Anticancer Effects of Bufalin on Human Hepatocellular Carcinoma HepG2 Cells: Roles of Apoptosis and Autophagy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
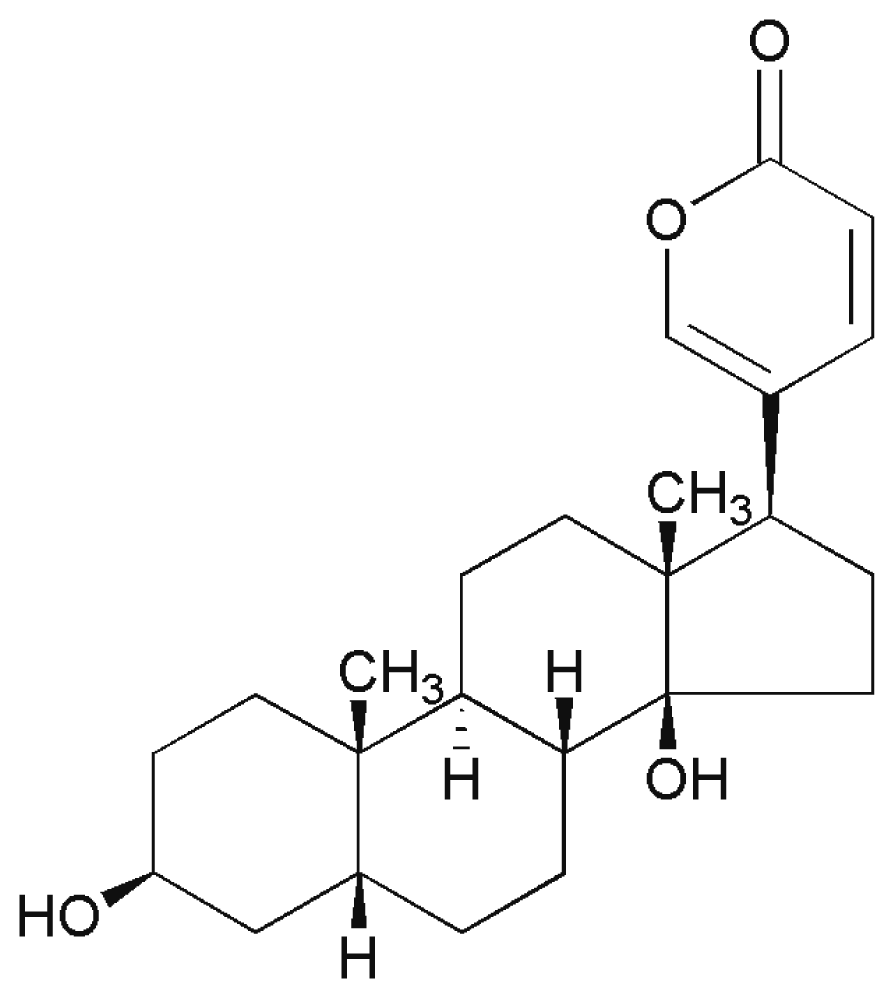
:1. Introduction
2. Results and Discussion
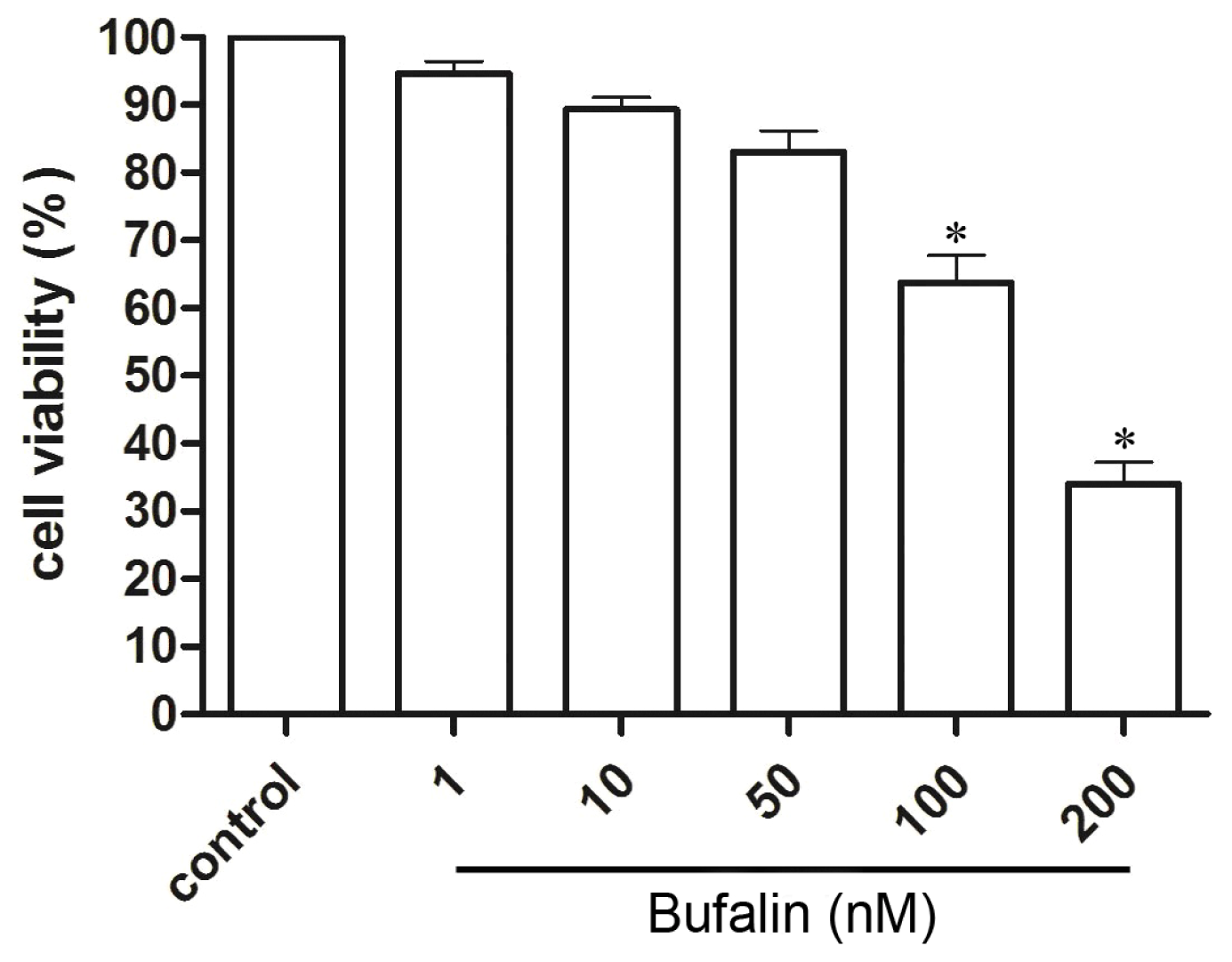
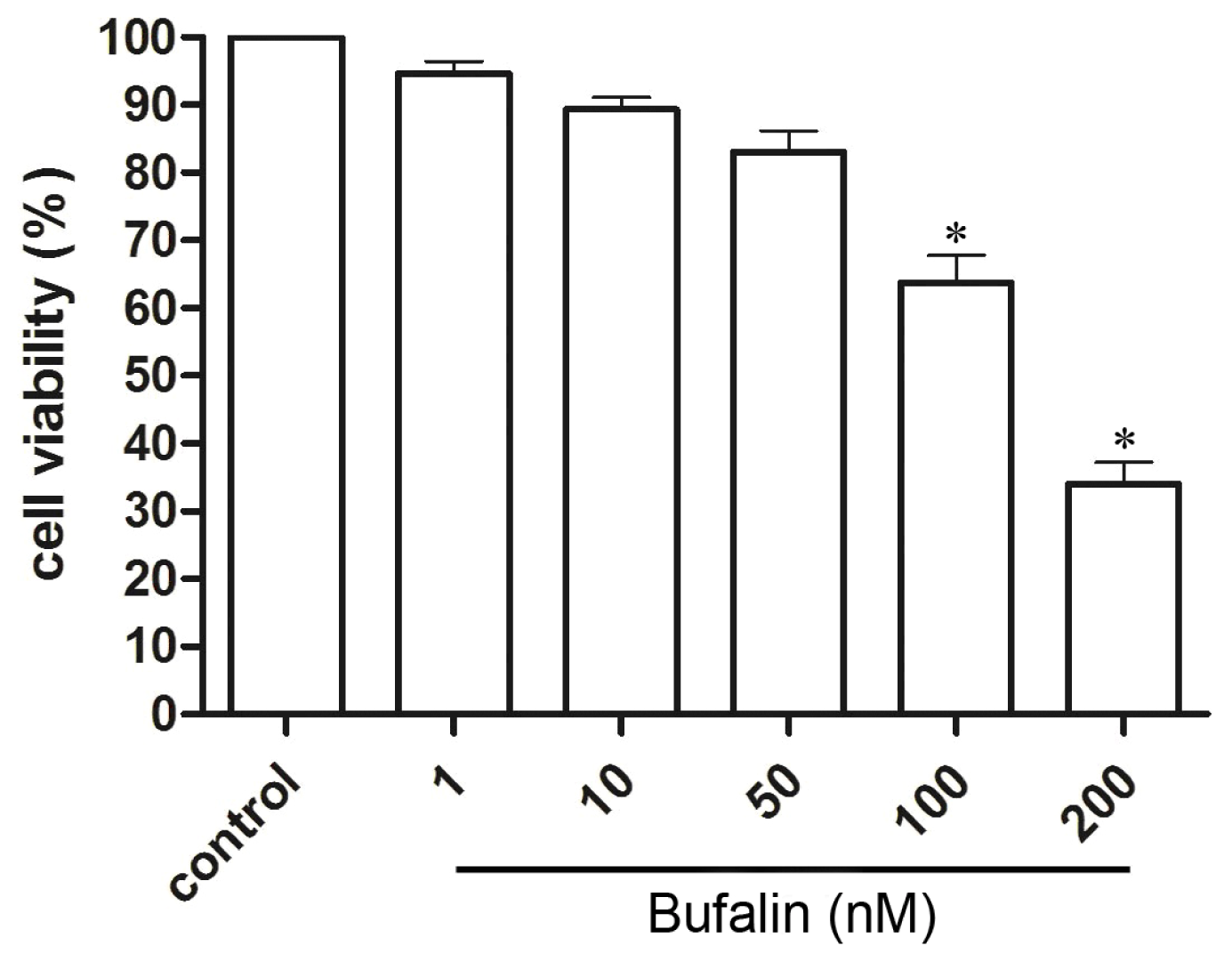
2.1. Bufalin Inhibits HepG2 Cell Proliferation
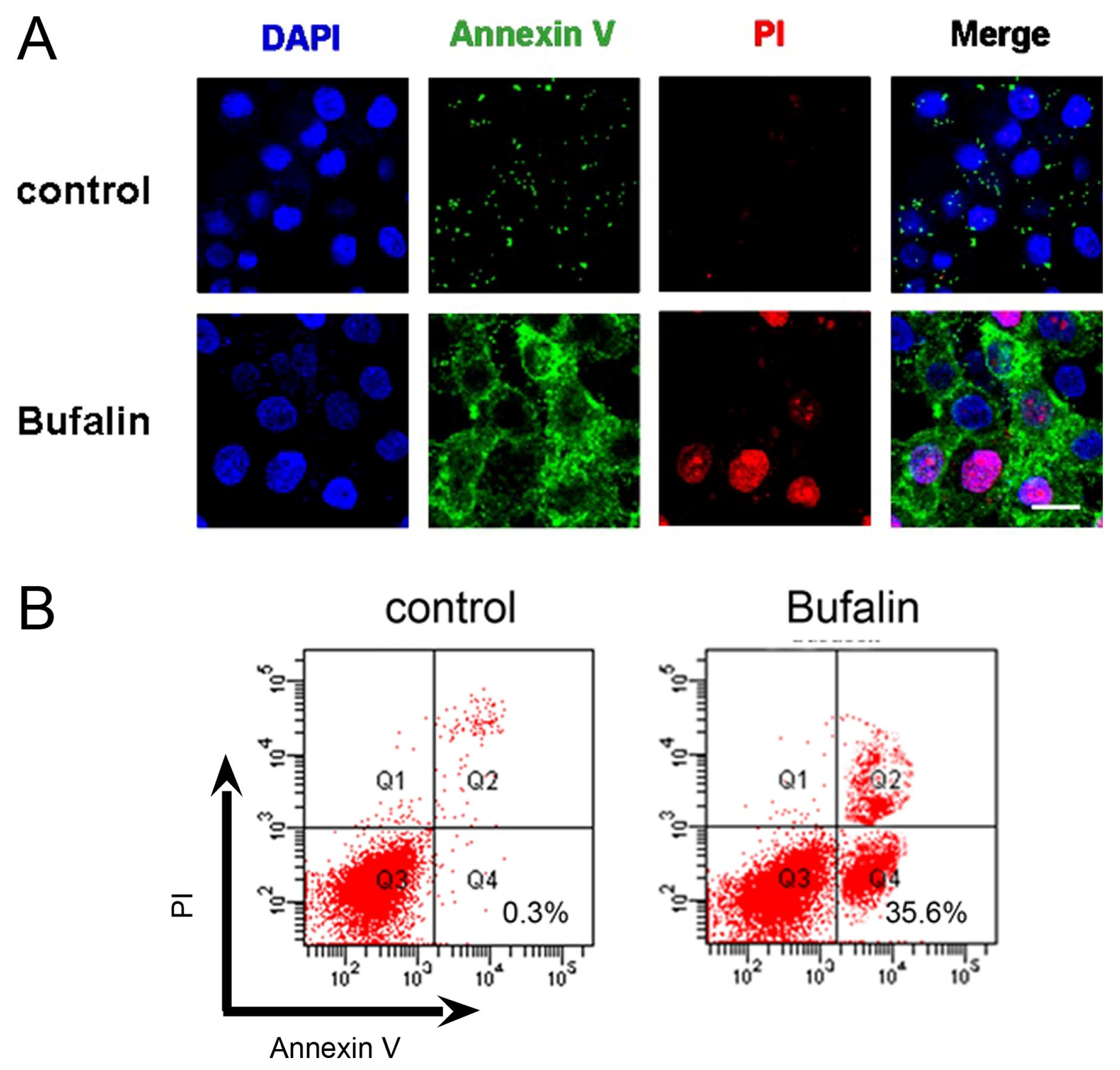
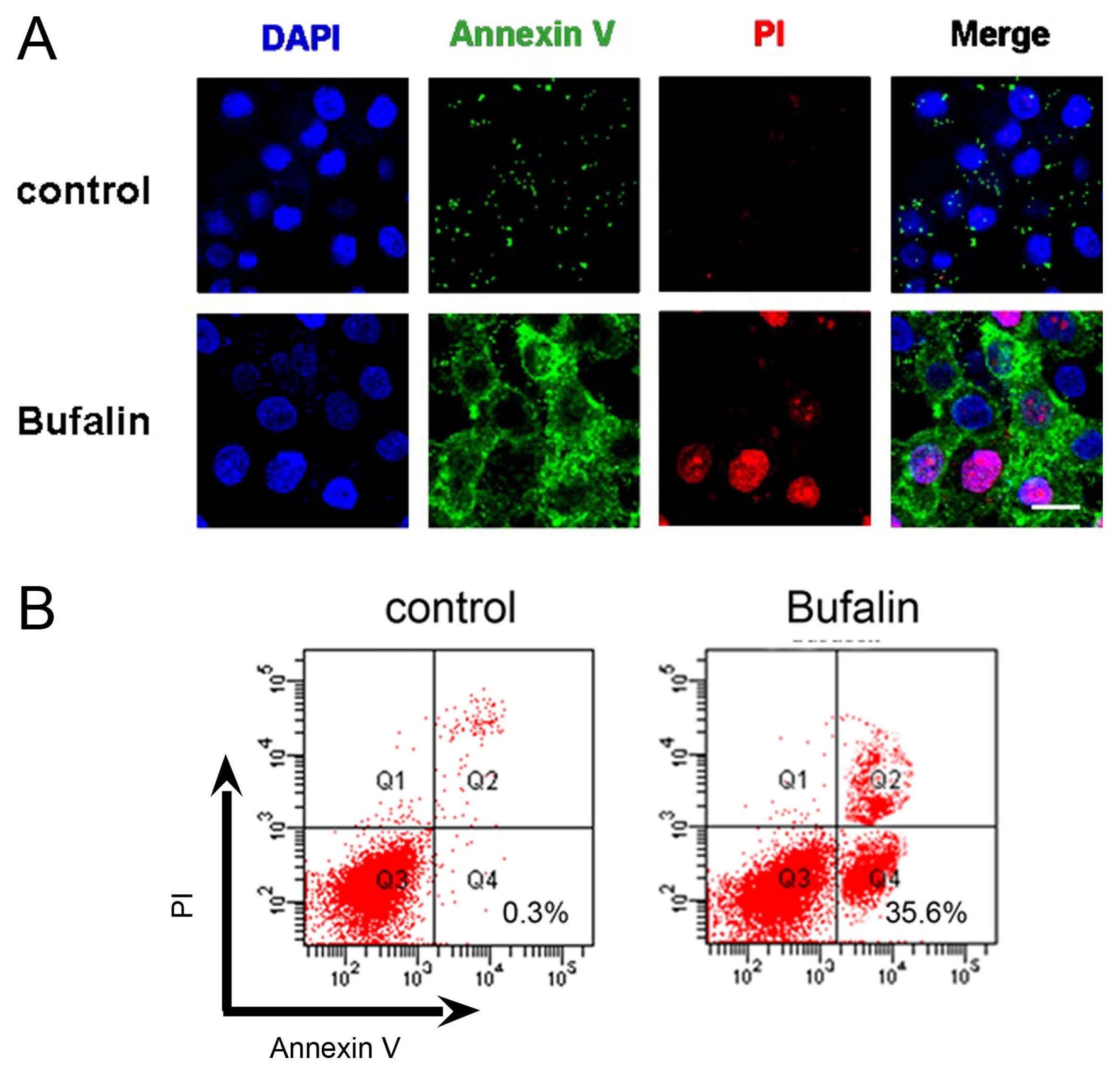
2.2. Bufalin Induces Apoptosis
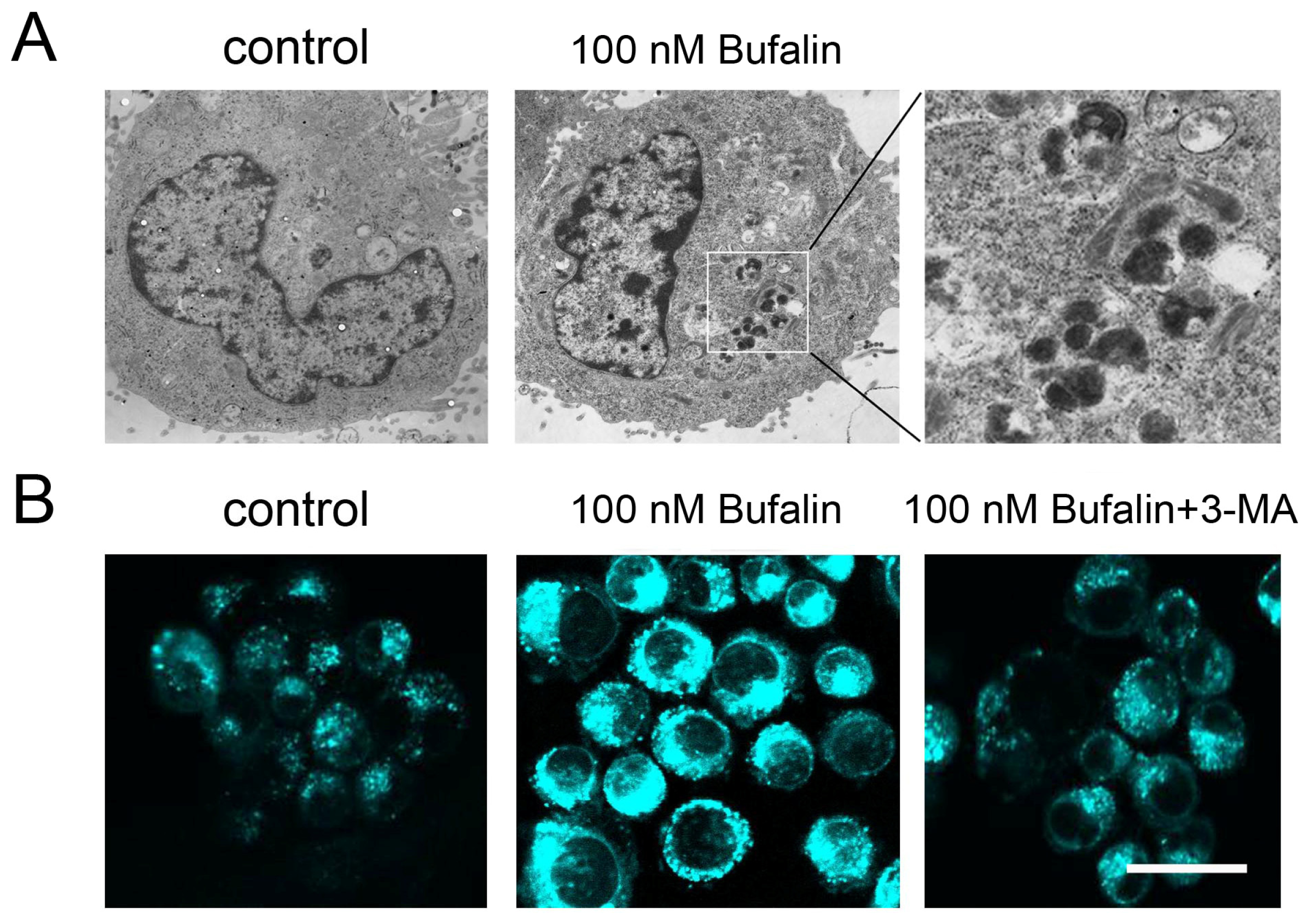
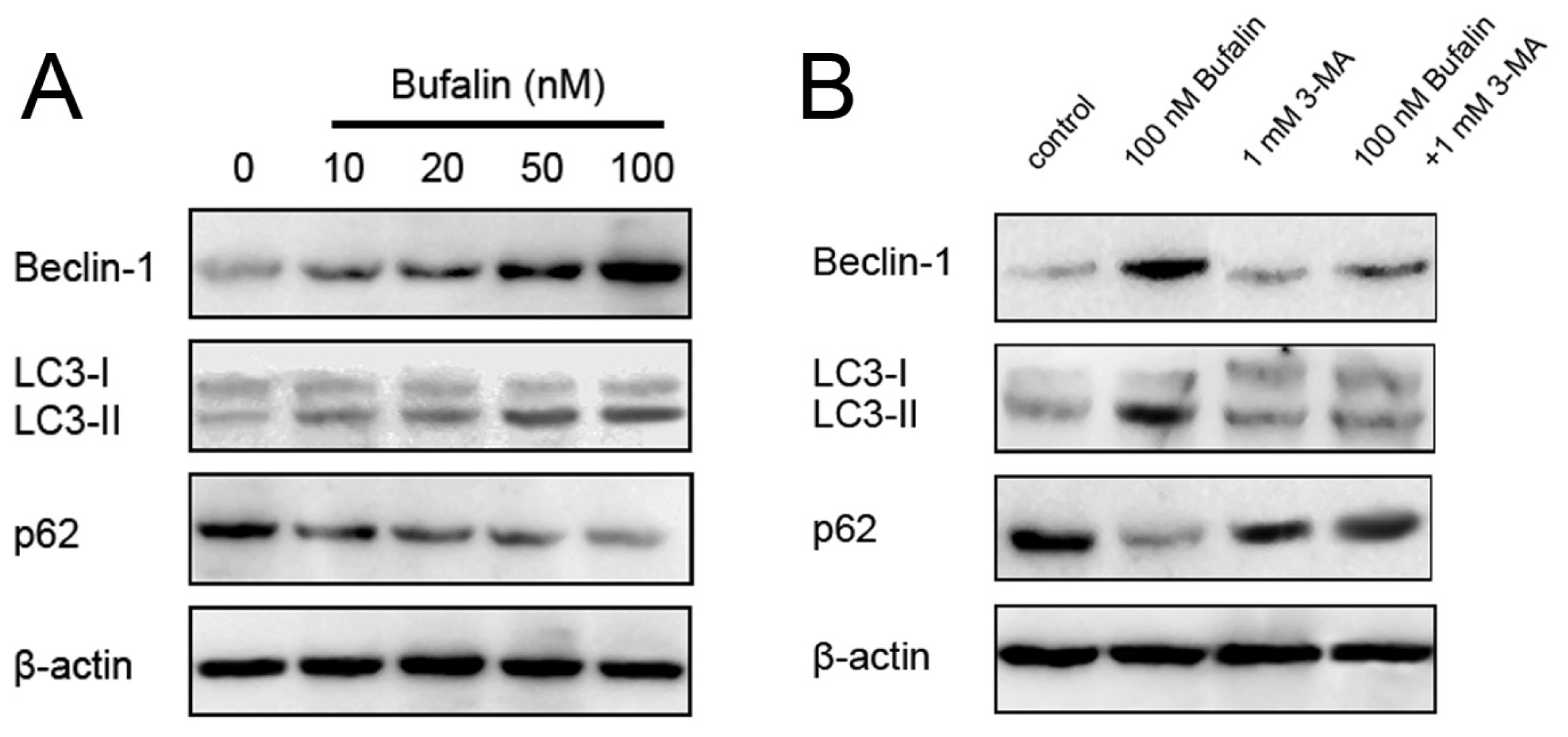
2.3. Bufalin Triggers Autophagy
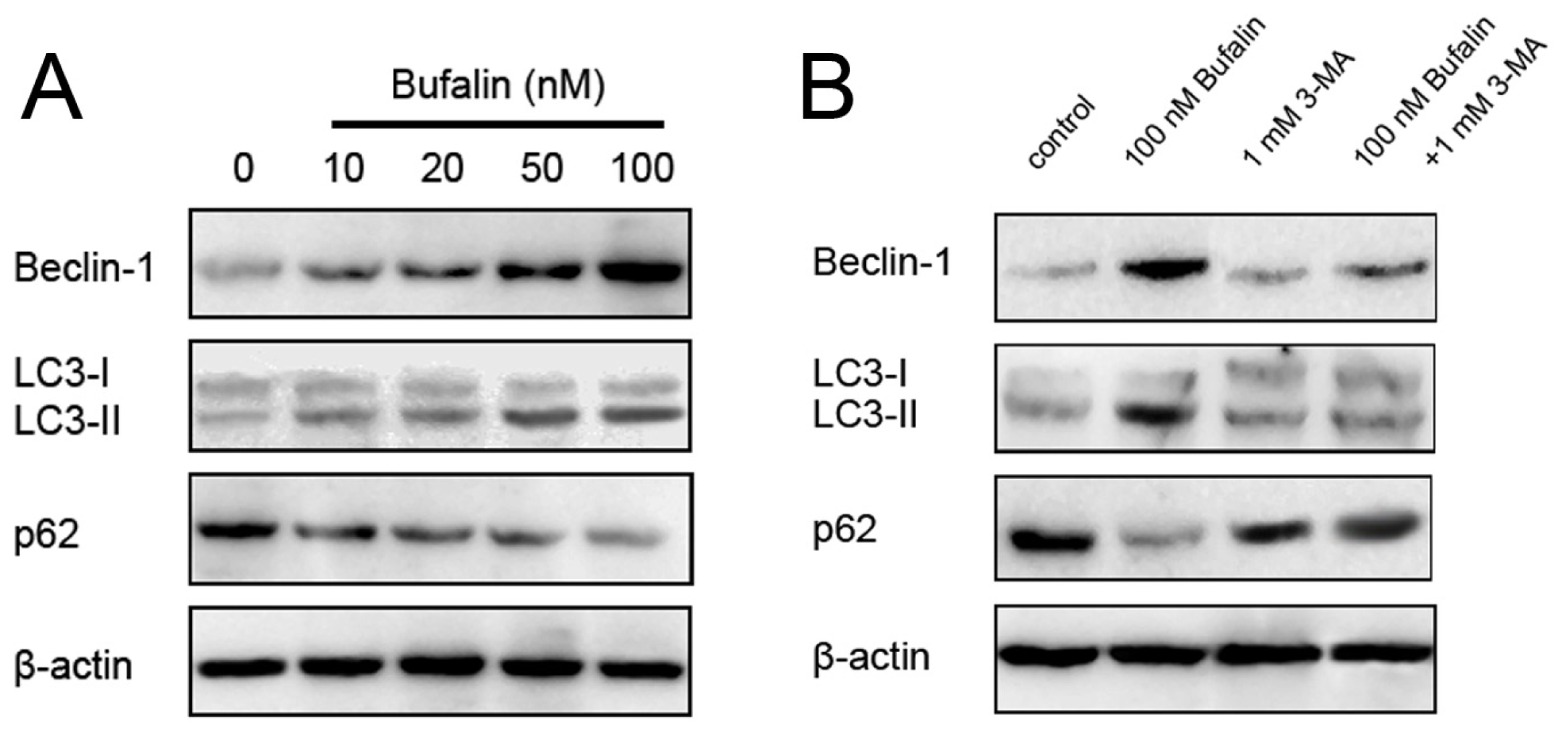
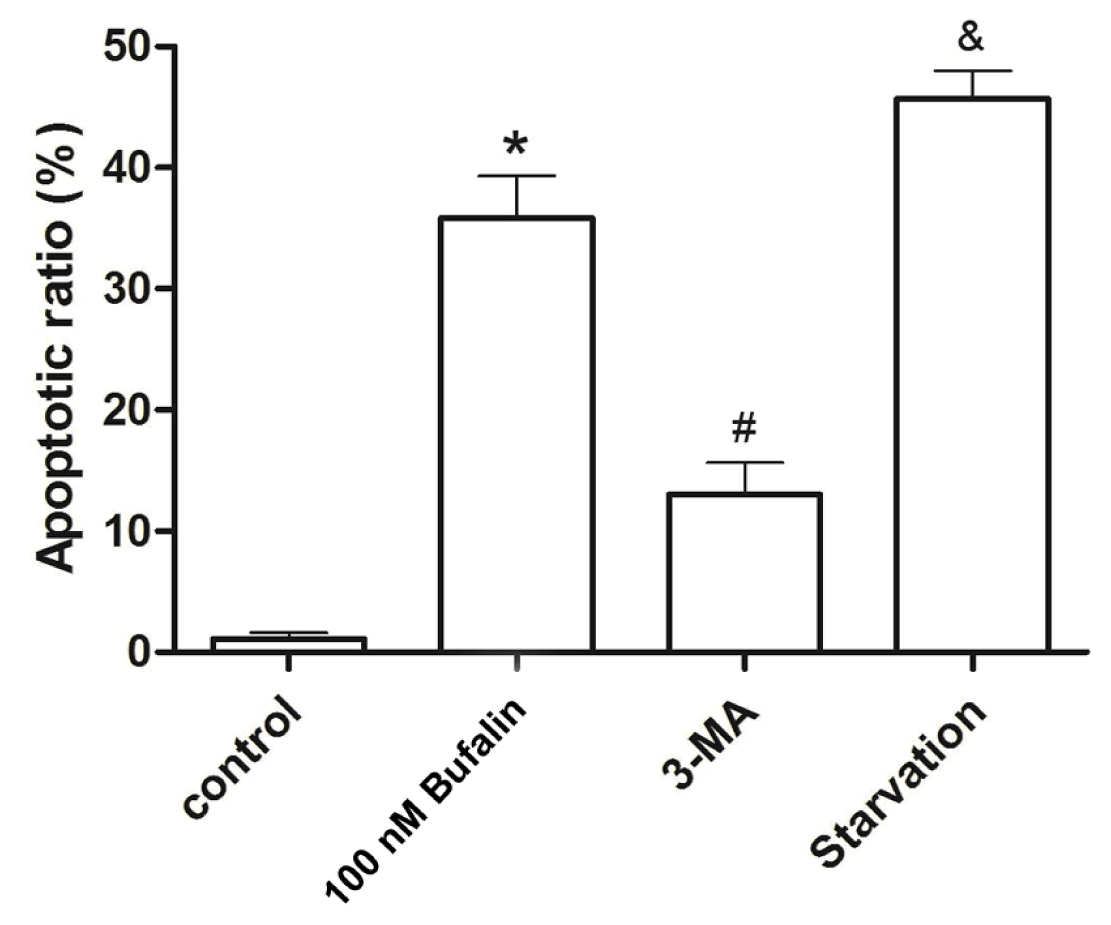
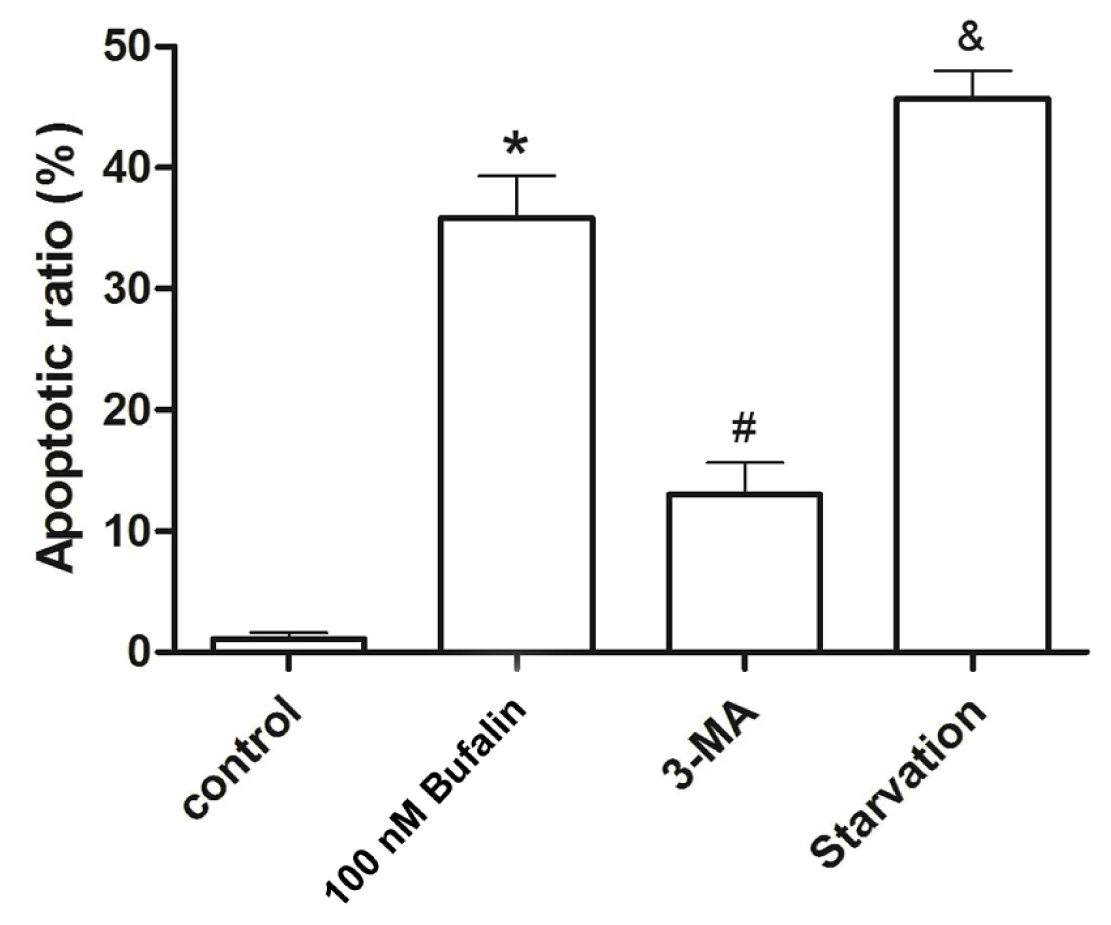
2.4. Bufalin-Induced Autophagy as a Proapoptotic Action
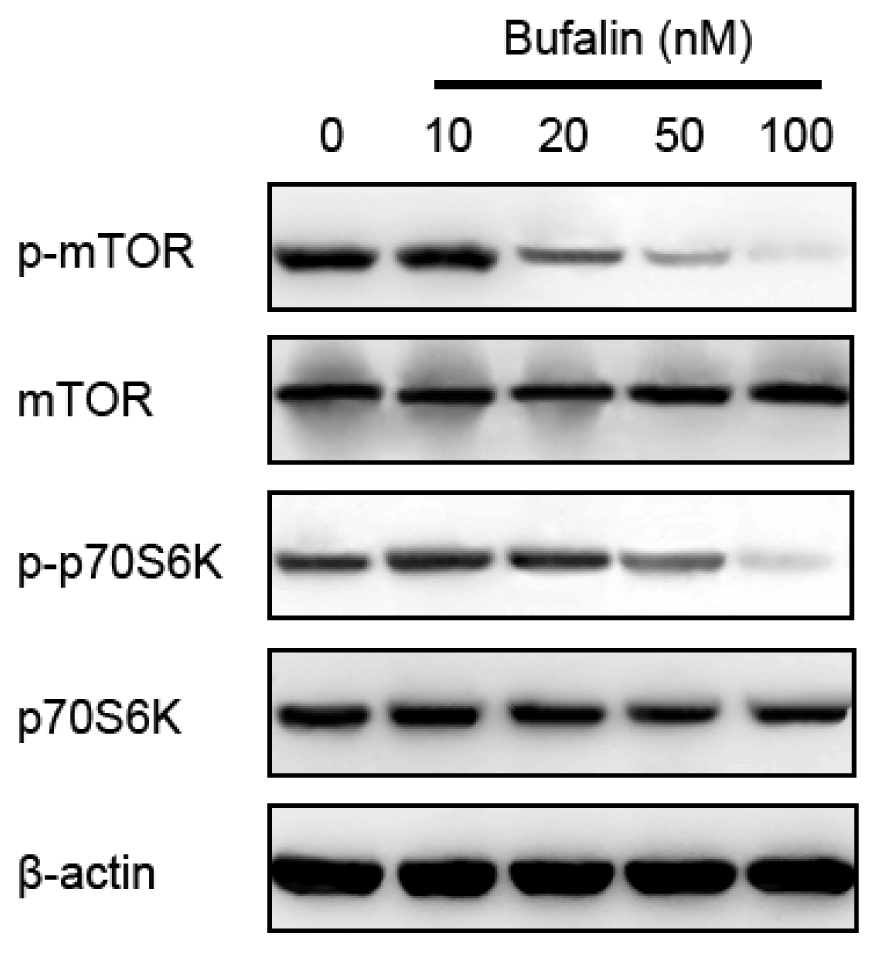
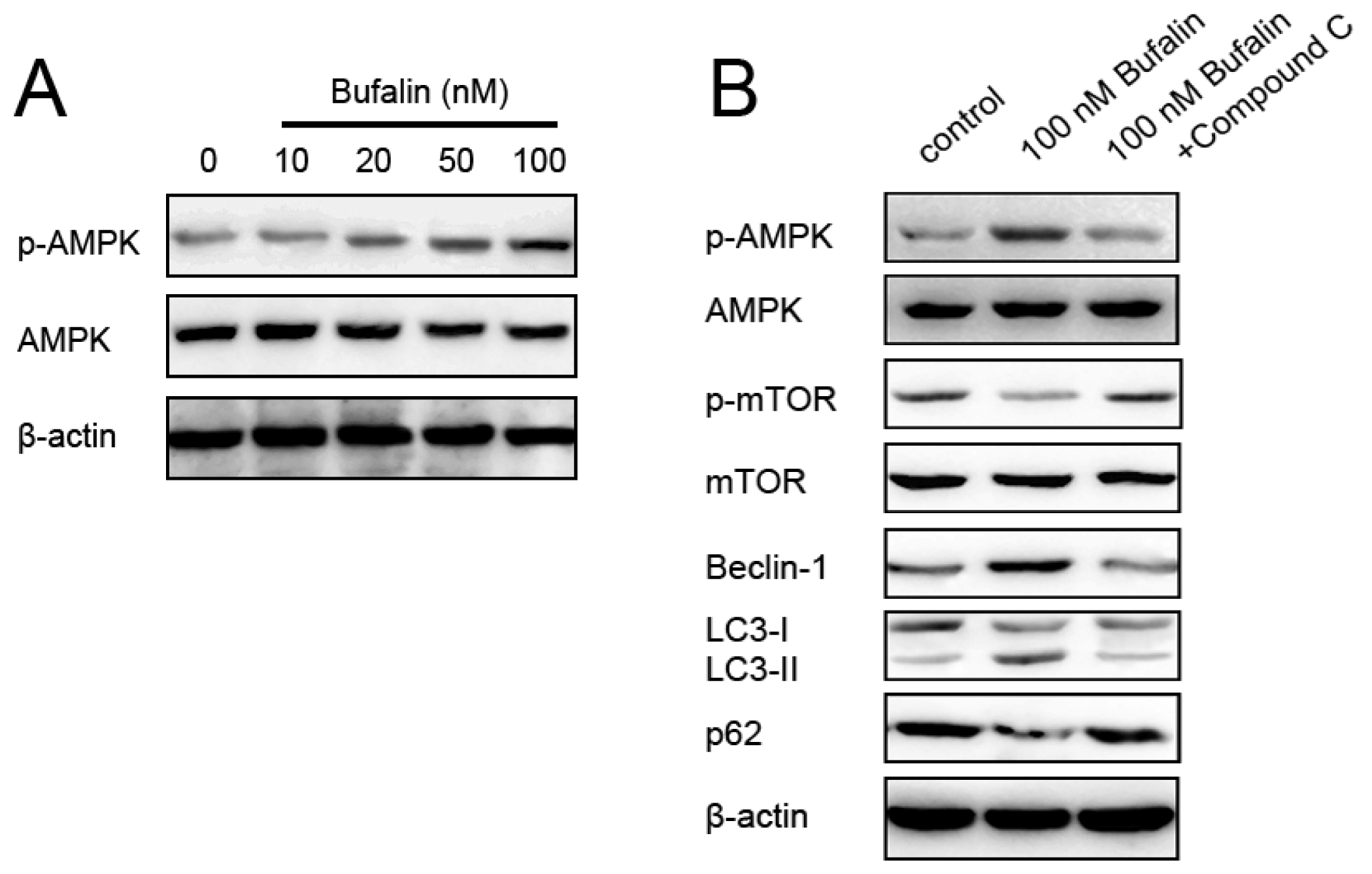
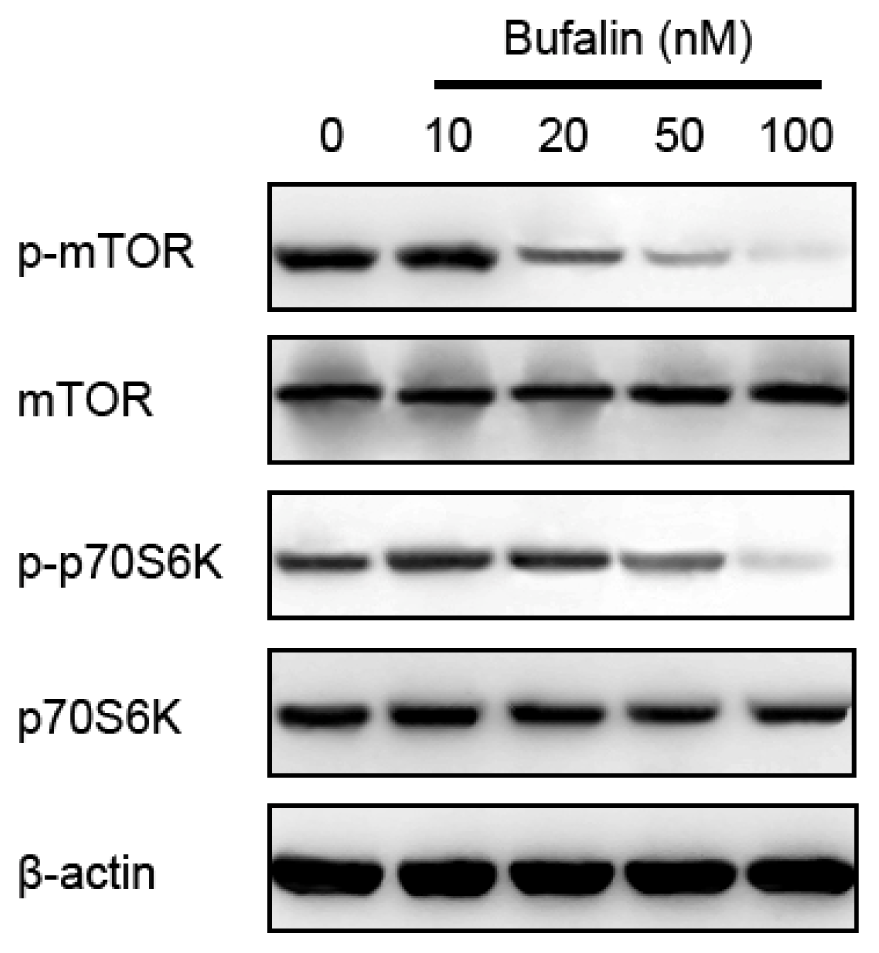
2.5. Bufalin-Induced Autophagy Is AMPK-mTOR Signaling-Dependent
3. Experimental Section
3.1. Reagents
3.2. Cell Culture
3.3. Cell Proliferation Assay
3.4. Measurement of Cell Apoptosis
3.5. Transmission Electron Microscopy of Autophagy
3.6. MDC Staining
3.7. Western Blot
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Fan, J.; Yang, G.S.; Fu, Z.R.; Peng, Z.H.; Xia, Q.; Peng, C.H.; Qian, J.M.; Zhou, J.; Xu, Y.; Qiu, S.J.; et al. Liver transplantation outcomes in 1078 hepatocellular carcinoma patients: A multi-center experience in Shanghai, China. J. Cancer Res. Clin. Oncol 2009, 135, 1403–1412. [Google Scholar]
- Yamamoto, J.; Okada, S.; Shimada, K.; Okusaka, T.; Yamasaki, S.; Ueno, H.; Kosuge, T. Treatment strategy for small hepatocellular carcinoma: Comparison of long-term results after percutaneous ethanol injection therapy and surgical resection. Hepatology 2001, 34, 707–713. [Google Scholar]
- Aravalli, R.N.; Steer, C.J.; Cressman, E.N. Molecular mechanisms of hepatocellular carcinoma. Hepatology 2008, 48, 2047–2063. [Google Scholar]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med 2008, 359, 378–390. [Google Scholar]
- Rimassa, L.; Santoro, A. Sorafenib therapy in advanced hepatocellular carcinoma: The SHARP trial. Expert Rev. Anticancer Ther 2009, 9, 739–745. [Google Scholar]
- Kelley, R.K.; Venook, A.P. Sorafenib in hepatocellular carcinoma: Separating the hype from the hope. J. Clin. Oncol 2008, 26, 5845–5848. [Google Scholar]
- Wang, P.S.; Yeh, J.Y.; Yu, C.H.; Wang, S.W. An Evidence-based Perspective of Bufo Gargarizans (Asiatic Toad) for Cancer Patients. In Evidence-Based Anticancer Materia Medica; Cho, W.C., Ed.; Springer: Berlin, Germany, 2011; pp. 389–407. [Google Scholar]
- Takai, N.; Kira, N.; Ishii, T.; Yoshida, T.; Nishida, M.; Nishida, Y.; Nasu, K.; Narahara, H. Bufalin, a traditional oriental medicine, induces apoptosis in human cancer cells. Asian Pac. J. Cancer Prev 2012, 13, 399–402. [Google Scholar]
- Takai, N.; Ueda, T.; Nishida, M.; Nasu, K.; Narahara, H. Bufalin induces growth inhibition, cell cycle arrest and apoptosis in human endometrial and ovarian cancer cells. Int. J. Mol. Med 2008, 21, 637–643. [Google Scholar]
- Li, D.; Qu, X.; Hou, K.; Zhang, Y.; Dong, Q.; Teng, Y.; Zhang, J.; Liu, Y. PI3K/Akt is involved in bufalin-induced apoptosis in gastric cancer cells. Anticancer Drugs 2009, 20, 59–64. [Google Scholar]
- Yeh, J.Y.; Huang, W.J.; Kan, S.F.; Wang, P.S. Effects of bufalin and cinobufagin on the proliferation of androgen dependent and independent prostate cancer cells. Prostate 2003, 54, 112–124. [Google Scholar]
- Tsai, S.C.; Yang, J.S.; Peng, S.F.; Lu, C.C.; Chiang, J.H.; Chung, J.G.; Lin, M.W.; Lin, J.K.; Amagaya, S.; Wai-Shan Chung, C.; et al. Bufalin increases sensitivity to AKT/mTOR-induced autophagic cell death in SK-HEP-1 human hepatocellular carcinoma cells. Int. J. Oncol 2012, 41, 1431–1442. [Google Scholar]
- Jing, Y.; Watabe, M.; Hashimoto, S.; Nakajo, S.; Nakaya, K. Cell cycle arrest and protein kinase modulating effect of bufalin on human leukemia ML1 cells. Anticancer Res 1994, 14, 1193–1198. [Google Scholar]
- Hsiao, Y.P.; Yu, C.S.; Yu, C.C.; Yang, J.S.; Chiang, J.H.; Lu, C.C.; Huang, H.Y.; Tang, N.Y.; Yang, J.H.; Huang, A.C.; et al. Triggering apoptotic death of human malignant melanoma a375.s2 cells by bufalin: Involvement of caspase cascade-dependent and independent mitochondrial signaling pathways. Evid. Based Complement. Alternat. Med 2012, 2012, 591241. [Google Scholar]
- Jiang, Y.; Zhang, Y.; Luan, J.; Duan, H.; Zhang, F.; Yagasaki, K.; Zhang, G. Effects of bufalin on the proliferation of human lung cancer cells and its molecular mechanisms of action. Cytotechnology 2010, 62, 573–583. [Google Scholar]
- Dong, Y.; Yin, S.; Li, J.; Jiang, C.; Ye, M.; Hu, H. Bufadienolide compounds sensitize human breast cancer cells to TRAIL-induced apoptosis via inhibition of STAT3/Mcl-1 pathway. Apoptosis 2011, 16, 394–403. [Google Scholar]
- Xie, C.M.; Chan, W.Y.; Yu, S.; Zhao, J.; Cheng, C.H. Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Radic. Biol. Med 2011, 51, 1365–1375. [Google Scholar]
- Xie, X.B.; Yin, J.Q.; Wen, L.L.; Gao, Z.H.; Zou, C.Y.; Wang, J.; Huang, G.; Tang, Q.L.; Colombo, C.; He, W.L.; et al. Critical role of heat shock protein 27 in bufalin-induced apoptosis in human osteosarcomas: A proteomic-based research. PLoS One 2012, 7, e47375. [Google Scholar]
- Yin, J.Q.; Shen, J.N.; Su, W.W.; Wang, J.; Huang, G.; Jin, S.; Guo, Q.C.; Zou, C.Y.; Li, H.M.; Li, F.B. Bufalin induces apoptosis in human osteosarcoma U-2OS and U-2OS methotrexate300-resistant cell lines. Acta Pharmacol. Sin 2007, 28, 712–720. [Google Scholar]
- Cui, X.; Inagaki, Y.; Xu, H.; Wang, D.; Qi, F.; Kokudo, N.; Fang, D.; Tang, W. Anti-hepatitis B virus activities of cinobufacini and its active components bufalin and cinobufagin in HepG2.2.15 cells. Biol. Pharm. Bull 2010, 33, 1728–1732. [Google Scholar]
- Qi, F.; Inagaki, Y.; Gao, B.; Cui, X.; Xu, H.; Kokudo, N.; Li, A.; Tang, W. Bufalin and cinobufagin induce apoptosis of human hepatocellular carcinoma cells via Fas- and mitochondria-mediated pathways. Cancer Sci 2011, 102, 951–958. [Google Scholar]
- Ye, M.X.; Zhao, Y.L.; Li, Y.; Miao, Q.; Li, Z.K.; Ren, X.L.; Song, L.Q.; Yin, H.; Zhang, J. Curcumin reverses cis-platin resistance and promotes human lung adenocarcinoma A549/DDP cell apoptosis through HIF-1α and caspase-3 mechanisms. Phytomedicine 2012, 19, 779–787. [Google Scholar]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar]
- Mizushima, N.; Ohsumi, Y.; Yoshimori, T. Autophagosome formation in mammalian cells. Cell Struct. Funct 2002, 27, 421–429. [Google Scholar]
- Liu, Y.L.; Yang, P.M.; Shun, C.T.; Wu, M.S.; Weng, J.R.; Chen, C.C. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010, 6, 1057–1065. [Google Scholar]
- Nahimana, A.; Attinger, A.; Aubry, D.; Greaney, P.; Ireson, C.; Thougaard, A.V.; Tjørnelund, J.; Dawson, K.M.; Dupuis, M.; Duchosal, M.A. The NAD biosynthesis inhibitor APO866 has potent antitumor activity against hematologic malignancies. Blood 2009, 113, 3276–3286. [Google Scholar]
- Kim, D.K.; Yang, J.S.; Maiti, K.; Hwang, J.I.; Kim, K.; Seen, D.; Ahn, Y.; Lee, C.; Kang, B.C.; Kwon, H.B.; et al. A gonadotropin-releasing hormone-II antagonist induces autophagy of prostate cancer cells. Cancer Res 2009, 69, 923–931. [Google Scholar]
- Kim, J.Y.; Cho, T.J.; Woo, B.H.; Choi, K.U.; Lee, C.H.; Ryu, M.H.; Park, H.R. Curcumin-induced autophagy contributes to the decreased survival of oral cancer cells. Arch. Oral Biol 2012, 57, 1018–1025. [Google Scholar]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar]
- Apel, A.; Zentgraf, H.; Büchler, M.W.; Herr, I. Autophagy—A double-edged sword in oncology. Int. J. Cancer 2009, 125, 991–995. [Google Scholar]
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ 2009, 16, 966–975. [Google Scholar]
- Meijer, A.J.; Codogno, P. Regulation and role of autophagy in mammalian cells. Int. J. Biochem. Cell Biol 2004, 36, 2445–2462. [Google Scholar]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 2005, 1, 15–25. [Google Scholar]
- Hay, N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell 2005, 8, 179–183. [Google Scholar]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network organization of the human autophagy system. Nature 2010, 466, 68–76. [Google Scholar]
- Herrero-Martín, G.; Høyer-Hansen, M.; García-García, C.; Fumarola, C.; Farkas, T.; López-Rivas, A.; Jäättelä, M. TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J 2009, 28, 677–685. [Google Scholar]
- Høyer-Hansen, M.; Jäättelä, M. AMP-activated protein kinase: A universal regulator of autophagy? Autophagy 2007, 3, 381–383. [Google Scholar]
- Misirkic, M.; Janjetovic, K.; Vucicevic, L.; Tovilovic, G.; Ristic, B.; Vilimanovich, U.; Harhaji-Trajkovic, L.; Sumarac-Dumanovic, M.; Micic, D.; Bumbasirevic, V.; et al. Inhibition of AMPK-dependent autophagy enhances in vitro antiglioma effect of simvastatin. Pharmacol. Res 2012, 65, 111–119. [Google Scholar]


© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Miao, Q.; Bi, L.-L.; Li, X.; Miao, S.; Zhang, J.; Zhang, S.; Yang, Q.; Xie, Y.-H.; Zhang, J.; Wang, S.-W. Anticancer Effects of Bufalin on Human Hepatocellular Carcinoma HepG2 Cells: Roles of Apoptosis and Autophagy. Int. J. Mol. Sci. 2013, 14, 1370-1382. https://doi.org/10.3390/ijms14011370
Miao Q, Bi L-L, Li X, Miao S, Zhang J, Zhang S, Yang Q, Xie Y-H, Zhang J, Wang S-W. Anticancer Effects of Bufalin on Human Hepatocellular Carcinoma HepG2 Cells: Roles of Apoptosis and Autophagy. International Journal of Molecular Sciences. 2013; 14(1):1370-1382. https://doi.org/10.3390/ijms14011370
Chicago/Turabian StyleMiao, Qing, Lin-Lin Bi, Xin Li, Shan Miao, Jin Zhang, Song Zhang, Qian Yang, Yan-Hua Xie, Jian Zhang, and Si-Wang Wang. 2013. "Anticancer Effects of Bufalin on Human Hepatocellular Carcinoma HepG2 Cells: Roles of Apoptosis and Autophagy" International Journal of Molecular Sciences 14, no. 1: 1370-1382. https://doi.org/10.3390/ijms14011370
APA StyleMiao, Q., Bi, L.-L., Li, X., Miao, S., Zhang, J., Zhang, S., Yang, Q., Xie, Y.-H., Zhang, J., & Wang, S.-W. (2013). Anticancer Effects of Bufalin on Human Hepatocellular Carcinoma HepG2 Cells: Roles of Apoptosis and Autophagy. International Journal of Molecular Sciences, 14(1), 1370-1382. https://doi.org/10.3390/ijms14011370