Recent Insights and Novel Bioinformatics Tools to Understand the Role of MicroRNAs Binding to 5' Untranslated Region

Abstract
:1. Introduction
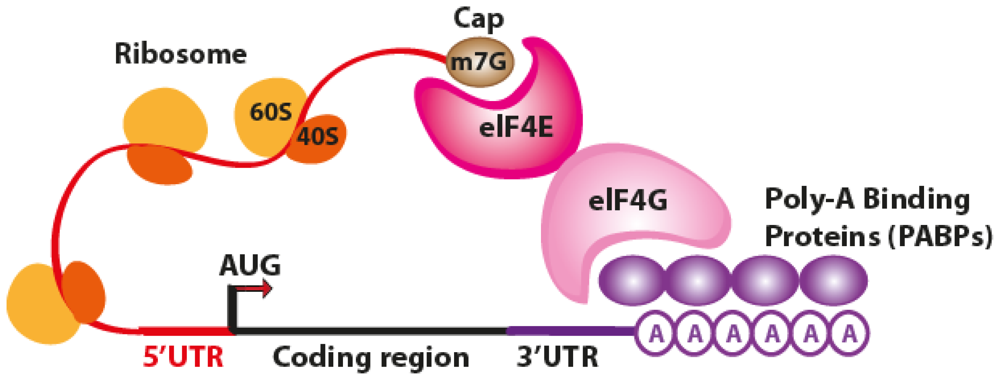
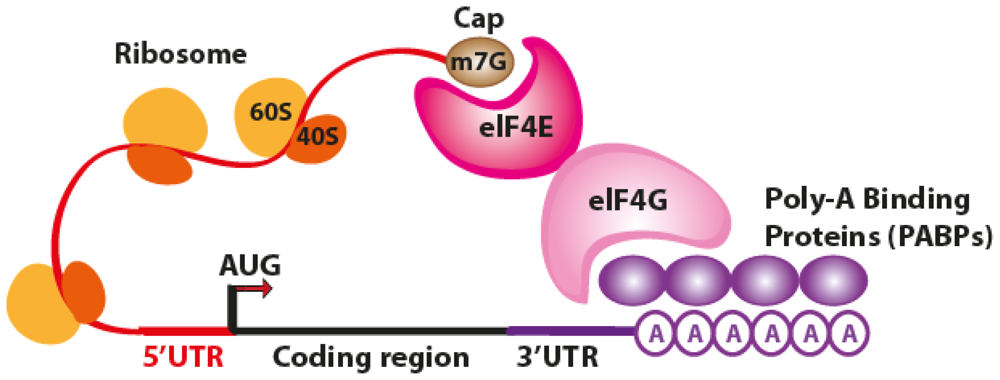
2. The Translation Process
3. Post-Transcriptional Gene Regulation by miRNAs Binding to the 3′UTR of Target Genes
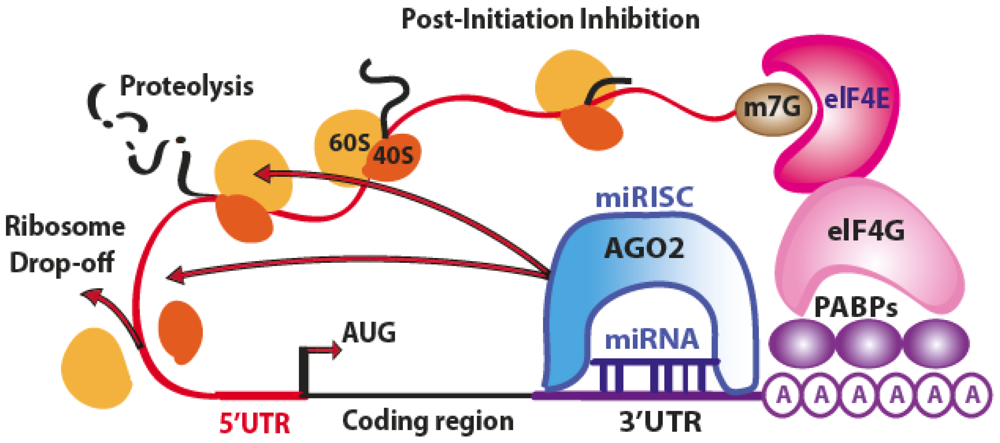
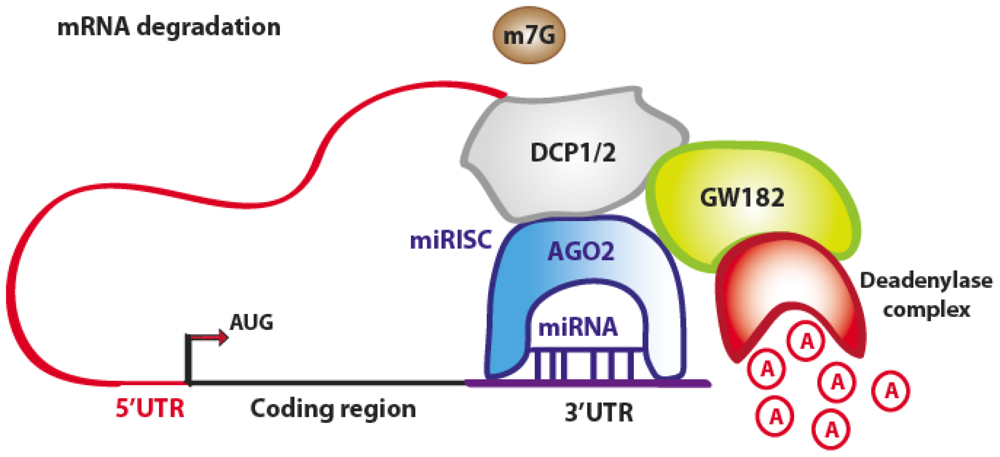
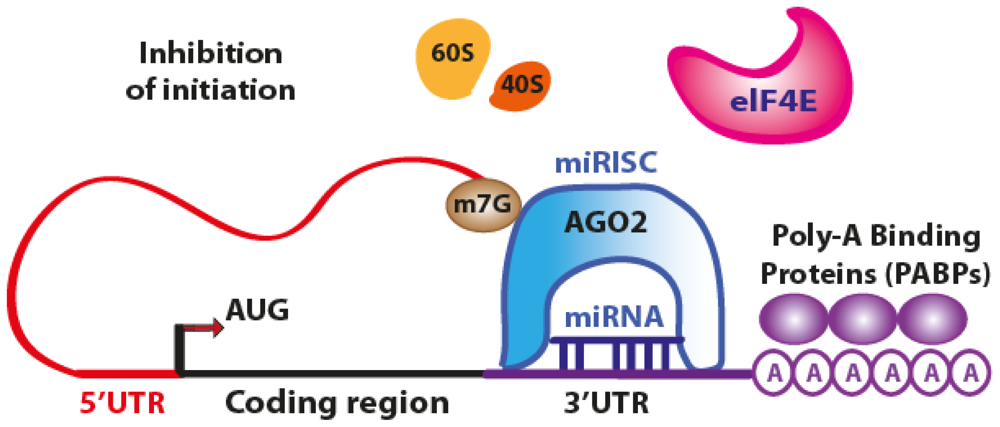
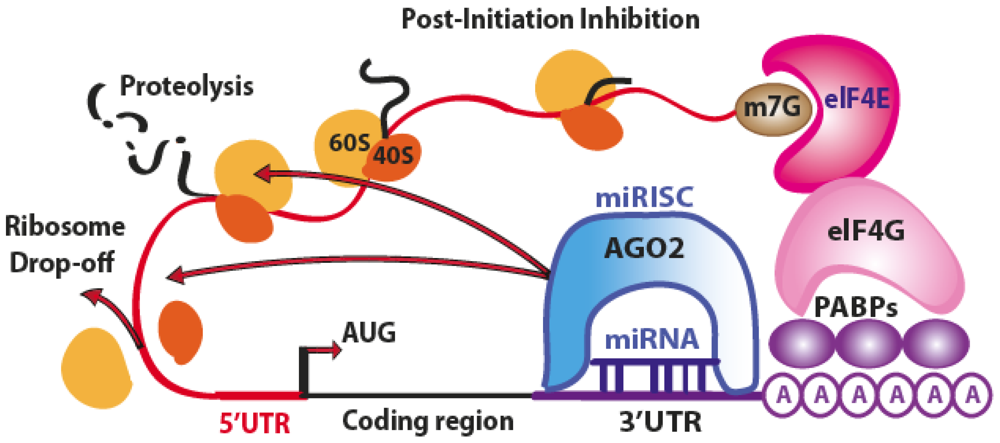
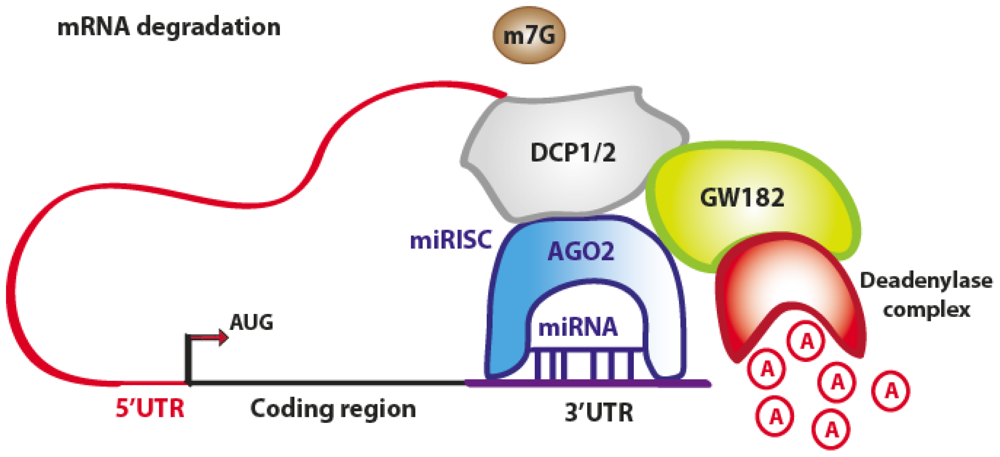
3.1. Translation Repression
3.2. Translation Activation
4. Post-Transcriptional Gene Regulation by miRNAs Binding to the 5′UTR of Target Genes
4.1. Translation Repression
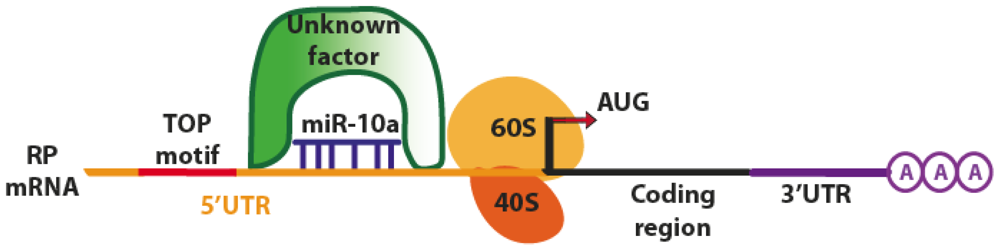
4.2. Translation Activation
5. Bioinformatics
5.1. miBridge
5.2. miRTar
5.3. miRWalk
5.4. Sfold-STarMirDB
5.5. miRNA_Targets
6. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Mattick, J.S.; Makunin, I.V. Small regulatory RNAs in mammals. Hum. Mol. Genet 2005, 14, R121–R132. [Google Scholar]
- Wienholds, E.; Plasterk, R.H. MicroRNA function in animal development. FEBS Lett 2005, 579, 5911–5922. [Google Scholar]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar]
- Alvarez-Garcia, I.; Miska, E.A. MicroRNA functions in animal development and human disease. Development 2005, 132, 4653–4662. [Google Scholar]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem 2010, 79, 351–379. [Google Scholar]
- Vasudevan, S. Posttranscriptional upregulation by microRNAs. Wiley Interdiscip. Rev. RNA 2012, 3, 311–330. [Google Scholar]
- Pillai, R.S.; Bhattacharyya, S.N.; Filipowicz, W. Repression of protein synthesis by miRNAs: How many mechanisms? Trends Cell Biol 2007, 17, 118–126. [Google Scholar]
- Shatsky, I.N.; Dmitriev, S.E.; Terenin, I.M.; Andreev, D.E. Cap- and IRES-independent scanning mechanism of translation initiation as an alternative to the concept of cellular IRESs. Mol. Cells 2010, 30, 285–293. [Google Scholar]
- Kieft, J.S. Viral IRES RNA structures and ribosome interactions. Trends Biochem. Sci 2008, 33, 274–283. [Google Scholar]
- Stefani, G.; Slack, F.J. Small non-coding RNAs in animal development. Nat. Rev. Mol. Cell Biol 2008, 9, 219–230. [Google Scholar]
- Hellen, C.U.; Sarnow, P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev 2001, 15, 1593–1612. [Google Scholar]
- Pisarev, A.V.; Shirokikh, N.E.; Hellen, C.U. Translation initiation by factor-independent binding of eukaryotic ribosomes to internal ribosomal entry sites. CR Biol 2005, 328, 589–605. [Google Scholar]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar]
- Rana, T.M. Illuminating the silence: Understanding the structure and function of small RNAs. Nat. Rev. Mol. Cell Biol 2007, 8, 23–36. [Google Scholar]
- Petersen, C.P.; Bordeleau, M.E.; Pelletier, J.; Sharp, P.A. Short RNAs repress translation after initiation in mammalian cells. Mol. Cell 2006, 21, 533–542. [Google Scholar]
- Parker, R.; Song, H. The enzymes and control of eukaryotic mRNA turnover. Nat. Struct. Mol. Biol 2004, 11, 121–127. [Google Scholar]
- Wu, L.; Fan, J.; Belasco, J.G. MicroRNAs direct rapid deadenylation of mRNA. Proc. Natl. Acad. Sci. USA 2006, 103, 4034–4039. [Google Scholar]
- Gibbings, D.J.; Ciaudo, C.; Erhardt, M.; Voinnet, O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat. Cell Biol 2009, 11, 1143–1149. [Google Scholar]
- Brengues, M.; Teixeira, D.; Parker, R. Movement of eukaryotic mRNAs between polysomes and cytoplasmic processing bodies. Science 2005, 310, 486–489. [Google Scholar]
- Truesdell, S.S.; Mortensen, R.D.; Seo, M.; Schroeder, J.C.; Lee, J.H.; Letonqueze, O.; Vasudevan, S. MicroRNA-mediated mRNA translation activation in quiescent cells and oocytes involves recruitment of a nuclear microRNP. Sci. Rep 2012, 2, 842. [Google Scholar]
- Mortensen, R.D.; Serra, M.; Steitz, J.A.; Vasudevan, S. Posttranscriptional activation of gene expression in Xenopus laevis oocytes by microRNA-protein complexes (microRNPs). Proc. Natl. Acad. Sci. USA 2011, 108, 8281–8286. [Google Scholar]
- Lytle, J.R.; Yario, T.A.; Steitz, J.A. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′ UTR as in the 3′ UTR. Proc. Natl. Acad. Sci. USA 2007, 104, 9667–9672. [Google Scholar]
- Lee, I.; Ajay, S.S.; Yook, J.I.; Kim, H.S.; Hong, S.H.; Kim, N.H.; Dhanasekaran, S.M.; Chinnaiyan, A.M.; Athey, B.D. New class of microRNA targets containing simultaneous 5′-UTR and 3′-UTR interaction sites. Genome Res 2009, 19, 1175–1183. [Google Scholar]
- Zhou, X.; Duan, X.; Qian, J.; Li, F. Abundant conserved microRNA target sites in the 5′-untranslated region and coding sequence. Genetica 2009, 137, 159–164. [Google Scholar]
- Moretti, F.; Thermann, R.; Hentze, M.W. Mechanism of translational regulation by miR-2 from sites in the 5′ untranslated region or the open reading frame. RNA 2010, 16, 2493–2502. [Google Scholar]
- Jin, Y.; Wang, C.; Liu, X.; Mu, W.; Chen, Z.; Yu, D.; Wang, A.; Dai, Y.; Zhou, X. Molecular characterization of the microRNA-138-Fos-like antigen 1 (FOSL1) regulatory module in squamous cell carcinoma. J. Biol. Chem 2011, 286, 40104–40109. [Google Scholar]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar]
- Jopling, C.L.; Schutz, S.; Sarnow, P. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell. Host Microbe 2008, 4, 77–85. [Google Scholar]
- Henke, J.I.; Goergen, D.; Zheng, J.; Song, Y.; Schuttler, C.G.; Fehr, C.; Junemann, C.; Niepmann, M. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J 2008, 27, 3300–3310. [Google Scholar]
- Jangra, R.K.; Yi, M.; Lemon, S.M. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J. Virol 2010, 84, 6615–6625. [Google Scholar]
- Villanueva, R.A.; Jangra, R.K.; Yi, M.; Pyles, R.; Bourne, N.; Lemon, S.M. miR-122 does not modulate the elongation phase of hepatitis C virus RNA synthesis in isolated replicase complexes. Antiviral Res 2010, 88, 119–123. [Google Scholar]
- Norman, K.L.; Sarnow, P. Modulation of hepatitis C virus RNA abundance and the isoprenoid biosynthesis pathway by microRNA miR-122 involves distinct mechanisms. J. Virol 2010, 84, 666–670. [Google Scholar]
- Roberts, A.P.; Lewis, A.P.; Jopling, C.L. miR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res 2011, 39, 7716–7729. [Google Scholar]
- Shimakami, T.; Yamane, D.; Jangra, R.K.; Kempf, B.J.; Spaniel, C.; Barton, D.J.; Lemon, S.M. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc. Natl. Acad. Sci. USA 2012, 109, 941–946. [Google Scholar]
- Orom, U.A.; Nielsen, F.C.; Lund, A.H. MicroRNA-10a binds the 5′UTR of ribosomal protein mRNAs and enhances their translation. Mol. Cell 2008, 30, 460–471. [Google Scholar]
- Tsai, N.P.; Lin, Y.L.; Wei, L.N. MicroRNA mir-346 targets the 5′-untranslated region of receptor-interacting protein 140 (RIP140) mRNA and up-regulates its protein expression. Biochem. J 2009, 424, 411–418. [Google Scholar]
- Rigoutsos, I. New tricks for animal microRNAS: Targeting of amino acid coding regions at conserved and nonconserved sites. Cancer Res 2009, 69, 3245–3248. [Google Scholar]
- Easow, G.; Teleman, A.A.; Cohen, S.M. Isolation of microRNA targets by miRNP immunopurification. RNA 2007, 13, 1198–1204. [Google Scholar]
- Jopling, C.L. Regulation of hepatitis C virus by microRNA-122. Biochem. Soc. Trans 2008, 36, 1220–1223. [Google Scholar]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab 2006, 3, 87–98. [Google Scholar]
- Meyuhas, O. Synthesis of the translational apparatus is regulated at the translational level. Eur. J. Biochem 2000, 267, 6321–6330. [Google Scholar]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol 2010, 11, R90. [Google Scholar]
- Maragkakis, M.; Vergoulis, T.; Alexiou, P.; Reczko, M.; Plomaritou, K.; Gousis, M.; Kourtis, K.; Koziris, N.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-microT Web server upgrade supports Fly and Worm miRNA target prediction and bibliographic miRNA to disease association. Nucleic Acids Res 2011, 39, W145–W148. [Google Scholar]
- Rehmsmeier, M.; Steffen, P.; Hochsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 2004, 10, 1507–1517. [Google Scholar]
- Grimson, A.; Farh, K.K.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar]
- Rusinov, V.; Baev, V.; Minkov, I.N.; Tabler, M. MicroInspector: A web tool for detection of miRNA binding sites in an RNA sequence. Nucleic Acids Res 2005, 33, W696–W700. [Google Scholar]
- Krek, A.; Grun, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet 2005, 37, 495–500. [Google Scholar]
- Kim, S.K.; Nam, J.W.; Rhee, J.K.; Lee, W.J.; Zhang, B.T. miTarget: microRNA target gene prediction using a support vector machine. BMC Bioinforma 2006, 7, 411. [Google Scholar]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.S.; Tam, W.L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet 2007, 39, 1278–1284. [Google Scholar]
- Thadani, R.; Tammi, M.T. MicroTar: Predicting microRNA targets from RNA duplexes. BMC Bioinforma 2006, 7, S20. [Google Scholar]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of microRNA-target recognition. PLoS Biol 2005, 3, e85. [Google Scholar]
- Hsu, J.B.; Chiu, C.M.; Hsu, S.D.; Huang, W.Y.; Chien, C.H.; Lee, T.Y.; Huang, H.D. miRTar: An integrated system for identifying miRNA-target interactions in human. BMC Bioinforma 2011, 12, 300. [Google Scholar]
- Dweep, H.; Sticht, C.; Pandey, P.; Gretz, N. miRWalk-database: Prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform 2011, 44, 839–847. [Google Scholar]
- Ding, Y.; Chan, C.Y.; Lawrence, C.E. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res 2004, 32, W135–W141. [Google Scholar]
- Long, D.; Lee, R.; Williams, P.; Chan, C.Y.; Ambros, V.; Ding, Y. Potent effect of target structure on microRNA function. Nat. Struct. Mol. Biol 2007, 14, 287–294. [Google Scholar]
- Kumar, A.; Wong, A.K.; Tizard, M.L.; Moore, R.J.; Lefevre, C. miRNA_Targets: A database for miRNA target predictions in coding and non-coding regions of mRNAs. Genomics 2012, 100, 352–356. [Google Scholar]
- Zhang, X.; Rice, K.; Wang, Y.; Chen, W.; Zhong, Y.; Nakayama, Y.; Zhou, Y.; Klibanski, A. Maternally expressed gene 3 (MEG3) noncoding ribonucleic acid: Isoform structure, expression, and functions. Endocrinology 2010, 151, 939–947. [Google Scholar]
- Baldassarre, A.; Masotti, A. Long non-coding RNAs and p53 regulation. Int. J. Mol. Sci 2012, 13, 16708–16717. [Google Scholar]
- Da Sacco, L.; Baldassarre, A.; Masotti, A. Bioinformatics tools and novel challenges in long non-coding RNAs (lncRNAs) functional analysis. Int. J. Mol. Sci 2012, 13, 97–114. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA Name | Target mRNA or protein | Function/Process/Interaction | Reference |
|---|---|---|---|
| Translation repression | |||
| miR-2 (Drosophila) | luciferase reporter gene | Deadenylation and formation of pseudopolysomes and reduction of 80S complex formation | [26] |
| miR-138 (Human) | FOSL1 | Tumor suppressor microRNA. Down regulation of FOS-like antigen-1 (FOSL1) affects snail homolog 2 (SNAI2) and repression of E-cadherin. Contribution to tumorigenesis, cancer initiation and progression | [27] |
| miR-34a (Human) | AXIN2 luciferase reporter assay | miBridge interaction (miRNA binding both 5′ and 3′ UTRs) | [24] |
| miR-605 (Human) | SEC24D luciferase reporter assay | miBridge interaction (miRNA binding both 5′ and 3′ UTRs) | [24] |
| Translation activation | |||
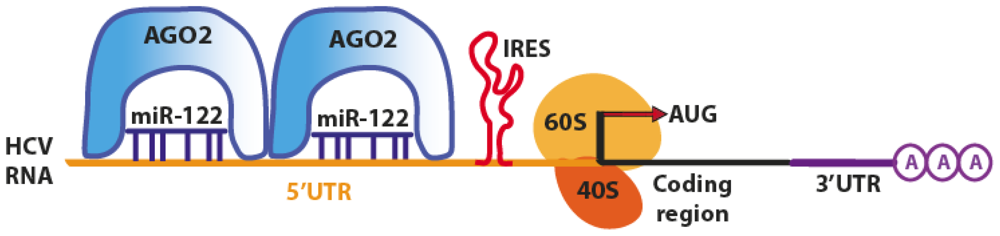
| miR-122 (Human) | HCV viral genome | Activation of translation of HCV mRNA, increase of 48s association in the initiation complex at the 5′UTR of viral genome. Accumulation of viral RNA and more efficient replication of HCV. | [28–35] |
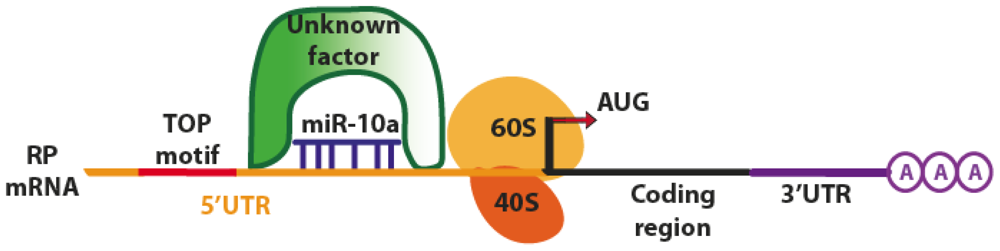
| miR-10a (Human) | mRNA Ribosomal Proteins | Binding to 5′ TOP of mRNAs stimulating translation and decreasing the repression induced by amino acid starvation. Control of ribosome biogenesis and global protein synthesis, oncogenic potential in transforming cells. 5′TOP regulation of cellular stress response. | [36] |
| miR-346 (Human) | RIP140 | Increase of the RIP140 level and direct increase of its repression activity. MiRNA is involved in the regulatory network to maintain the homeostasis in hormonal responses and metabolism. | [37] |
| Bioinformatics Resources | Year | Description | Web Link | Reference |
|---|---|---|---|---|
| miBridge | 2009 | Algorithm and Database | http://sitemaker.umich.edu/mibridge/home | [24] |
| miRTar | 2011 | Bioinformatics Tools integrated with KEGG pathways | http://miRTar.mbc.nctu.edu.tw | [54] |
| miRWalk | 2011 | Searchable database | http://mirwalk.uni-hd.de | [55] |
| Sfold-STarMirDB | 2007 | Algorithm and Database | http://sfold.wadsworth.org/cgi-bin/index.pl http://sfold.wadsworth.org/starmirDB.php | [56,57] |
| MiRNA_Targets | 2012 | Database and GO classification | http://mamsap.it.deakin.edu.au/mirna_targets/ | [58] |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sacco, L.D.; Masotti, A. Recent Insights and Novel Bioinformatics Tools to Understand the Role of MicroRNAs Binding to 5' Untranslated Region. Int. J. Mol. Sci. 2013, 14, 480-495. https://doi.org/10.3390/ijms14010480
Sacco LD, Masotti A. Recent Insights and Novel Bioinformatics Tools to Understand the Role of MicroRNAs Binding to 5' Untranslated Region. International Journal of Molecular Sciences. 2013; 14(1):480-495. https://doi.org/10.3390/ijms14010480
Chicago/Turabian StyleSacco, Letizia Da, and Andrea Masotti. 2013. "Recent Insights and Novel Bioinformatics Tools to Understand the Role of MicroRNAs Binding to 5' Untranslated Region" International Journal of Molecular Sciences 14, no. 1: 480-495. https://doi.org/10.3390/ijms14010480