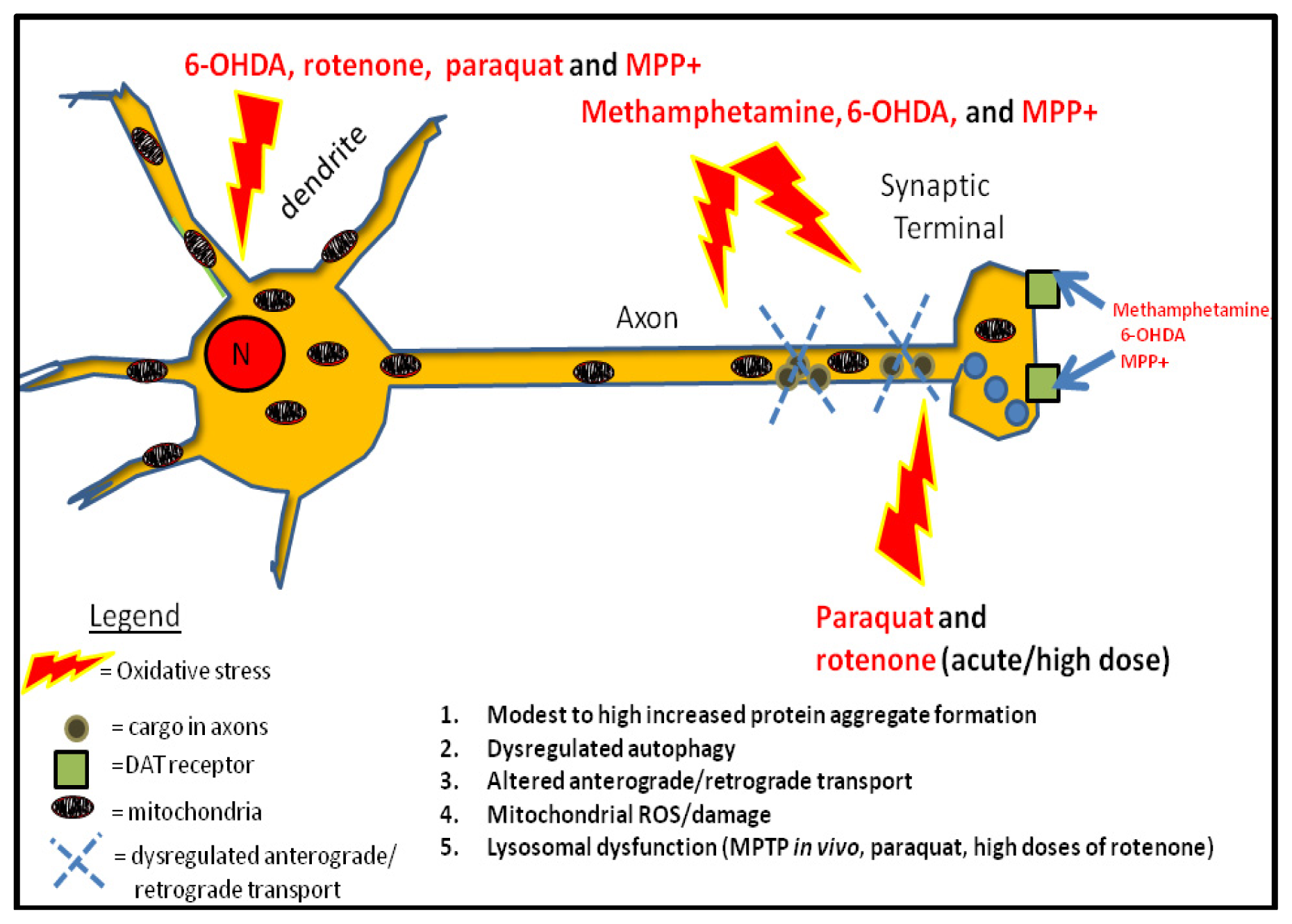
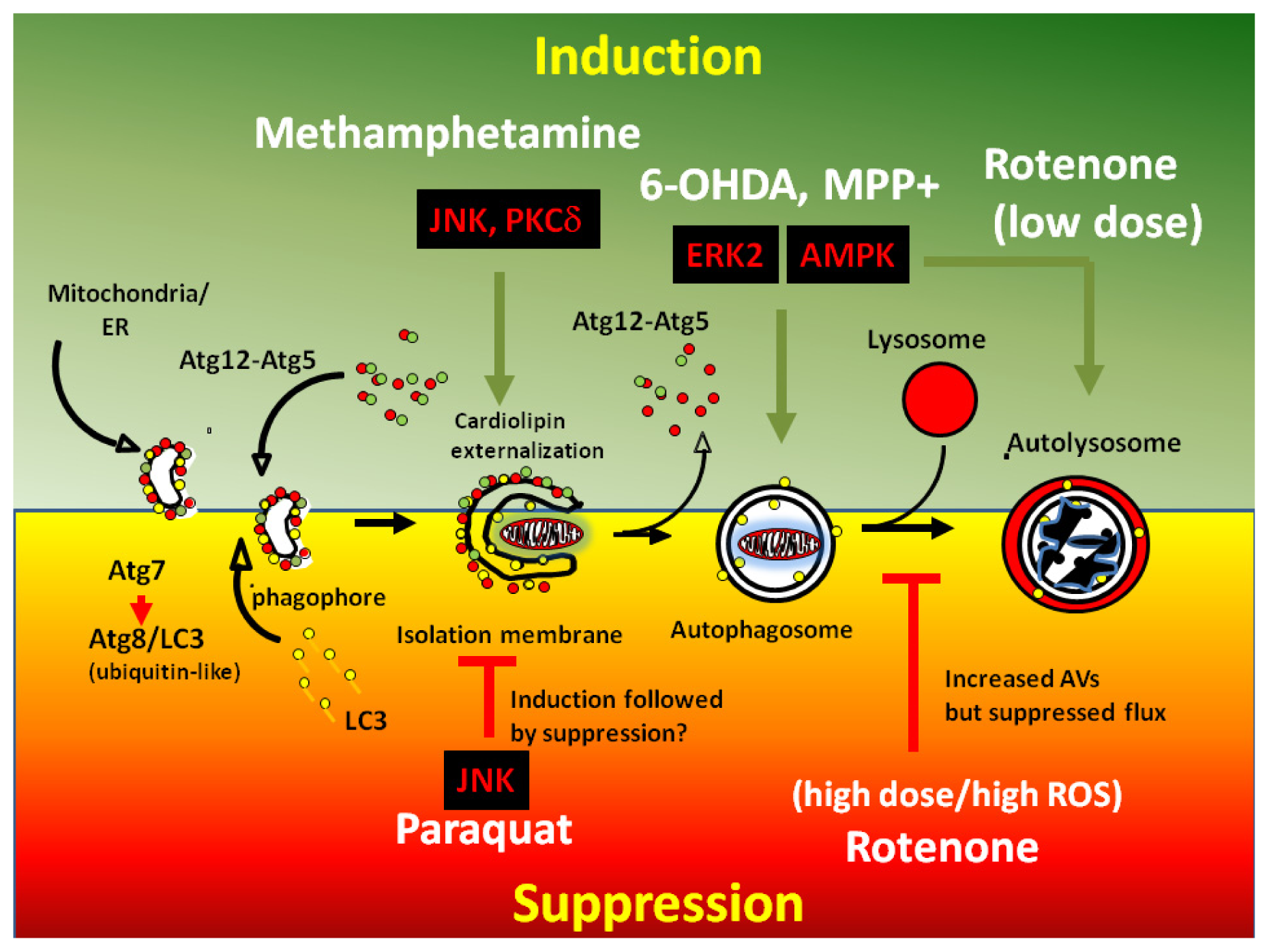
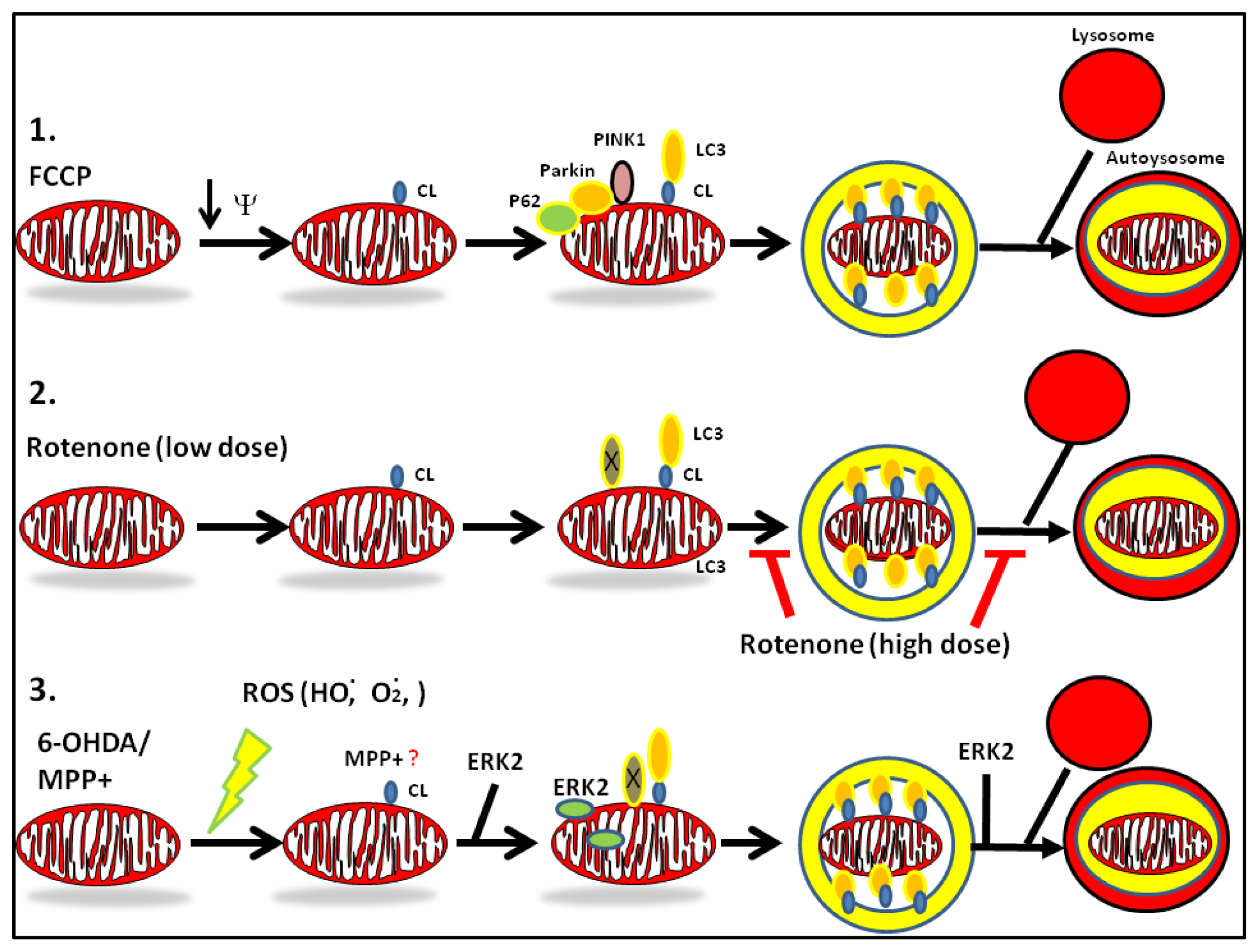
2. Mechanisms of Dysregulated Autophagy and Mitophagy by Parkinsonian Toxins
3. Convergence of PD Toxins and Genetic Models of PD: Role of ROS
4. PD Toxins and Their Mechanisms of Autophagy
4.1. MPTP
Mechanisms of Dysregulation of Autophagy by MPP+
4.2. Rotenone
Mechanisms of Dysregulation of Autophagy by Rotenone
4.3. 6-Hydroxydopamine (6-OHDA)
Mechanisms of Dysregulation of Autophagy by 6-OHDA
4.4. Paraquat
4.5. Methamphetamine
5. Environmental Exposure and Prevention
6. Concluding Remarks

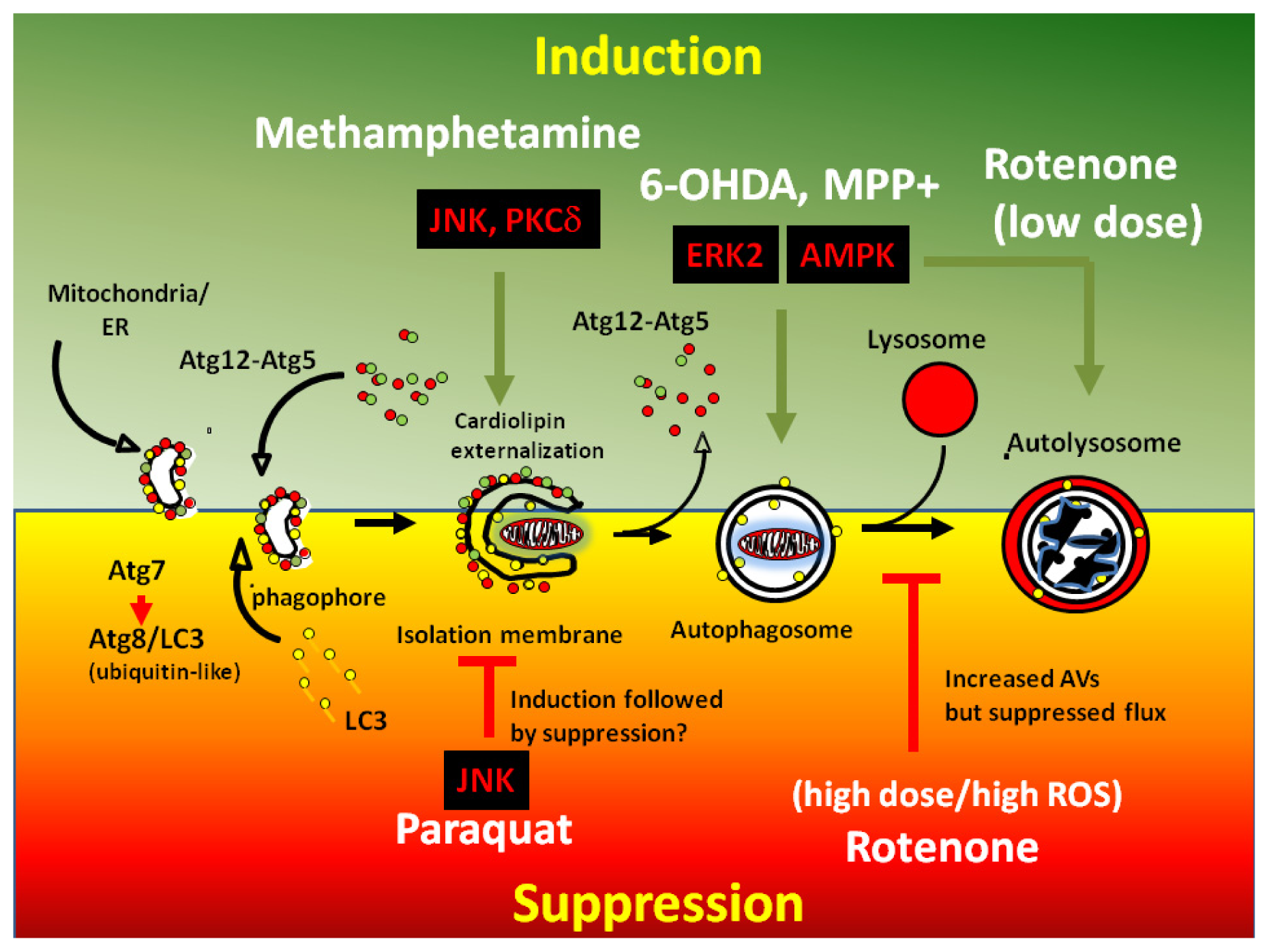

Acknowledgments
Conflicts of Interest
References
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar]
- Farre, J.C.; Subramani, S. Peroxisome turnover by micropexophagy: An autophagy-related process. Trends Cell Biol 2004, 14, 515–523. [Google Scholar]
- Narendra, D.; Kane, L.A.; Hauser, D.N.; Fearnley, I.M.; Youle, R.J. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010, 6, 1090–1106. [Google Scholar]
- Bernales, S.; Schuck, S.; Walter, P. ER-phagy: Selective autophagy of the endoplasmic reticulum. Autophagy 2007, 3, 285–287. [Google Scholar]
- Dagda, R.K.; Zhu, J.; Kulich, S.M.; Chu, C.T. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: Implications for Parkinson’s disease. Autophagy 2008, 4, 770–782. [Google Scholar]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol 2010, 12, 119–131. [Google Scholar]
- Gusdon, A.M.; Chu, C.T. To eat or not to eat: Neuronal metabolism, mitophagy, and Parkinson’s disease. Antioxid. Redox Signal 2011, 14, 1979–1987. [Google Scholar]
- Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway: A mitochondrial quality control system? J. Bioenerg. Biomembr 2009, 41, 499–503. [Google Scholar]
- Komatsu, M.; Wang, Q.J.; Holstein, G.R.; Friedrich, V.L., Jr.; Iwata, J.; Kominami, E.; Chait, B.T.; Tanaka, K.; Yue, Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 14489–14494. [Google Scholar]
- Nishiyama, J.; Miura, E.; Mizushima, N.; Watanabe, M.; Yuzaki, M. Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy 2007, 3, 591–596. [Google Scholar]
- Cherra, S.J.; Chu, C.T. Autophagy in neuroprotection and neurodegeneration: A question of balance. Future Neurol 2008, 3, 309–323. [Google Scholar]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet 2002, 11, 1107–1117. [Google Scholar]
- Yu, W.H.; Kumar, A.; Peterhoff, C.; Shapiro Kulnane, L.; Uchiyama, Y.; Lamb, B.T.; Cuervo, A.M.; Nixon, R.A. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: Implications for beta-amyloid peptide over-production and localization in Alzheimer’s disease. Int. J. Biochem. Cell Biol 2004, 36, 2531–2540. [Google Scholar]
- Zhu, J.-H.; Guo, F.; Shelburne, J.; Watkins, S.; Chu, C.T. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol 2003, 13, 473–481. [Google Scholar]
- Wong, A.S.; Lee, R.H.; Cheung, A.Y.; Yeung, P.K.; Chung, S.K.; Cheung, Z.H.; Ip, N.Y. Cdk5-mediated phosphorylation of endophilin B1 is required for induced autophagy in models of Parkinson’s disease. Nat. Cell Biol 2011, 13, 568–579. [Google Scholar]
- Zhu, J.H.; Horbinski, C.; Guo, F.; Watkins, S.; Uchiyama, Y.; Chu, C.T. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am. J. Pathol 2007, 170, 75–86. [Google Scholar]
- Zhu, J.H.; Gusdon, A.M.; Cimen, H.; van Houten, B.; Koc, E.; Chu, C.T. Impaired mitochondrial biogenesis contributes to depletion of functional mitochondria in chronic MPP+ toxicity: Dual roles for ERK1/2. Cell Death Dis 2012, 3, e312. [Google Scholar]
- Wills, J.; Credle, J.; Oaks, A.W.; Duka, V.; Lee, J.H.; Jones, J.; Sidhu, A. Paraquat, but not maneb, induces synucleinopathy and tauopathy in striata of mice through inhibition of proteasomal and autophagic pathways. PLoS One 2012, 7, e30745. [Google Scholar]
- Mak, S.K.; McCormack, A.L.; Manning-Bog, A.B.; Cuervo, A.M.; di Monte, D.A. Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem. 2010, 285, 13621–13629. [Google Scholar]
- Zhu, J.; Wang, K.Z.; Chu, C.T. After the banquet: Mitochondrial biogenesis, mitophagy and cell survival. Autophagy 2013, in press. [Google Scholar]
- Abeliovich, A.; Flint Beal, M. Parkinsonism genes: Culprits and clues. J. Neurochem 2006, 99, 1062–1072. [Google Scholar]
- Gelb, D.J.; Oliver, E.; Gilman, S. Diagnostic criteria for Parkinson disease. Arch. Neurol 1999, 56, 33–39. [Google Scholar]
- Gandhi, S.; Muqit, M.M.; Stanyer, L.; Healy, D.G.; Abou-Sleiman, P.M.; Hargreaves, I.; Heales, S.; Ganguly, M.; Parsons, L.; Lees, A.J.; et al. PINK1 protein in normal human brain and Parkinson’s disease. Brain 2006, 129, 1720–1731. [Google Scholar]
- Cookson, M.R. The biochemistry of Parkinson’s disease. Annu. Rev. Biochem 2005, 74, 29–52. [Google Scholar]
- Dagda, R.K.; Chu, C.T. Mitochondrial quality control: Insights on how Parkinson’s disease related genes PINK1, parkin, and Omi/HtrA2 interact to maintain mitochondrial homeostasis. J. Bioenerg. Biomembr 2009, 41, 473–479. [Google Scholar]
- Liang, C.L.; Wang, T.T.; Luby-Phelps, K.; German, D.C. Mitochondria mass is low in mouse substantia nigra dopamine neurons: Implications for Parkinson’s disease. Exp. Neurol 2007, 203, 370–380. [Google Scholar]
- Cherra, S.J., III; Dagda, R.K.; Tandon, A.; Chu, C.T. Mitochondrial autophagy as a compensatory response to PINK1 deficiency. Autophagy 2009, 5, 1213–1214. [Google Scholar]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A., Jr.; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 2003, 5, 539–545. [Google Scholar]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar]
- Nazarko, V.Y.; Zhong, Q. ULK1 targets Beclin-1 in autophagy. Nat. Cell Biol 2013, 15, 727–728. [Google Scholar]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev 2011, 12, 9–14. [Google Scholar]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010, 8, e1000298. [Google Scholar]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol 2008, 183, 795–803. [Google Scholar]
- Kawajiri, S.; Saiki, S.; Sato, S.; Sato, F.; Hatano, T.; Eguchi, H.; Hattori, N. PINK1 is recruited to mitochondria with parkin and associates with LC3 in mitophagy. FEBS Lett 2010, 584, 1073–1079. [Google Scholar]
- Van Laar, V.S.; Berman, S.B. Mitochondrial dynamics in Parkinson’s disease. Exp. Neurol 2009, 218, 247–256. [Google Scholar]
- Grenier, K.; McLelland, G.L.; Fon, E.A. Parkin- and PINK1-dependent mitophagy in neurons: Will the real pathway please stand up? Front. Neurol 2013, 4, 100. [Google Scholar]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol 2013. [Google Scholar] [CrossRef]
- Dagda, R.K.; Cherra, S.J., III; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem 2009, 284, 13843–13855. [Google Scholar]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet 2011, 20, 867–879. [Google Scholar]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar]
- Dehay, B.; Bove, J.; Rodriguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci 2010, 30, 12535–12544. [Google Scholar]
- Gonzalez-Polo, R.A.; Niso-Santano, M.; Ortiz-Ortiz, M.A.; Gomez-Martin, A.; Moran, J.M.; Garcia-Rubio, L.; Francisco-Morcillo, J.; Zaragoza, C.; Soler, G.; Fuentes, J.M. Inhibition of paraquat-induced autophagy accelerates the apoptotic cell death in neuroblastoma SH-SY5Y cells. Toxicol. Sci 2007, 97, 448–458. [Google Scholar]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neuro-Signals 2011, 19, 163–174. [Google Scholar]
- Wu, Y.; Li, X.; Xie, W.; Jankovic, J.; Le, W.; Pan, T. Neuroprotection of deferoxamine on rotenone-induced injury via accumulation of HIF-1 alpha and induction of autophagy in SH-SY5Y cells. Neurochem. Int 2011, 57, 198–205. [Google Scholar]
- Pan, T.; Rawal, P.; Wu, Y.; Xie, W.; Jankovic, J.; Le, W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 2009, 164, 541–551. [Google Scholar]
- Dadakhujaev, S.; Noh, H.S.; Jung, E.J.; Cha, J.Y.; Baek, S.M.; Ha, J.H.; Kim, D.R. Autophagy protects the rotenone-induced cell death in alpha-synuclein overexpressing SH-SY5Y cells. Neurosci. Lett 2010, 472, 47–52. [Google Scholar]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J. Cell Sci 2007, 120, 4155–4166. [Google Scholar]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys 2007, 462, 245–253. [Google Scholar]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 2005, 8, 3–5. [Google Scholar]
- Chu, C.T. Diversity in the regulation of autophagy and mitophagy: lessons from Parkinson’s disease. Parkinsons Dis 2011. [Google Scholar] [CrossRef]
- Plowey, E.D.; Cherra, S.J., III; Liu, Y.J.; Chu, C.T. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J. Neurochem 2008, 105, 1048–1056. [Google Scholar]
- Krebiehl, G.; Ruckerbauer, S.; Burbulla, L.F.; Kieper, N.; Maurer, B.; Waak, J.; Wolburg, H.; Gizatullina, Z.; Gellerich, F.N.; Woitalla, D.; et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS One 2010, 5, e9367. [Google Scholar]
- Grunewald, A.; Arns, B.; Seibler, P.; Rakovic, A.; Munchau, A.; Ramirez, A.; Sue, C.M.; Klein, C. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol. Aging 2012, 33, e1841–e1847. [Google Scholar]
- Gusdon, A.M.; Zhu, J.; van Houten, B.; Chu, C.T. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol. Dis 2012, 45, 962–972. [Google Scholar]
- Janda, E.; Isidoro, C.; Carresi, C.; Mollace, V. Defective autophagy in Parkinson’s disease: Role of oxidative stress. Mol. Neurobiol 2012, 46, 639–661. [Google Scholar]
- Chu, C.T.; Levinthal, D.J.; Kulich, S.M.; Chalovich, E.M.; DeFranco, D.B. Oxidative neuronal injury. The dark side of ERK1/2. Eur. J. Biochem 2004, 271, 2060–2066. [Google Scholar]
- Kulich, S.M.; Horbinski, C.; Patel, M.; Chu, C.T. 6-Hydroxydopamine induces mitochondrial ERK activation. Free Radic. Biol. Med 2007, 43, 372–383. [Google Scholar]
- Siegel, G.; Agranoff, R.; Albers, W.; Fisher, S.K.; Uhler, M. Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed; Lippincot-Raven: Philadelphia, PA, USA, 1999; pp. 761–780. [Google Scholar]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar]
- Bove, J.; Prou, D.; Perier, C.; Przedborski, S. Toxin-induced models of Parkinson’s disease. NeuroRx 2005, 2, 484–494. [Google Scholar]
- Buckman, T.D. Toxicity of MPTP and structural analogs in clonal cell lines of neuronal origin expressing B type monoamine oxidase activity. Mol. Chem. Neuropathol 1991, 15, 87–102. [Google Scholar]
- Mihatsch, W.; Russ, H.; Przuntek, H. Intracerebroventricular administration of 1-methyl-4-phenylpyridinium ion in mice: Effects of simultaneously administered nomifensine, deprenyl, and 1-t-butyl-4,4-diphenylpiperidine. J. Neural. Transm 1988, 71, 177–188. [Google Scholar]
- Michel, P.P.; Hefti, F. Toxicity of 6-hydroxydopamine and dopamine for dopaminergic neurons in culture. J. Neurosci. Res 1990, 26, 428–435. [Google Scholar]
- Chiasson, K.; Daoust, B.; Levesque, D.; Martinoli, M.G. Dopamine D2 agonists, bromocriptine and quinpirole, increase MPP+-induced toxicity in PC12 cells. Neurotox. Res 2006, 10, 31–42. [Google Scholar]
- Choi, W.S.; Kruse, S.E.; Palmiter, R.D.; Xia, Z. Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP+, or paraquat. Proc. Natl. Acad. Sci. USA 2008, 105, 15136–15141. [Google Scholar]
- Singer, T.P.; Ramsay, R.R.; McKeown, K.; Revor, A.; Castagnoli, N.E.J. Mechanism of the neurotoxicity of 1-methyl-4-phenylpyridinium (MPP+), the toxic bioactivation product of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicology 1988, 49, 17–23. [Google Scholar]
- Holtz, W.A.; O’Malley, K.L. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J. Biol. Chem 2003, 278, 19367–19377. [Google Scholar]
- Kalivendi, S.V.; Cunningham, S.; Kotamraju, S.; Joseph, J.; Hillard, C.J.; Kalyanaraman, B. Alpha-synuclein up-regulation and aggregation during MPP+-induced apoptosis in neuroblastoma cells: Intermediacy of transferrin receptor iron and hydrogen peroxide. J. Biol. Chem 2004, 279, 15240–15247. [Google Scholar]
- Lee, C.S.; Park, W.J.; Ko, H.H.; Han, E.S. Differential involvement of mitochondrial permeability transition in cytotoxicity of 1-methyl-4-phenylpyridinium and 6-hydroxydopamine. Mol. Cell. Biochem 2006, 289, 193–200. [Google Scholar]
- Bove, J.; Martinez-Vicente, M.; Vila, M. Fighting neurodegeneration with rapamycin: Mechanistic insights. Nat. Rev. Neurosci 2011, 12, 437–452. [Google Scholar]
- Lim, J.; Kim, H.W.; Youdim, M.B.; Rhyu, I.J.; Choe, K.M.; Oh, Y.J. Binding preference of p62 towards LC3-ll during dopaminergic neurotoxin-induced impairment of autophagic flux. Autophagy 2011, 7, 51–60. [Google Scholar]
- Liu, K.; Shi, N.; Sun, Y.; Zhang, T.; Sun, X. Therapeutic effects of rapamycin on MPTP-induced Parkinsonism in mice. Neurochem. Res 2013, 38, 201–207. [Google Scholar]
- Cherra, S.J., 3rd; Kulich, S.M.; Uechi, G.; Balasubramani, M.; Mountzouris, J.; Day, B.W.; Chu, C.T. Regulation of the autophagy protein LC3 by phosphorylation. J. Cell Biol 2010, 190, 533–539. [Google Scholar]
- Jiang, H.; Cheng, D.; Liu, W.; Peng, J.; Feng, J. Protein kinase C inhibits autophagy and phosphorylates LC3. Biochem. Biophys. Res. Commun 2010, 395, 471–476. [Google Scholar]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci 2000, 3, 1301–1306. [Google Scholar]
- Sherer, T.B.; Betarbet, R.; Stout, A.K.; Lund, S.; Baptista, M.; Panov, A.V.; Cookson, M.R.; Greenamyre, J.T. An in vitro model of Parkinson’s disease: Linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J. Neurosci 2002, 22, 7006–7015. [Google Scholar]
- Metcalf, R. The mode of action of organic insecticides. Chem. Coordication Center Natl. Res. Counc 1948, 84, 29–35. [Google Scholar]
- Inden, M.; Kitamura, Y.; Abe, M.; Tamaki, A.; Takata, K.; Taniguchi, T. Parkinsonian rotenone mouse model: Reevaluation of long-term administration of rotenone in C57BL/6 mice. Biol. Pharm. Bull 2011, 34, 92–96. [Google Scholar]
- Pei, W.; Liou, A.K.; Chen, J. Two caspase-mediated apoptotic pathways induced by rotenone toxicity in cortical neuronal cells. FASEB J 2003, 17, 520–522. [Google Scholar]
- Sonia Angeline, M.; Chaterjee, P.; Anand, K.; Ambasta, R.K.; Kumar, P. Rotenone-induced parkinsonism elicits behavioral impairments and differential expression of parkin, heat shock proteins and caspases in the rat. Neuroscience 2011, 220, 291–301. [Google Scholar]
- Gao, H.M.; Hong, J.S.; Zhang, W.; Liu, B. Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J. Neurosci 2002, 22, 782–790. [Google Scholar]
- Filomeni, G.; Graziani, I.; de Zio, D.; Dini, L.; Centonze, D.; Rotilio, G.; Ciriolo, M.R. Neuroprotection of kaempferol by autophagy in models of rotenone-mediated acute toxicity: Possible implications for Parkinson’s disease. Neurobiol. Aging 2012, 33, 767–785. [Google Scholar]
- Mader, B.J.; Pivtoraiko, V.N.; Flippo, H.M.; Klocke, B.J.; Roth, K.A.; Mangieri, L.R.; Shacka, J.J. Rotenone inhibits autophagic flux prior to inducing cell death. ACS Chem. Neurosci 2012, 3, 1063–1072. [Google Scholar]
- Dranka, B.P.; Zielonka, J.; Kanthasamy, A.G.; Kalyanaraman, B. Alterations in bioenergetic function induced by Parkinson’s disease mimetic compounds: Lack of correlation with superoxide generation. J. Neurochem 2012, 122, 941–951. [Google Scholar]
- Storch, A.; Kaftan, A.; Burkhardt, K.; Schwarz, J. 6-Hydroxydopamine toxicity towards human SH-SY5Y dopaminergic neuroblastoma cells: Independent of mitochondrial energy metabolism. J. Neural. Transm 2000, 107, 281–293. [Google Scholar]
- Jameson, G.N.L.; Linert, W. 6-Hydroxydopamine, Dopamine, and Ferritin: A Cycle of Reactions Sustaining Parkinson’s Disease? In Free Radicals in Brain Pathophysiology; Poli, G., Cadenas, E., Packer, L., Eds.; Marcel Dekker, Inc: New York, NY, USA, 2000; Volume 5, pp. 247–272. [Google Scholar]
- Lotharius, J.; Dugan, L.L.; O’Malley, K.L. Distinct mechanisms underlie neurotoxin-mediated cell death in cultured dopaminergic neurons. J. Neurosci 1999, 19, 1284–1293. [Google Scholar]
- Kulich, S.M.; Chu, C.T. Sustained extracellular signal-regulated kinase activation by 6-hydroxydopamine: Implications for Parkinson’s disease. J. Neurochem 2001, 77, 1058–1066. [Google Scholar]
- Gomez-Lazaro, M.; Bonekamp, N.A.; Galindo, M.F.; Jordan, J.; Schrader, M. 6-Hydroxydopamine (6-OHDA) induces Drp1-dependent mitochondrial fragmentation in SH-SY5Y cells. Free Radic. Biol. Med 2008, 44, 1960–1969. [Google Scholar]
- Chalovich, E.M.; Zhu, J.H.; Caltagarone, J.; Bowser, R.; Chu, C.T. Functional repression of cAMP response element in 6-hydroxydopamine-treated neuronal cells. J. Biol. Chem 2006, 281, 17870–17881. [Google Scholar]
- Patel, V.P.; Defranco, D.B.; Chu, C.T. Altered transcription factor trafficking in oxidatively-stressed neuronal cells. Biochim. Biophys. Acta 2012, 1822, 1773–1782. [Google Scholar]
- Blum, D.; Torch, S.; Nissou, M.F.; Benabid, A.L.; Verna, J.M. Extracellular toxicity of 6-hydroxydopamine on PC12 cells. Neurosci. Lett 2000, 283, 193–196. [Google Scholar]
- Bensadoun, J.C.; Mirochnitchenko, O.; Inouye, M.; Aebischer, P.; Zurn, A.D. Attenuation of 6-OHDA-induced neurotoxicity in glutathione peroxidase transgenic mice. Eur. J. Neurosci 1998, 10, 3231–3236. [Google Scholar]
- Cheng, H.C.; Kim, S.R.; Oo, T.F.; Kareva, T.; Yarygina, O.; Rzhetskaya, M.; Wang, C.; During, M.; Talloczy, Z.; Tanaka, K.; et al. Akt suppresses retrograde degeneration of dopaminergic axons by inhibition of macroautophagy. J. Neurosci 2011, 31, 2125–2135. [Google Scholar]
- Arsikin, K.; Kravic-Stevovic, T.; Jovanovic, M.; Ristic, B.; Tovilovic, G.; Zogovic, N.; Bumbasirevic, V.; Trajkovic, V.; Harhaji-Trajkovic, L. Autophagy-dependent and -independent involvement of AMP-activated protein kinase in 6-hydroxydopamine toxicity to SH-SY5Y neuroblastoma cells. Biochim. Biophys. Acta 2012, 1822, 1826–1836. [Google Scholar]
- Tovilovic, G.; Zogovic, N.; Soskic, V.; Schrattenholz, A.; Kostic-Rajacic, S.; Misirkic-Marjanovic, M.; Janjetovic, K.; Vucicevic, L.; Arsikin, K.; Harhaji-Trajkovic, L.; et al. Arylpiperazine-mediated activation of Akt protects SH-SY5Y neuroblastoma cells from 6-hydroxydopamine-induced apoptotic and autophagic death. Neuropharmacology 2013, 72, 224–235. [Google Scholar]
- Liou, H.H.; Tsai, M.C.; Chen, C.J.; Jeng, J.S.; Chang, Y.C.; Chen, S.Y.; Chen, R.C. Environmental risk factors and Parkinson’s disease: A case-control study in Taiwan. Neurology 1997, 48, 1583–1588. [Google Scholar]
- Tawara, T.; Fukushima, T.; Hojo, N.; Isobe, A.; Shiwaku, K.; Setogawa, T.; Yamane, Y. Effects of paraquat on mitochondrial electron transport system and catecholamine contents in rat brain. Arch. Toxicol 1996, 70, 585–589. [Google Scholar]
- Takamura, A.; Higaki, K.; Kajimaki, K.; Otsuka, S.; Ninomiya, H.; Matsuda, J.; Ohno, K.; Suzuki, Y.; Nanba, E. Enhanced autophagy and mitochondrial aberrations in murine G(M1)-gangliosidosis. Biochem. Biophys. Res. Commun 2008, 367, 616–622. [Google Scholar]
- Manning-Bog, A.B.; McCormack, A.L.; Purisai, M.G.; Bolin, L.M.; di Monte, D.A. Alpha-synuclein overexpression protects against paraquat-induced neurodegeneration. J. Neurosci 2003, 23, 3095–3099. [Google Scholar]
- Manning-Bog, A.B.; McCormack, A.L.; Li, J.; Uversky, V.N.; Fink, A.L.; di Monte, D.A. The herbicide paraquat causes up-regulation and aggregation of α-synuclein in mice: Paraquat and α-synuclein. J. Biol. Chem 2002, 277, 1641–1644. [Google Scholar]
- Garcia-Garcia, A.; Annandurai, A.; Burns, M.; Chen, H.; Zhou, Y.; Franco, R. Impairment of Atg5-dependent autophagic flux promotes paraquat- and MPP+-induced apoptosis but not rotenone or 6-hydroxydopamine toxicity. Toxicol. Sci 2013. [Google Scholar] [CrossRef]
- Janda, E.; Parafati, M.; Aprigliano, S.; Carresi, C.; Visalli, V.; Sacco, I.; Ventrice, D.; Mega, T.; Vadala, N.; Rinaldi, S.; et al. The antidote effect of quinone oxidoreductase 2 inhibitor against paraquat-induced toxicity in vitro and in vivo. Br. J. Pharmacol. 2013, 168, 46–59. [Google Scholar]
- Ravikumar, B.; Berger, Z.; Vacher, C.; O’Kane, C.J.; Rubinsztein, D.C. Rapamycin pre-treatment protects against apoptosis. Hum. Mol. Genet 2006, 15, 1209–1216. [Google Scholar]
- Gonzalez-Polo, R.A.; Boya, P.; Pauleau, A.L.; Jalil, A.; Larochette, N.; Souquere, S.; Eskelinen, E.L.; Pierron, G.; Saftig, P.; Kroemer, G. The apoptosis/autophagy paradox: Autophagic vacuolization before apoptotic death. J. Cell Sci 2005, 118, 3091–3102. [Google Scholar]
- Wang, C.; Ko, H.S.; Thomas, B.; Tsang, F.; Chew, K.C.; Tay, S.P.; Ho, M.W.; Lim, T.M.; Soong, T.W.; Pletnikova, O.; et al. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin’s protective function. Hum. Mol. Genet 2005, 14, 3885–3897. [Google Scholar]
- Thrash, B.; Thiruchelvan, K.; Ahuja, M.; Suppiramaniam, V.; Dhanasekaran, M. Methamphetamine-induced neurotoxicity: The road to Parkinson’s disease. Pharmacol. Rep 2009, 61, 966–977. [Google Scholar]
- Xie, Z.; Miller, G.M. A receptor mechanism for methamphetamine action in dopamine transporter regulation in brain. J. Pharmacol. Exp. Ther 2009, 330, 316–325. [Google Scholar]
- Brown, J.M.; Hanson, G.R.; Fleckenstein, A.E. Regulation of the vesicular monoamine transporter-2: A novel mechanism for cocaine and other psychostimulants. J. Pharmacol. Exp. Ther 2001, 296, 762–767. [Google Scholar]
- Brown, J.M.; Hanson, G.R.; Fleckenstein, A.E. Methamphetamine rapidly decreases vesicular dopamine uptake. J. Neurochem 2000, 74, 2221–2223. [Google Scholar]
- Berman, S.B.; Hastings, T.G. Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: Implications for Parkinson’s disease. J. Neurochem 1999, 73, 1127–1137. [Google Scholar]
- Dawson, R., Jr.; Beal, M.F.; Bondy, S.C.; di Monte, D.A.; Isom, G.E. Excitotoxins, aging, and environmental neurotoxins: Implications for understanding human neurodegenerative diseases. Toxicol. Appl. Pharmacol 1995, 134, 1–17. [Google Scholar]
- Cubells, J.F.; Rayport, S.; Rajendran, G.; Sulzer, D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J. Neurosci 1994, 14, 2260–2271. [Google Scholar]
- Pasquali, L.; Lazzeri, G.; Isidoro, C.; Ruggieri, S.; Paparelli, A.; Fornai, F. Role of autophagy during methamphetamine neurotoxicity. Ann. N. Y. Acad. Sci 2008, 1139, 191–196. [Google Scholar]
- Castino, R.; Lazzeri, G.; Lenzi, P.; Bellio, N.; Follo, C.; Ferrucci, M.; Fornai, F.; Isidoro, C. Suppression of autophagy precipitates neuronal cell death following low doses of methamphetamine. J. Neurochem 2008, 106, 1426–1439. [Google Scholar]
- Kanthasamy, A.; Anantharam, V.; Ali, S.F.; Kanthasamy, A.G. Methamphetamine induces autophagy and apoptosis in a mesencephalic dopaminergic neuronal culture model: Role of cathepsin-D in methamphetamine-induced apoptotic cell death. Ann. N. Y. Acad. Sci 2006, 1074, 234–244. [Google Scholar]
- Larsen, K.E.; Fon, E.A.; Hastings, T.G.; Edwards, R.H.; Sulzer, D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J. Neurosci 2002, 22, 8951–8960. [Google Scholar]
- Nopparat, C.; Porter, J.E.; Ebadi, M.; Govitrapong, P. The mechanism for the neuroprotective effect of melatonin against methamphetamine-induced autophagy. J. Pineal Res 2010, 49, 382–389. [Google Scholar]
- Moszczynska, A.; Yamamoto, B.K. Methamphetamine oxidatively damages parkin and decreases the activity of 26S proteasome in vivo. J. Neurochem. 2011, 116, 1005–1017. [Google Scholar]
- Liu, B.; Traini, R.; Killinger, B.; Schneider, B.; Moszczynska, A. Overexpression of parkin in the rat nigrostriatal dopamine system protects against methamphetamine neurotoxicity. Exp. Neurol 2013, 247, 359–372. [Google Scholar]
- Lin, M.; Chandramani-Shivalingappa, P.; Jin, H.; Ghosh, A.; Anantharam, V.; Ali, S.; Kanthasamy, A.G.; Kanthasamy, A. Methamphetamine-induced neurotoxicity linked to ubiquitin-proteasome system dysfunction and autophagy-related changes that can be modulated by protein kinase C delta in dopaminergic neuronal cells. Neuroscience 2012, 210, 308–332. [Google Scholar]
- Ascherio, A.; Chen, H.; Weisskopf, M.G.; O’Reilly, E.; McCullough, M.L.; Calle, E.E.; Schwarzschild, M.A.; Thun, M.J. Pesticide exposure and risk for Parkinson’s disease. Ann. Neurol 2006, 60, 197–203. [Google Scholar]
- Costello, S.; Cockburn, M.; Bronstein, J.; Zhang, X.; Ritz, B. Parkinson’s disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am. J. Epidemiol 2009, 169, 919–926. [Google Scholar]
- Wang, A.; Costello, S.; Cockburn, M.; Zhang, X.; Bronstein, J.; Ritz, B. Parkinson’s disease risk from ambient exposure to pesticides. Eur. J. Epidemiol 2011, 26, 547–555. [Google Scholar]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect 2011, 119, 866–872. [Google Scholar]
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Kortsha, G.X.; Brown, G.G.; Richardson, R.J. Occupational exposures to metals as risk factors for Parkinson’s disease. Neurology 1997, 48, 650–658. [Google Scholar]
- Coon, S.; Stark, A.; Peterson, E.; Gloi, A.; Kortsha, G.; Pounds, J.; Chettle, D.; Gorell, J. Whole-body lifetime occupational lead exposure and risk of Parkinson’s disease. Environ. Health Perspect 2006, 114, 1872–1876. [Google Scholar]
- Firestone, J.A.; Lundin, J.I.; Powers, K.M.; Smith-Weller, T.; Franklin, G.M.; Swanson, P.D.; Longstreth, W.T., Jr.; Checkoway, H. Occupational factors and risk of Parkinson’s disease: A population-based case-control study. Am. J. Ind. Med 2010, 53, 217–223. [Google Scholar]
- Afeseh Ngwa, H.; Kanthasamy, A.; Gu, Y.; Fang, N.; Anantharam, V.; Kanthasamy, A.G. Manganese nanoparticle activates mitochondrial dependent apoptotic signaling and autophagy in dopaminergic neuronal cells. Toxicol. Appl. Pharmacol 2011, 256, 227–240. [Google Scholar]
- Trejo-Solis, C.; Jimenez-Farfan, D.; Rodriguez-Enriquez, S.; Fernandez-Valverde, F.; Cruz-Salgado, A.; Ruiz-Azuara, L.; Sotelo, J. Copper compound induces autophagy and apoptosis of glioma cells by reactive oxygen species and JNK activation. BMC Cancer 2012, 12, 156. [Google Scholar]
- Sun, T.; Yan, Y.; Zhao, Y.; Guo, F.; Jiang, C. Copper oxide nanoparticles induce autophagic cell death in A549 cells. PLoS One 2012, 7, e43442. [Google Scholar]
- Fleming, A.; Noda, T.; Yoshimori, T.; Rubinsztein, D.C. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Biol 2011, 7, 9–17. [Google Scholar]
- Garcia-Arencibia, M.; Hochfeld, W.E.; Toh, P.P.; Rubinsztein, D.C. Autophagy, a guardian against neurodegeneration. Semin. Cell Dev. Biol 2010, 21, 691–698. [Google Scholar]
- Horan, M.P.; Pichaud, N.; Ballard, J.W. Review: Quantifying mitochondrial dysfunction in complex diseases of aging. J. Gerontol 2012, 67, 1022–1035. [Google Scholar]
- Zharikov, S.; Shiva, S. Platelet mitochondrial function: From regulation of thrombosis to biomarker of disease. Biochem. Soc. Trans 2013, 41, 118–123. [Google Scholar]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol 2008, 4, 295–305. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dagda, R.K.; Banerjee, T.D.; Janda, E. How Parkinsonian Toxins Dysregulate the Autophagy Machinery. Int. J. Mol. Sci. 2013, 14, 22163-22189. https://doi.org/10.3390/ijms141122163
Dagda RK, Banerjee TD, Janda E. How Parkinsonian Toxins Dysregulate the Autophagy Machinery. International Journal of Molecular Sciences. 2013; 14(11):22163-22189. https://doi.org/10.3390/ijms141122163
Chicago/Turabian StyleDagda, Ruben K., Tania Das Banerjee, and Elzbieta Janda. 2013. "How Parkinsonian Toxins Dysregulate the Autophagy Machinery" International Journal of Molecular Sciences 14, no. 11: 22163-22189. https://doi.org/10.3390/ijms141122163
APA StyleDagda, R. K., Banerjee, T. D., & Janda, E. (2013). How Parkinsonian Toxins Dysregulate the Autophagy Machinery. International Journal of Molecular Sciences, 14(11), 22163-22189. https://doi.org/10.3390/ijms141122163