Chemical Constituents from Andrographis echioides and Their Anti-Inflammatory Activity

Abstract
:1. Introduction
2. Results and Discussion
2.1. Purification and Characterization
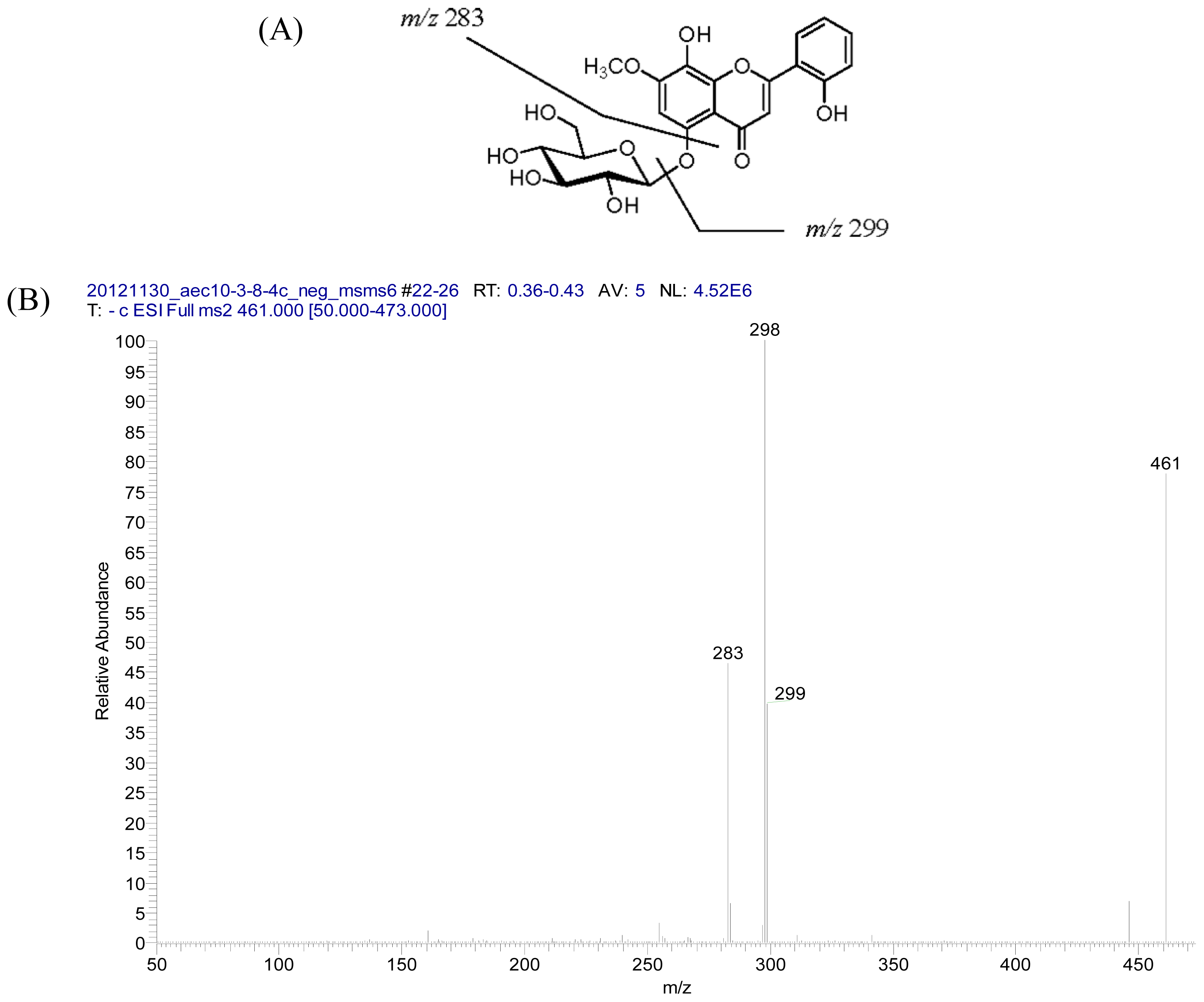
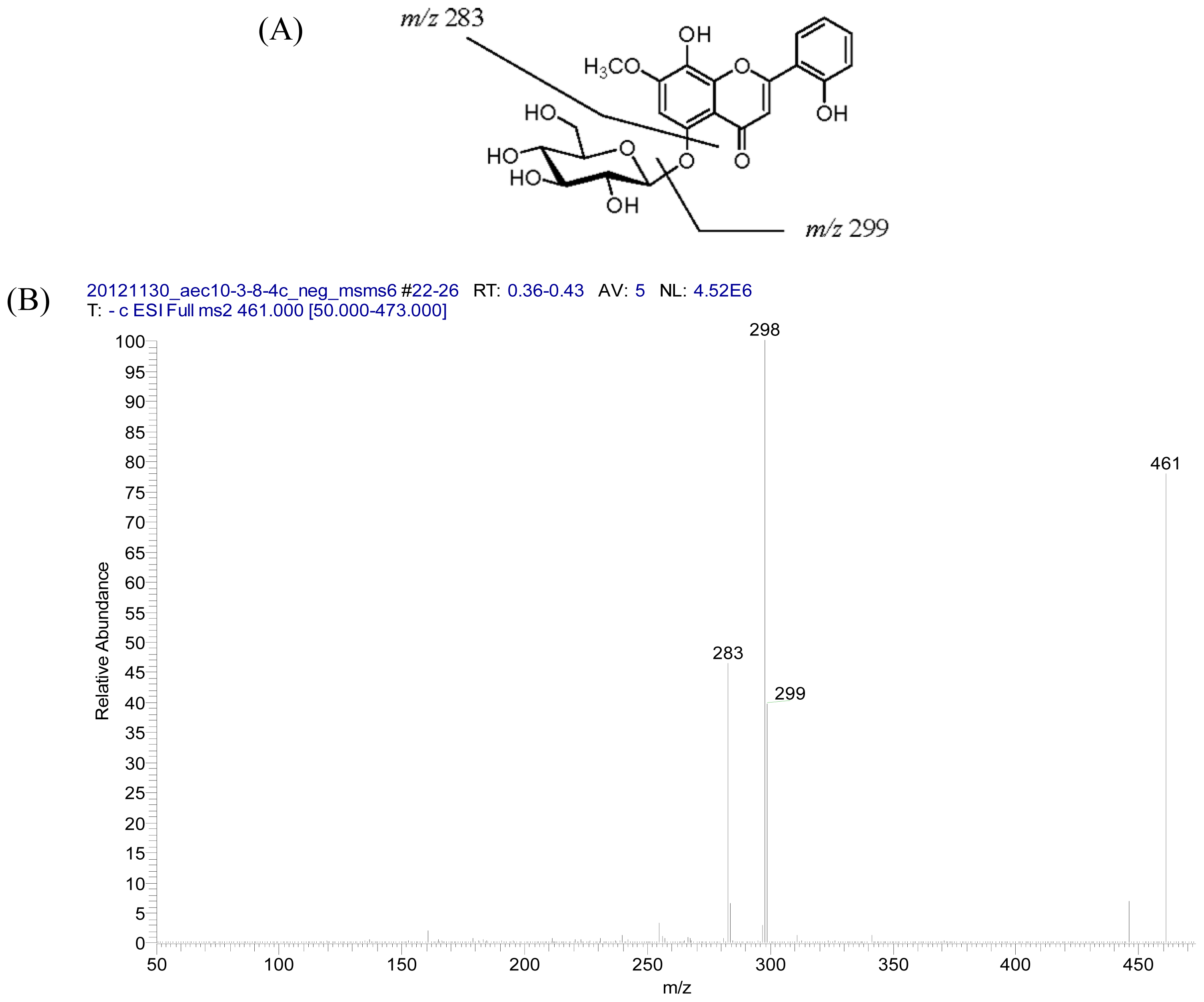
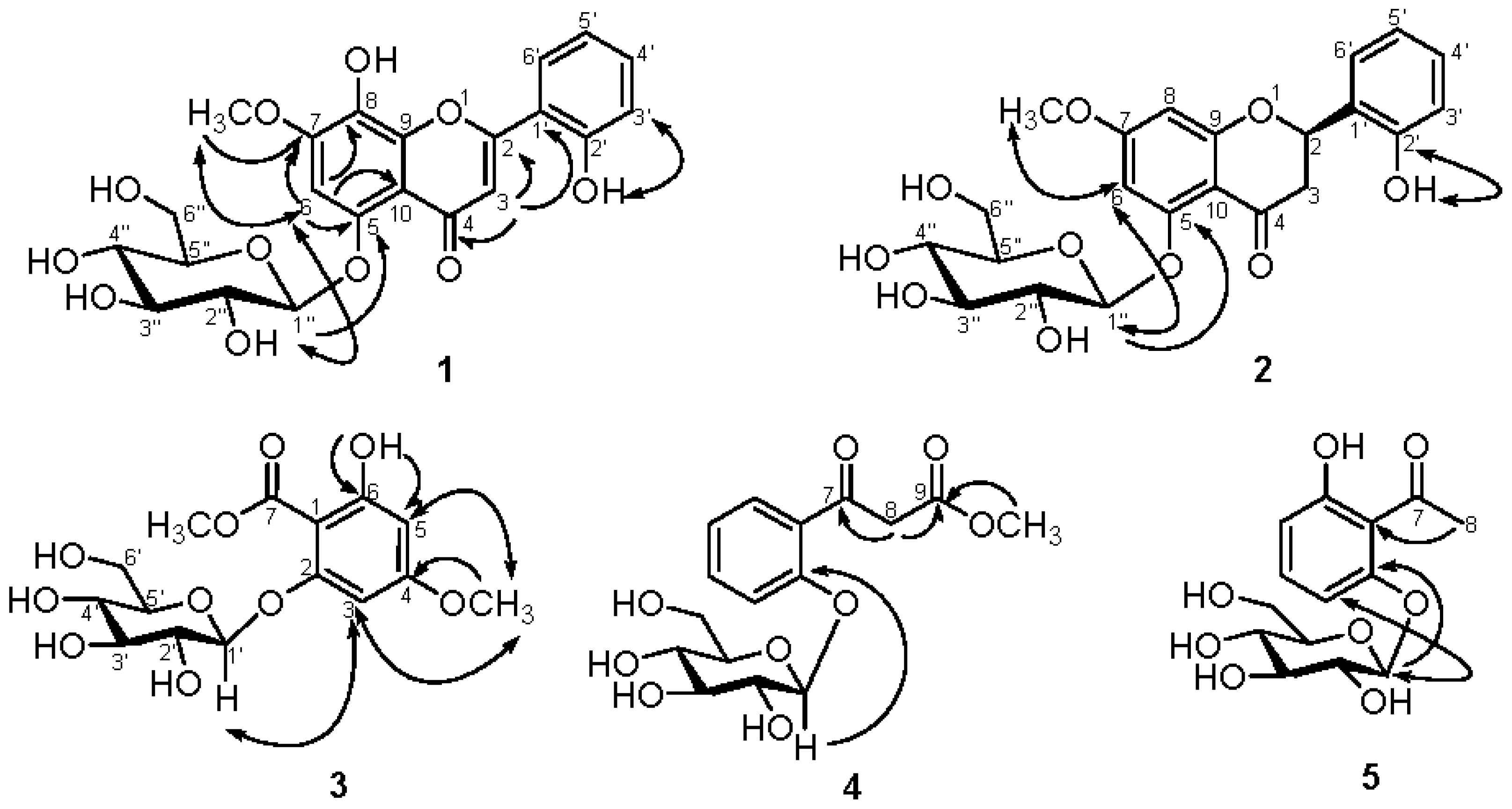
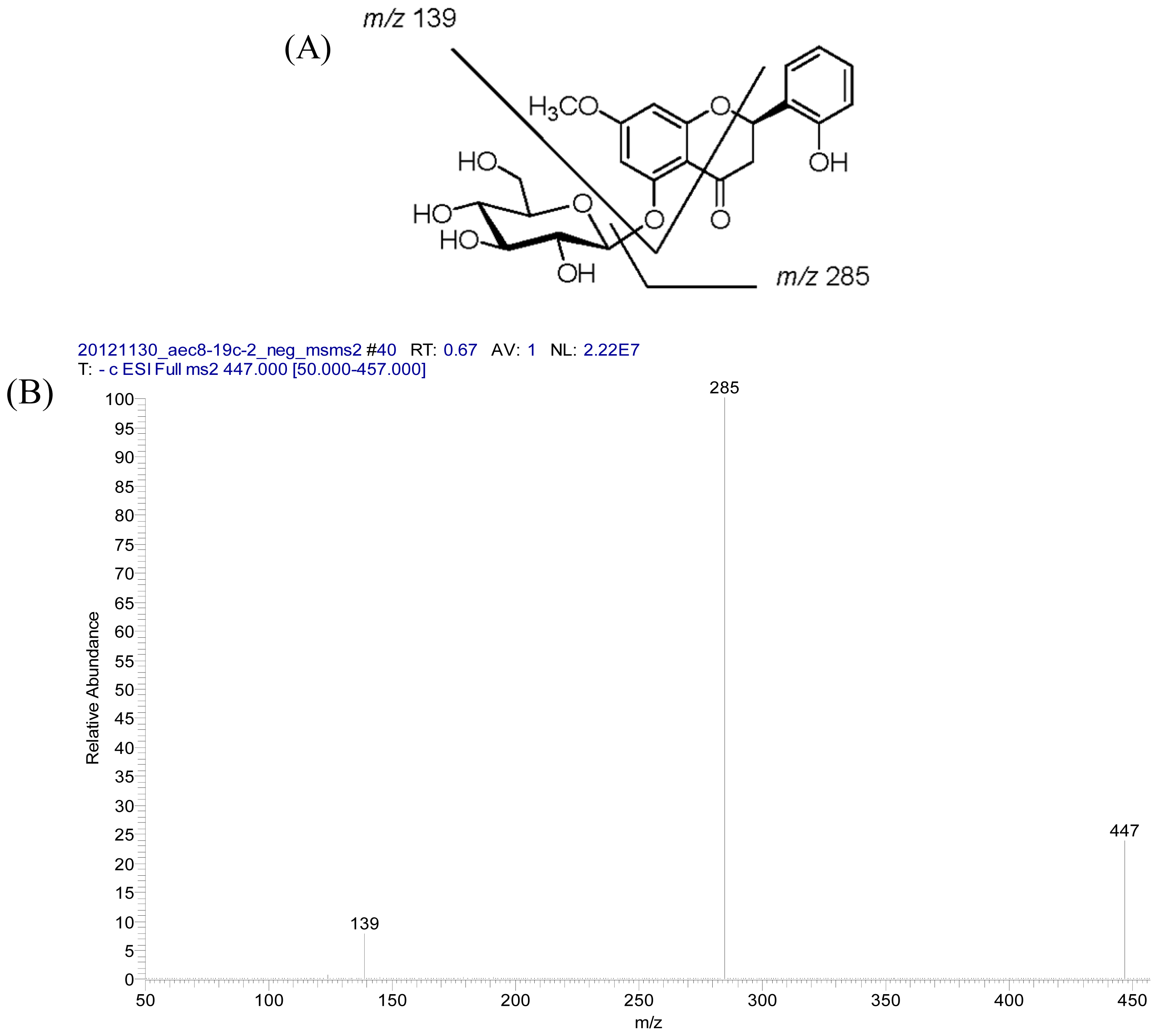
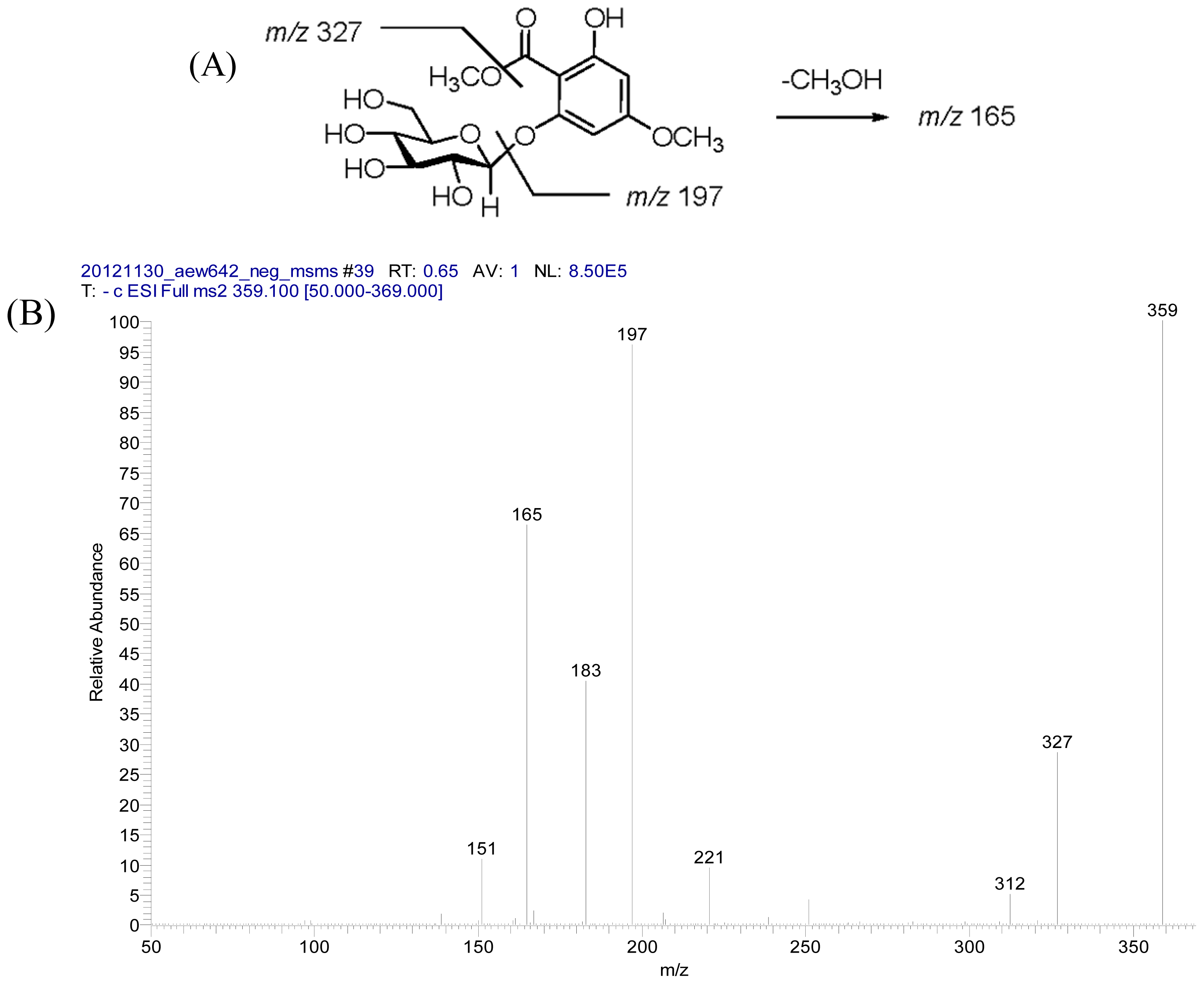
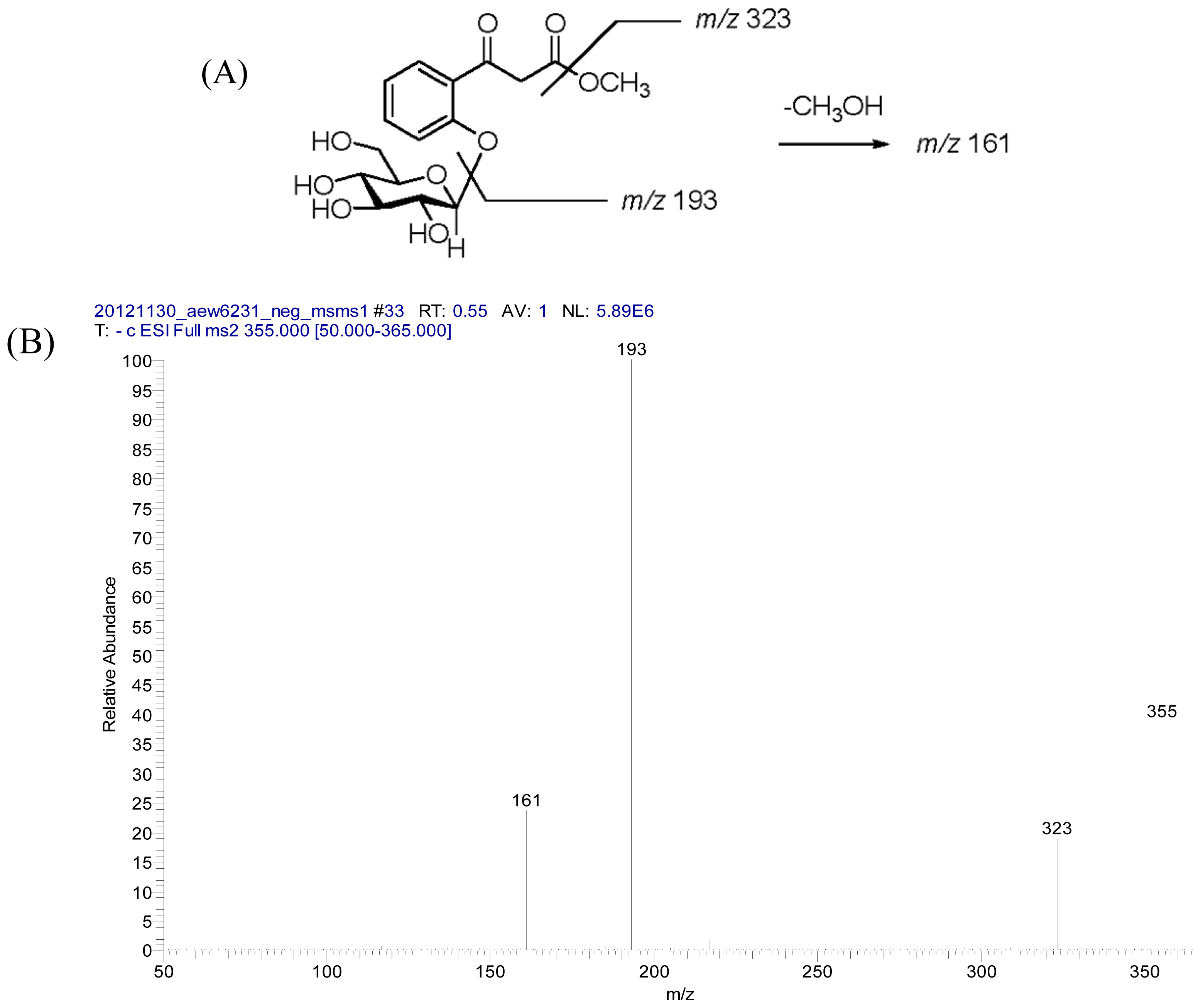
2.2. Structural Elucidation of Compounds 1–5
2.3. Anti-Inflammatory Activity
3. Experimental Section
3.1. General
3.2. Plant Materials
3.3. Extraction and Isolation
3.3.1. Androgechoside A (1)
3.3.2. Androgechoside B (2)
3.3.3. Androechioside A (3)
3.3.4. Androechioside B (4)
3.3.5. 2′,6′-Dihydroxyacetophenone 2′-O-β-d-glucopyranoside (5)
3.4. Determination of Aldose Configuration
3.5. Determination of iNOS Inhibitory Effects
3.5.1. Cell Culture
3.5.2. Cell Viability
3.5.3. Measurement of Nitric Oxide/Nitrite
3.5.4. Statistical Analysis
4. Conclusions
Acknowledgments
- Conflict of InterestThe authors have no conflict of interest.
References
- Kirtikar, K.R.; Basu, B.D. Indian Medicinal Plants; Periodical Experts Book Agency: New Delhi, India, 1975; Volume 3, pp. 1884–1886. [Google Scholar]
- Chopra, R.N.; Nayer, S.L.; Chopra, I.C. Glossary of Indian Medicinal Plants; Council of Scientific and Industrial Research: New Delhi, India, 1980; p. 18. [Google Scholar]
- Harborne, J.B. The Flavonoids: Advances in Research Since 1986; Chapman and Hall: London, UK, 1994; pp. 280–290. [Google Scholar]
- Iinuma, M.; Mizuno, M. Natural occurrence and synthesis of 2′-oxygenated flavones, flavonols, flavanones and chalcones. Phytochemistry 1989, 28, 681–694. [Google Scholar]
- Kleipool, R.J.C. Constituents of Andrographis paniculata Nees. Nature 1952, 169, 33–34. [Google Scholar]
- Chan, W.R.; Taylor, C.R.; Willis, R.L.; Bodden, R.L. The structure and stereochemistry of neoandrographolide, a diterpene glucoside from Andrographis paniculata Nees. Tetrahedron 1971, 27, 5081–5091. [Google Scholar]
- Balmain, A.; Connolly, J.D. Minor diterpenoid constituents of Andrographis paniculata Nees. J. Chem. Soc., Perkin Trans 1 1973, 1, 1247–1251. [Google Scholar]
- Fujita, T.; Fujitani, R.; Takeda, Y.; Takaishi, Y.; Yamada, T.; Kido, M.; Miura, I. On the diterpenoids of Andrographis paniculata: X-ray crystallographic analysis of andrographolide and structure determination of new minor diterpenoids. Chem. Pharm. Bull 1984, 32, 2117–2125. [Google Scholar]
- Matsuda, T.; Kuroyanagi, M.; Sugiyama, S.; Umehara, K.; Ueno, A.; Nishi, K. Cell differentiation-inducing diterpenes from Andrographis paniculata Nees. Chem. Pharm. Bull 1994, 42, 1216–1225. [Google Scholar]
- Reddy, M.K.; Reddy, M.V.; Gunasekar, D.; Murthy, M.M.; Caux, C.; Bodo, B. A flavone and an unusual 23-carbon terpenoid from Andrographis paniculata. Phytochemistry 2003, 62, 1271–1275. [Google Scholar]
- Govindachari, T.R.; Parthasarathy, P.C.; Pai, B.R.; Subramaniam, P.S. Andrographis echioides. I. Structure and synthesis of echioidinin. Tetrahedron 1965, 21, 2633–2640. [Google Scholar]
- Govindachari, T.R.; Parthasarathy, P.C.; Pai, B.R.; Subramaniam, P.S. Andrographis echioides. II. Structure and synthesis of echoidin. Tetrahedron 1965, 21, 3715–3720. [Google Scholar]
- Jayaprakasam, B.; Damu, A.G.; Gunasekar, D.; Blond, A.; Bodo, B. Dihydroechioidinin, a flavanone from Andrographis echioides. Phytochemistry 1999, 52, 935–937. [Google Scholar]
- Jayaprakasam, B.; Gunasekar, D.; Rao, K.V.; Blond, A.; Bodo, B. Androechin, a new chalcone glucoside from Andrographis echioides. J. Asian Nat. Prod. Res 2001, 3, 43–48. [Google Scholar]
- Rao, Y.K.; Damu, A.G.; Rao, A.J.; Venkatesan, S.; Kuo, P.C.; Rao, C.V.; Wu, T.S. Flavonoids from Andrographis viscosula. Chem. Pharm. Bull 2003, 51, 1374–1376. [Google Scholar]
- Wu, T.S.; Chern, H.J.; Damu, A.G.; Kuo, P.C.; Su, C.R.; Lee, E.J.; Teng, C.M. Flavonoids and ent-labdane diterpenoids from Andrographis paniculata and their antiplatelet aggregatory and vasorelaxing effects. J. Asian Nat. Prod. Res 2008, 10, 17–24. [Google Scholar]
- Gupte, A.; Buolamwini, J.K. Synthesis and biological evaluation of phloridzin analogs as human concentrative nucleoside transporter 3 (hCNT3) inhibitors. Bioorg. Med. Chem. Lett 2009, 19, 917–921. [Google Scholar]
- Chou, T.-H.; Chen, J.-J.; Lee, S.-J.; Chiang, M.Y.; Yang, C.-W.; Chen, I.-S. Cytotoxic flavonoids from the leaves of Cryptocarya chinensis. J. Nat. Prod 2010, 73, 1470–1475. [Google Scholar]
- Kuroyanagi, M.; Sato, M.; Ueno, A.; Nishi, K. Flavonoids from Andrographis paniculata. Chem. Pharm. Bull 1987, 35, 4429–4435. [Google Scholar]
- Geibel, M.; Geiger, H.; Treutter, D. Tectochrysin 5- and genistein 5-glucosides from the bark of Prunus cerasus. Phytochemistry 1990, 29, 1351–1353. [Google Scholar]
- Uchiyama, T.; Miyase, T.; Ueno, A.; Usmanghani, K. Terpenic glycosides from Pluchea indica. Phytochemistry 1989, 28, 3369–3372. [Google Scholar]
- Gupta, K.K.; Taneja, S.C.; Dhar, K.L.; Atal, C.K. Flavonoids of Andrographis paniculata. Phytochemistry 1983, 22, 314–315. [Google Scholar]
- Mesquita, A.A.L.; Corrêa, D.D.B.; de Pádua, A.P.; Guedes, M.L.O.; Gottlieb, O.R. Flavonoids from four compositae species. Phytochemistry 1986, 25, 1255–1256. [Google Scholar]
- Lendl, A.; Werner, I.; Glasl, S.; Kletter, C.; Mucaji, P.; Presser, A.; Reznicek, G.; Jurenitsch, J.; Taylor, D.W. Phenolic and terpenoid compounds from Chione venosa (sw.) urban var. venosa (Bois Bandé). Phytochemistry 2005, 66, 2381–2387. [Google Scholar]
- Reddy, B.A.K.; Reddy, M.V.B.; Gunasekar, D.; Murthy, M.M.; Caux, C.; Bodo, B. Two new flavonoids from Andrographis macrobotrys. Indian J. Chem. B 2005, 44B, 1966–1969. [Google Scholar]
- Reddy, M.K.; Reddy, M.V.B.; Jayakrishna, G.; Gunasekar, D.; Caux, C.; Bodo, B. Two new flavonoids from Andrographis rothii. Chem. Pharm. Bull 2003, 51, 191–193. [Google Scholar]
- Reddy, M.V.B.; Kishore, P.H.; Rao, C.V.; Gunasekar, D.; Caux, C.; Bodo, B. New 2′-oxygenated flavonoids from Andrographis affinis. J. Nat. Prod 2003, 66, 295–297. [Google Scholar]
- Rao, Y.K.; Fang, S.-H.; Tzeng, Y.-M. Synthesis, growth inhibition, and cell cycle evaluations of novel flavonoid derivatives. Bioorg. Med. Chem 2005, 13, 6850–6855. [Google Scholar]
- Fan, Y.; Xing-Cong, L.; Han-Qing, W.; Chong-Ren, Y. Flavonoid glycosides from Colebrookea oppositifolia. Phytochemistry 1996, 42, 867–869. [Google Scholar]
- Wallace, G.; Fry, S.C. In vitro peroxidase-catalysed oxidation of ferulic acid esters. Phytochemistry 1995, 39, 1293–1299. [Google Scholar]
- Choy, J.; Jaime-Figueroa, S.; Lara-Jaime, T. A novel practical cleavage of tert-butyl esters and carbonates using fluorinated alcohols. Tetrahedron Lett 2010, 51, 2244–2246. [Google Scholar]
- Machida, K.; Kikuchi, M. Norisoprenoids from Viburnum dilatatum. Phytochemistry 1996, 41, 1333–1336. [Google Scholar]
- Reynertson, K.A.; Wallace, A.M.; Adachi, S.; Gil, R.R.; Yang, H.; Basile, M.J.; D’Armiento, J.; Weinstein, I.B.; Kennelly, E.J. Bioactive depsides and anthocyanins from Jaboticaba (Myrciaria cauliflora). J. Nat. Prod 2006, 69, 1228–1230. [Google Scholar]
- Barbosa-Filho, J.M.; Yoshida, M.; Gottlieb, O.R.; de Barbosa, R.C.S.B.C.; Giesbrecht, A.M.; Young, M.C.M. Benzoyl esters and amides, styrylpyrones and neolignans from the fruits of Aniba riparia. Phytochemistry 1987, 26, 2615–2617. [Google Scholar]
- Sakdarat, S.; Shuyprom, A.; Pientong, C. Bioactive constituents from the leaves of Clinacanthus nutans Lindau. Biorg. Med. Chem 2009, 17, 1857–1860. [Google Scholar]
- Rajab, M.S.; Cantrell, C.L.; Franzblau, S.G.; Fischer, N.H. Antimycobacterial activity of (E)-phytol and derivatives: A preliminary structure-activity study. Planta medica 1998, 64, 2–4. [Google Scholar]
- Brown, G.D. Phytene-1,2-diol from Artemisia annua. Phytochemistry 1994, 36, 1553–1554. [Google Scholar]
- Brown, G.D.; Liang, G.-Y.; Sy, L.-K. Terpenoids from the seeds of Artemisia annua. Phytochemistry 2003, 64, 303–323. [Google Scholar]
- Heilmann, J.; Müller, E.; Merfort, I. Flavonoid glucosides and dicaffeoylquinic acids from flowerheads of Buphthalmum salicifolium. Phytochemistry 1999, 51, 713–718. [Google Scholar]
- Nakabayashi, R.; Kusano, M.; Kobayashi, M.; Tohge, T.; Yonekura-Sakakibara, K.; Kogure, N.; Yamazaki, M.; Kitajima, M.; Saito, K.; Takayama, H. Metabolomics-oriented isolation and structure elucidation of 37 compounds including two anthocyanins from Arabidopsis thaliana. Phytochemistry 2009, 70, 1017–1029. [Google Scholar]
- Nes, W.D.; Norton, R.A.; Benson, M. Carbon-13 nmr studies on sitosterol biosynthesized from [13C]mevalonates. Phytochemistry 1992, 31, 805–811. [Google Scholar]
- Chan, H.-H.; Hwang, T.-L.; Reddy, M.V.B.; Li, D.-T.; Qian, K.; Bastow, K.F.; Lee, K.-H.; Wu, T.-S. Bioactive constituents from the roots of Panax japonicus var. major and development of a LC-MS/MS method for distinguishing between natural and artifactual compounds. J. Nat. Prod 2011, 74, 796–802. [Google Scholar]
- Fan, J.-T.; Kuang, B.; Zeng, G.-Z.; Zhao, S.-M.; Ji, C.-J.; Zhang, Y.-M.; Tan, N.-H. Biologically active arborinane-type triterpenoids and anthraquinones from Rubia yunnanensis. J. Nat. Prod 2011, 74, 2069–2080. [Google Scholar]
- Wu, S.J.; Fotso, S.; Li, F.; Qin, S.; Laatsch, H. Amorphane sesquiterpenes from a Marine Streptomyces sp. J. Nat. Prod 2007, 70, 304–306. [Google Scholar]
- Cheng, S.-Y.; Huang, K.-J.; Wang, S.-K.; Wen, Z.-H.; Chen, P.-W.; Duh, C.-Y. Antiviral and anti-inflammatory metabolites from the soft coral Sinularia capillosa. J. Nat. Prod 2010, 73, 771–775. [Google Scholar]
- Tomimori, T.; Miyaichi, Y.; Imoto, Y.; Kizu, H.; Namba, T. Studies on the nepese crude drugs. VI: On the flavonoid constituents of the root of Scutellaria discolor COLEBR. (2). Chem. Pharm. Bull 1986, 34, 406–408. [Google Scholar]
- Tanaka, T.; Iinuma, M.; Mizuno, M. Spectral properties of 2′-oxygenated flavones. Chem. Pharm. Bull 1986, 34, 1667–1671. [Google Scholar]
- Takagi, S.; Yamaki, M.; Inoue, K. Studies on the water-soluble constituents of the roots of Scutellaria baicalensis GEORGI (Wogon). Yakugaku Zasshi 1980, 100, 1220–1224. [Google Scholar]
- Harborne, J.B.; Mabry, T.J. The Flavonoids; Chapman and Hall: London, UK, 1975; pp. 45–77. [Google Scholar]
- Matsuda, H.; Nishida, N.; Yoshikawa, M. Antidiabetic principles of natural medicines. V.(1) Aldose reductase inhibitors from Myrcia multiflora DC. (2): Structures of myrciacitrins III, IV, and V. Chem. Pharm. Bull 2002, 50, 429–431. [Google Scholar]
- Gaffield, W. Circular dichroism, optical rotatory dispersion and absolute configuration of flavanones, 3-hydroxyflavanones and their glycosides. Tetrahedron 1970, 26, 4093–4108. [Google Scholar]
- Tian, L.-W.; Zhang, Y.-J.; Qu, C.; Wang, Y.-F.; Yang, C.-R. Phloroglucinol glycosides from the fresh fruits of Eucalyptus maideni. J. Nat. Prod 2010, 73, 160–163. [Google Scholar]
- Matsuda, H.; Morikawa, T.; Ando, S.; Toguchida, I.; Yoshikawa, M. Structural requirements of flavonoids for nitric oxide production inhibitory activity and mechanism of action. Biorg. Med. Chem 2003, 11, 1995–2000. [Google Scholar]
- Kim, H.K.; Cheon, B.S.; Kim, Y.H.; Kim, S.Y.; Kim, H.P. Effects of naturally occurring flavonoids on nitric oxide production in the macrophage cell Line RAW 264.7 and their structure-activity relationships. Biochem. Pharmacol 1999, 58, 759–765. [Google Scholar]
- Chang, C.-T.; Huang, S.-S.; Lin, S.-S.; Amagaya, S.; Ho, H.-y.; Hou, W.-C.; Shie, P.-H.; Wu, J.-B.; Huang, G.-J. Anti-inflammatory activities of tormentic acid from suspension cells of Eriobotrya Japonica ex vivo and in vivo. Food Chem 2011, 127, 1131–1137. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | |||
|---|---|---|---|---|
| Position | δH (J, Hz) | δC | δH (J, Hz) | δC |
| 2 | 159.1 | 5.70, dd (12.5, 3.0) | 74.2 | |
| 3 | 7.05, s | 111.5 | 2.64, dd (16.0, 3.0) | 43.9 |
| 3.04, dd (16.0, 12.5) | ||||
| 4 | 178.3 | 189.6 | ||
| 5 | 149.5 | 159.7 | ||
| 6 | 7.17, s | 102.1 | 6.46, d (2.5) | 97.7 |
| 7 | 151.6 | 165.4 | ||
| 8 | 130.9 | 6.32, d (2.5) | 95.9 | |
| 9 | 146.1 | 164.3 | ||
| 10 | 109.5 | 106.3 | ||
| 1′ | 117.1 | 125.0 | ||
| 2′ | 156.8 | 154.3 | ||
| 3′ | 7.04, d (8.0) | 117.1 | 6.85, d (6.5) | 115.6 |
| 4′ | 7.38, dd (8.0, 8.0) | 132.7 | 7.17, dd (6.5, 6.5) | 129.4 |
| 5′ | 6.99, ddd (8.0, 8.0, 1.6) | 119.6 | 6.84, dd (6.5, 6.5) | 119.2 |
| 6′ | 8.00, dd (8.0, 1.6) | 128.7 | 7.41, d (6.5) | 126.9 |
| 1″ | 4.60, d (7.6) | 105.5 | 4.83, d (7.5) | 101.9 |
| 2″ | 3.27–3.37, m | 73.8 | 3.26–3.43, m | 73.5 |
| 3″ | 3.27–3.37, m | 77.8 | 3.15, m | 76.4 |
| 4″ | 3.13, m | 70.4 | 3.26–3.43, m | 70.0 |
| 5″ | 3.27–3.37, m | 76.1 | 3.26–3.43, m | 77.6 |
| 6″ | 3.46, m | 61.3 | 3.26–3.43, m | 61.0 |
| 3.73, m | 3.64, m | |||
| OH-2′b | 10.75, br s | 9.80, br s | ||
| OH-8b | 9.30, br s | |||
| OCH3-7 | 3.90, s | 56.3 | 3.79, s | 55.9 |
| Position | 3a | 4b | 5a | |||
|---|---|---|---|---|---|---|
| δC | δH mult (J, Hz) | δC | δH mult (J, Hz) | δC | δH mult (J, Hz) | |
| 1 | 98.5 | 127.3 | 111.8 | |||
| 2 | 161.0 | 156.9 | 159.5 | |||
| 3 | 95.4 | 6.36, d (2.4) | 116.0 | 7.33, d (7.8) | 105.3 | 6.75, dd (8.4, 1.2) |
| 4 | 166.1 | 134.3 | 7.54, dd (7.8, 7.8) | 136.1 | 7.38, dd (8.4, 8.4) | |
| 5 | 96.1 | 6.14, d (2.4) | 122.0 | 7.12, dd (7.8, 7.8) | 111.2 | 6.55, dd (8.4, 1.2) |
| 6 | 165.2 | 129.9 | 7.75, d (7.8) | 163.8 | ||
| 7 | 171.7 | 193.7 | 205.0 | |||
| 8 | 49.8 | 4.23, d (16.4) | 33.8 | 2.77, s | ||
| 4.07, d (16.4) | ||||||
| 9 | 168.5 | |||||
| 1′ | 102.5 | 4.96, d (7.6) | 101.1 | 5.15, d ( 7.2) | 101.2 | 5.12, d (8.8) |
| 2′ | 74.7 | 3.60–3.40, m | 73.4 | 3.60–3.40, m | 73.6 | 3.59, dd ( 8.8, 8.8) |
| 3′ | 77.7 | 3.60–3.40, m | 77.0 | 3.60–3.40, m | 77.3 | 3.46, dd ( 8.8, 8.8) |
| 4′ | 71.2 | 3.60–3.40, m | 70.1 | 3.60–3.40, m | 70.2 | 3.54, dd (8.8, 8.8) |
| 5′ | 78.0 | 3.60–3.40, m | 77.0 | 3.60–3.40, m | 77.1 | 3.55, m |
| 6′a | 62.6 | 3.91, m | 61.4 | 3.87, m | 61.6 | 3.88, dd (12.0, 2.0) |
| 6′b | 3.69, m | 3.72, m | 3.70, dd (12.0, 5.2) | |||
| OCH3-4 | 55.9 | 3.81, s | ||||
| OCH3-7 | 52.5 | 3.88, s | ||||
| OCH3-9 | 51.1 | 3.66, s | ||||
| OH-6 | 11.4, s | 13.0, br s | ||||
| Dose (μM) | Cell viability (% of control) | NO level | NO inhibition (% of control) | IC50 (μM) | |
|---|---|---|---|---|---|
| control | (−) | 100.0 ± 3.3 | 0.2 ± 0.9 | (−) | |
| LPS | (+) | 96.9 ± 6.1 | 59.4 ± 0.8 ### | (−) | |
| 1 | 5.25 | 92.1 ± 4.2 | 48.1 ± 1.5 | 19.0 ± 2.5 | >42 |
| 10.5 | 86.6 ± 1.5 | 48.1 ± 1.7 | 19.1 ± 2.9 | ||
| 21 | 83.9 ± 3.5 | 45.3 ± 1.4 | 23.7 ± 2.3 | ||
| 42 | 80.1 ± 3.7 | 42.7 ± 1.1 * | 28.1 ± 1.8 | ||
| 2 | 5.63 | 103.6 ± 2.3 | 49.8 ± 4.6 | 16.1 ± 7.7 | >45 |
| 11.25 | 99.2 ± 6.8 | 46.2 ± 2.5 | 22.2 ± 4.2 | ||
| 22.5 | 99.2 ± 4.2 | 41.8 ± 1.6 * | 29.6 ± 2.7 | ||
| 45 | 99.8 ± 6.0 | 39.9 ± 0.7 ** | 32.8 ± 1.1 | ||
| 6 | 5.63 | 92.6 ± 3.0 | 45.0 ± 2.7 | 24.2 ± 4.6 | >45 |
| 11.25 | 85.7 ± 4.6 | 46.2 ± 1.8 | 22.3 ± 3.0 | ||
| 22.5 | 84.6 ± 3.5 | 44.4 ± 2.1 * | 25.2 ± 3.5 | ||
| 45 | 81.4 ± 2.1 | 43.2 ± 1.8 * | 27.2 ± 3.0 | ||
| 7 | 8.75 | 90.5 ± 4.0 | 45.2 ± 1.2 | 23.9 ± 2.0 | >70 |
| 17.5 | 88.2 ± 1.7 | 44.6 ± 2.6 | 25.0 ± 4.3 | ||
| 35 | 84.9 ± 2.2 | 43.8 ± 2.1 * | 26.2 ± 3.5 | ||
| 70 | 83.2 ± 5.6 | 43.5 ± 2.7 * | 26.8 ± 4.6 | ||
| 8 | 9.25 | 90.1 ± 7.0 | 51.7 ± 1.9 | 12.9 ± 3.2 | >74 |
| 18.5 | 86.4 ± 6.5 | 47.2 ± 2.1 | 20.5 ± 3.6 | ||
| 37 | 82.0 ± 4.4 | 47.1 ± 1.8 | 20.7 ± 3.1 | ||
| 74 | 79.3 ± 1.7 | 42.2 ± 2.5 * | 28.9 ± 4.1 | ||
| 9 | 5.5 | 89.7 ± 2.7 | 42.7 ± 4.0 * | 28.1 ± 6.8 | >44 |
| 11 | 84.8 ± 1.8 | 41.1 ± 1.6 * | 30.8 ± 2.7 | ||
| 22 | 83.4 ± 1.9 | 38.7 ± 3.0 ** | 34.8 ± 5.0 | ||
| 44 | 80.2 ± 1.7 | 38.3 ± 2.0 ** | 35.6 ± 3.3 | ||
| 10 | 8.75 | 89.4 ± 8.3 | 41.3 ± 1.3 | 30.4 ± 2.3 | 37.6 ± 1.2 |
| 17.5 | 84.9 ± 2.0 | 39.6 ± 1.5 ** | 33.3 ± 2.5 | ||
| 35 | 82.1 ± 3.7 | 30.7 ± 1.5 ** | 48.3 ± 2.6 | ||
| 70 | 80.1 ± 2.9 | 21.2 ± 2.7 *** | 64.2 ± 4.5 | ||
| 11 | 5.75 | 98.9 ± 4.4 | 50.6 ± 0.6 | 14.9 ± 1.0 | >46 |
| 11.5 | 96.2 ± 4.2 | 47.5 ± 4.0 | 20.0 ± 6.7 | ||
| 23 | 96.5 ± 4.5 | 46.7 ± 0.9 | 21.4 ± 1.4 | ||
| 46 | 93.0 ± 3.4 | 46.5 ± 1.9 | 21.7 ± 3.2 | ||
| 12 | 8 | 100 ± 5.4 | 50.0 ± 2.3 | 15.8 ± 3.9 | >64 |
| 16 | 96.5 ± 9.2 | 49.5 ± 1.0 | 16.7 ± 1.7 | ||
| 32 | 96.3 ± 4.2 | 48.0 ± 1.0 | 19.2 ± 1.6 | ||
| 64 | 94.3 ± 2.1 | 45.5 ± 2.0 | 23.4 ± 3.4 | ||
| 13 | 8.5 | 98.1 ± 6.6 | 52.2 ± 1.3 | 12.1 ± 2.2 | >68 |
| 17 | 97.7 ± 1.7 | 48.8 ± 0.7 | 17.8 ± 1.3 | ||
| 34 | 90.8 ± 2.9 | 47.9 ± 3.9 | 19.4 ± 6.6 | ||
| 68 | 90.8 ± 8.5 | 47.5 ± 1.0 | 20.1 ± 1.7 | ||
| 14 | 8 | 98.9 ± 3.5 | 40.8 ± 2.7 * | 31.4 ± 4.5 | 39.1 ± 1.3 |
| 16 | 97.9 ± 3.8 | 34.3 ± 2.0 ** | 42.3 ± 3.4 | ||
| 32 | 94.3 ± 4.6 | 32.2 ± 1.5 ** | 45.8 ± 7.5 | ||
| 64 | 92.2 ± 3.2 | 23.1 ± 2.3 *** | 61.1 ± 3.8 | ||
| 15 | 7.5 | 99.4 ± 4.3 | 45.6 ± 1.1 | 23.3 ± 1.8 | >60 |
| 15 | 99.2 ± 2.9 | 43.0 ± 3.5 * | 27.7 ± 5.9 | ||
| 30 | 98.9 ± 1.8 | 42.0 ± 4.0 * | 29.3 ± 6.7 | ||
| 60 | 98.8 ± 6.9 | 41.2 ± 1.9 * | 30.6 ± 3.1 | ||
| 16 | 8.5 | 101.5 ± 4.3 | 56.5 ± 2.0 | 4.9 ± 3.4 | >68 |
| 17 | 100.3 ± 1.0 | 51.7 ± 2.6 | 13.0 ± 4.4 | ||
| 34 | 99.6 ± 1.0 | 50.4 ± 4.6 | 15.1 ± 7.8 | ||
| 68 | 93.7 ± 0.9 | 44.6 ± 0.5 * | 24.9 ± 0.9 | ||
| 17 | 5.63 | 100.8 ± 0.8 | 52.4 ± 2.9 | 11.8 ± 4.9 | >45 |
| 11.25 | 98.8 ± 3.5 | 51.7 ± 4.6 | 12.9 ± 7.7 | ||
| 22.5 | 97.8 ± 3.6 | 50.8 ± 3.8 | 14.5 ± 6.4 | ||
| 45 | 98.3 ± 1.9 | 46.4 ± 4.0 | 21.8 ± 6.7 | ||
| 18 | 5.25 | 101.4 ± 1.2 | 53.5 ± 4.4 | 9.9 ± 7.4 | >42 |
| 10.5 | 100.5 ± 1.8 | 52.1 ± 3.7 | 12.3 ± 6.2 | ||
| 21 | 95.6 ± 0.8 | 50.7 ± 4.5 | 14.6 ± 7.6 | ||
| 42 | 74.1 ± 6.6 | (−) | (−) | ||
| 19 | 9.25 | 103.5 ± 4.8 | 52.7 ± 4.7 | 11.2 ± 8.0 | >74 |
| 18.5 | 98.2 ± 11.3 | 47.5 ± 4.8 | 20.1 ± 8.0 | ||
| 37 | 92.9 ± 7.6 | 46.7 ± 4.0 | 21.4 ± 6.8 | ||
| 74 | 81.6 ± 6.5 | 44.4 ± 2.8 * | 25.2 ± 4.7 | ||
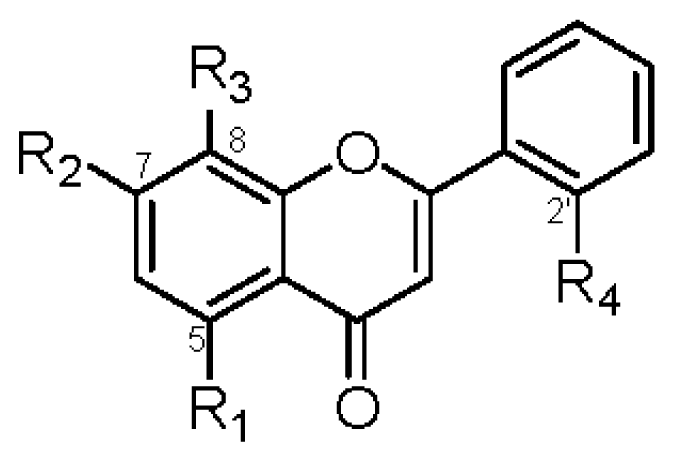 | 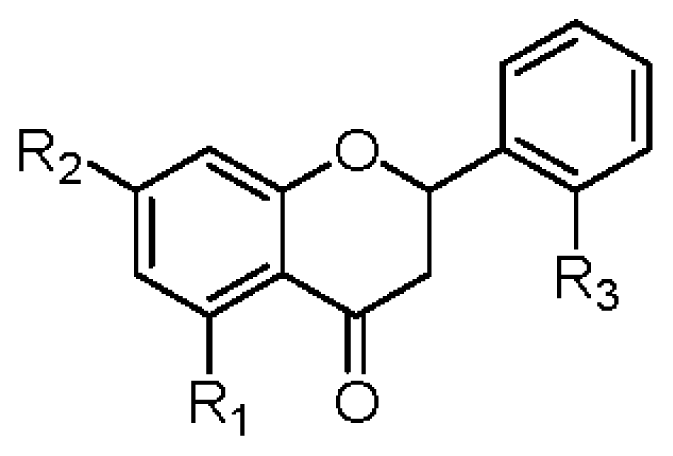 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
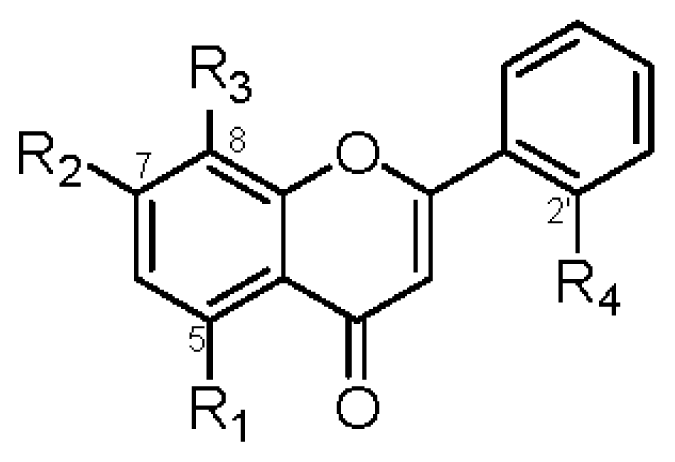
| R1 | R2 | R3 | R4 | IC50 (μM) | R1 | R2 | R3 | IC50 (μM) | ||
| 1 | OGlc | OCH3 | OH | OH | >42 | 2 | OGlc | OCH3 | OH | >45 |
| 6 | OGlc | OCH3 | H | OH | >45 | 8 | OH | OCH3 | H | >74 |
| 7 | OH | OCH3 | H | OH | >70 | 10 | OH | OCH3 | OH | 37.6 ± 1.2 b |
| 9 | OGlc | OCH3 | OCH3 | H | >44 | |||||
| 11 | OGlc | OCH3 | H | H | >46 | |||||
| 13 | OH | OCH3 | OCH3 | H | >64 | |||||
| 14 | OCH3 | OCH3 | OCH3 | H | 39.1 ± 1.3 a | |||||
| 15 | OH | OCH3 | OCH3 | OCH3 | >60 | |||||
| 18 | OH | OCH3 | OCH3 | OGlc | >42 | |||||
| 19 | OH | OCH3 | H | H | >74 | |||||
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shen, D.-Y.; Juang, S.-H.; Kuo, P.-C.; Huang, G.-J.; Chan, Y.-Y.; Damu, A.G.; Wu, T.-S. Chemical Constituents from Andrographis echioides and Their Anti-Inflammatory Activity. Int. J. Mol. Sci. 2013, 14, 496-514. https://doi.org/10.3390/ijms14010496
Shen D-Y, Juang S-H, Kuo P-C, Huang G-J, Chan Y-Y, Damu AG, Wu T-S. Chemical Constituents from Andrographis echioides and Their Anti-Inflammatory Activity. International Journal of Molecular Sciences. 2013; 14(1):496-514. https://doi.org/10.3390/ijms14010496
Chicago/Turabian StyleShen, De-Yang, Shin-Hun Juang, Ping-Chung Kuo, Guan-Jhong Huang, Yu-Yi Chan, Amooru G. Damu, and Tian-Shung Wu. 2013. "Chemical Constituents from Andrographis echioides and Their Anti-Inflammatory Activity" International Journal of Molecular Sciences 14, no. 1: 496-514. https://doi.org/10.3390/ijms14010496