HGF–MET Cascade, a Key Target for Inhibiting Cancer Metastasis: The Impact of NK4 Discovery on Cancer Biology and Therapeutics

Abstract
:1. Introduction
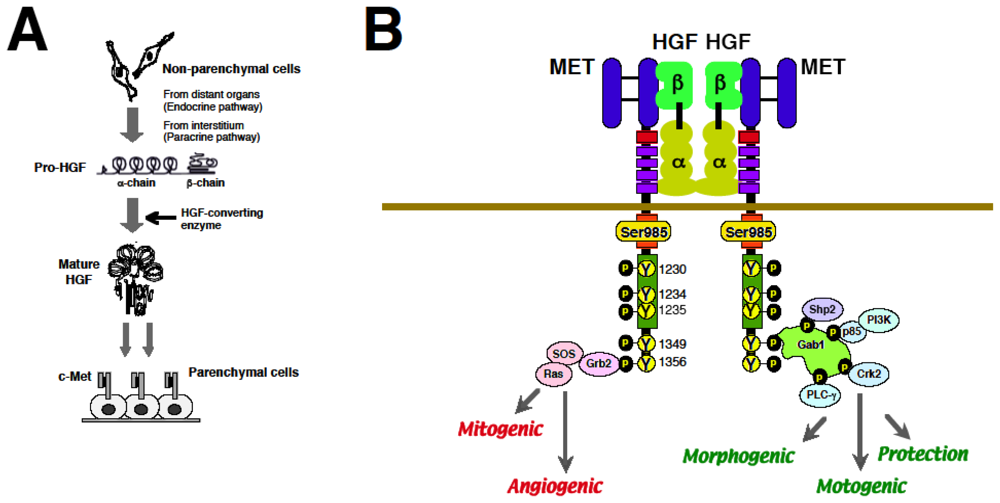
2. Discovery of HGF and MET
2.1. Discovery of HGF and Scatter Factor
2.2. Identification of MET as a Functional Receptor for HGF
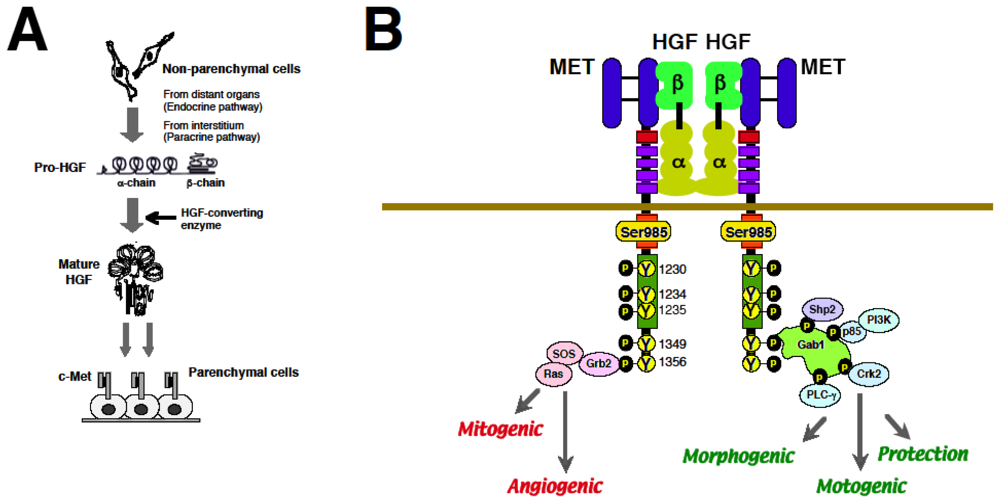
2.3. Biological Properties of HGF via MET
2.4. HGF–MET System for Organ Development and Regeneration
3. Roles of HGF–MET Axis in Tumorigenesis
3.1. Roles of MET Mutations for Oncogenesis
3.1.1. In Vitro Study
3.1.2. Animal Study
3.1.3. Human Studies
3.2. Molecular Basis of MET-Mediated Cancer Development
4. HGF-Mediated Cancer Metastasis
4.1. Functions of HGF for Tumor Cell Scattering and Invasion
4.1.1. Tumor Cell Dissociation
4.1.2. Cell Movement
4.1.3. Basement Membrane Breakdown
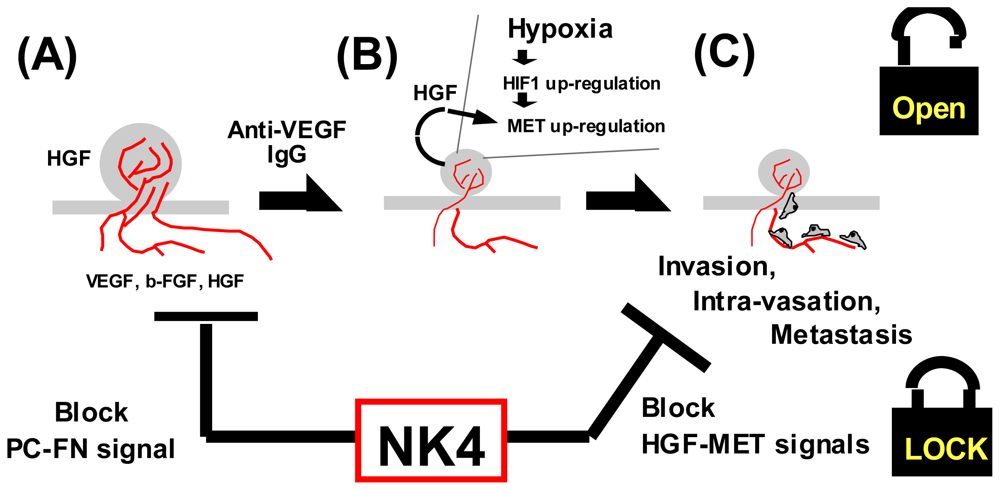
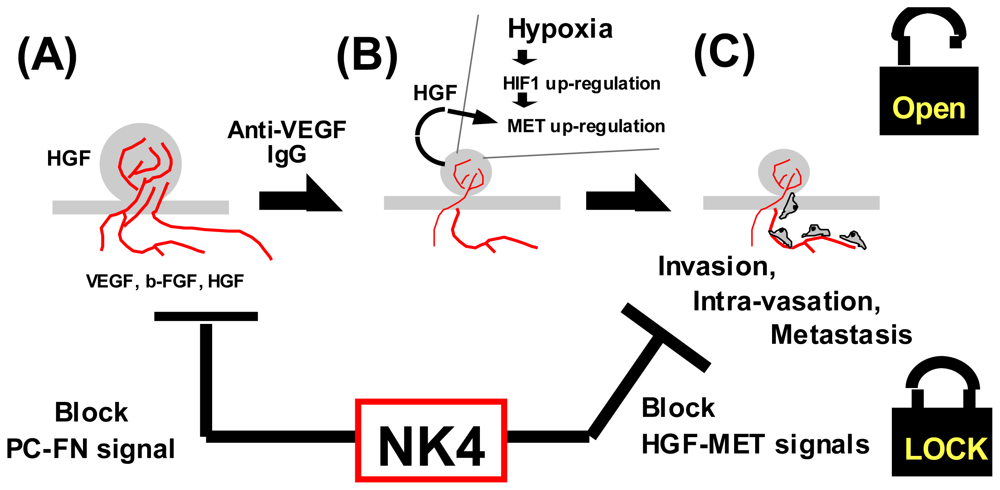
4.1.4. Hypoxia as a Trigger for Invasion
4.2. Roles of HGF during Metastatic Cascade
4.2.1. Angiogenic Events
4.2.2. Anti-Anoikis
4.2.3. Homing
4.2.4. Immune Tolerance
4.3. Regulation of HGF Production during Cancer Progression
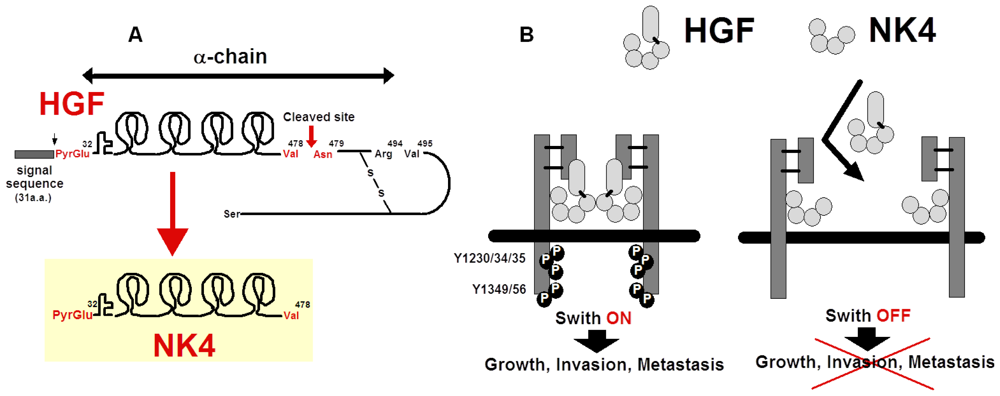
5. Preparation of NK4 as HGF-Antagonist
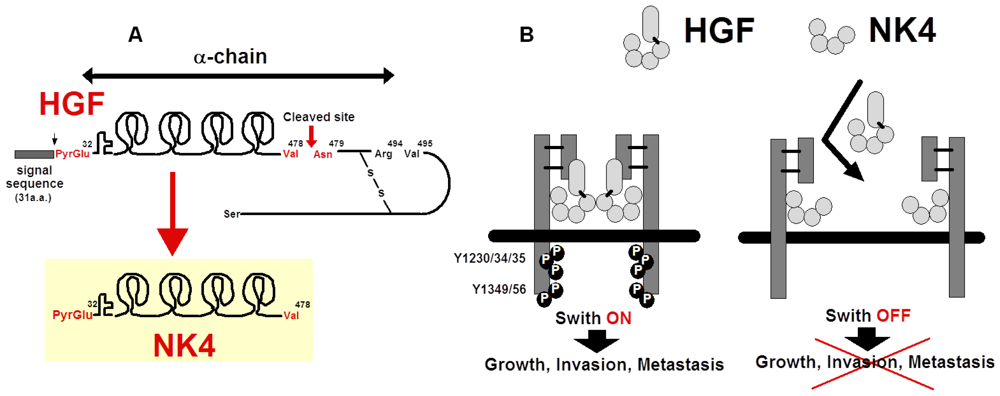
5.1. Structure and Anti-Invasive Function of NK4
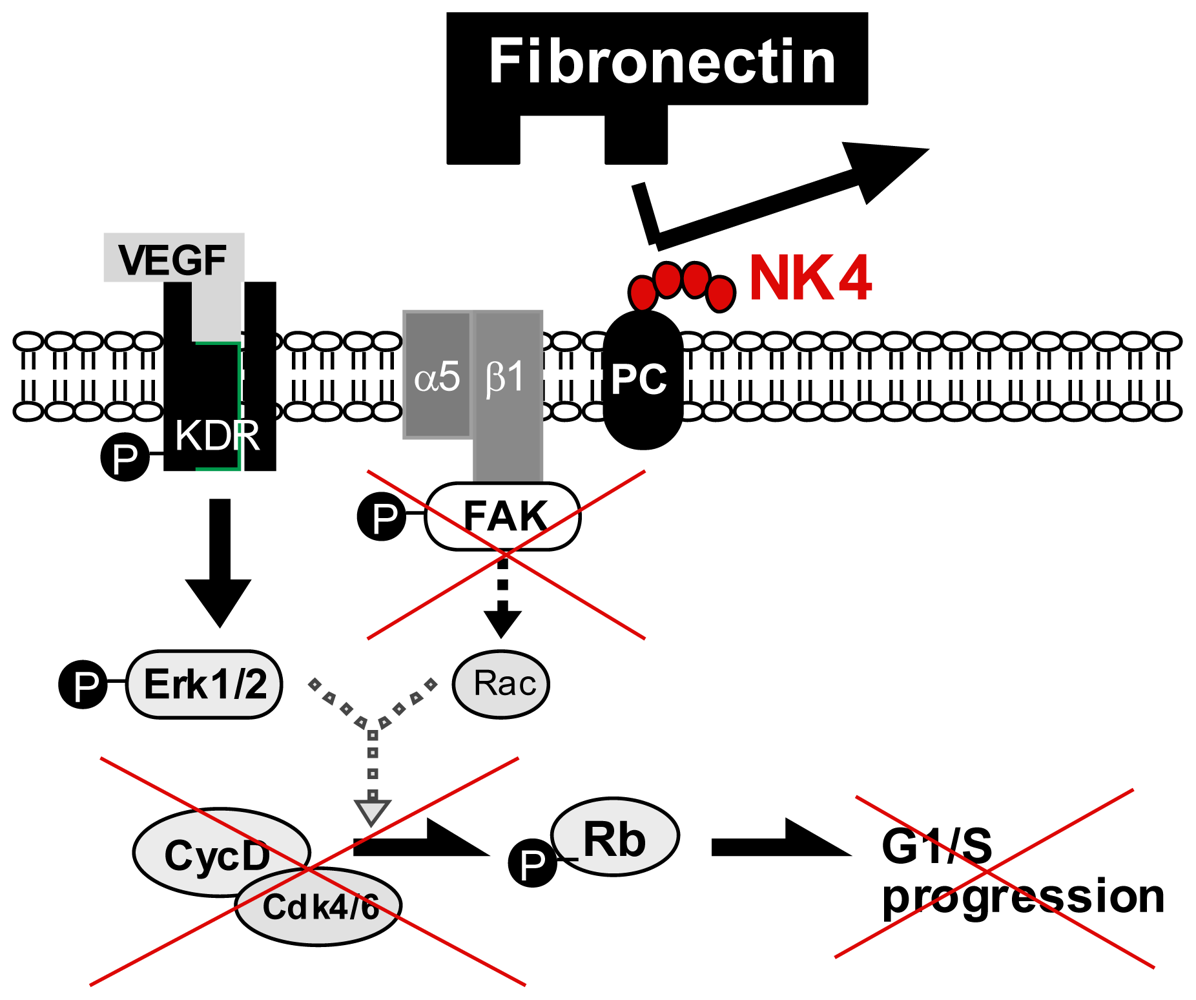
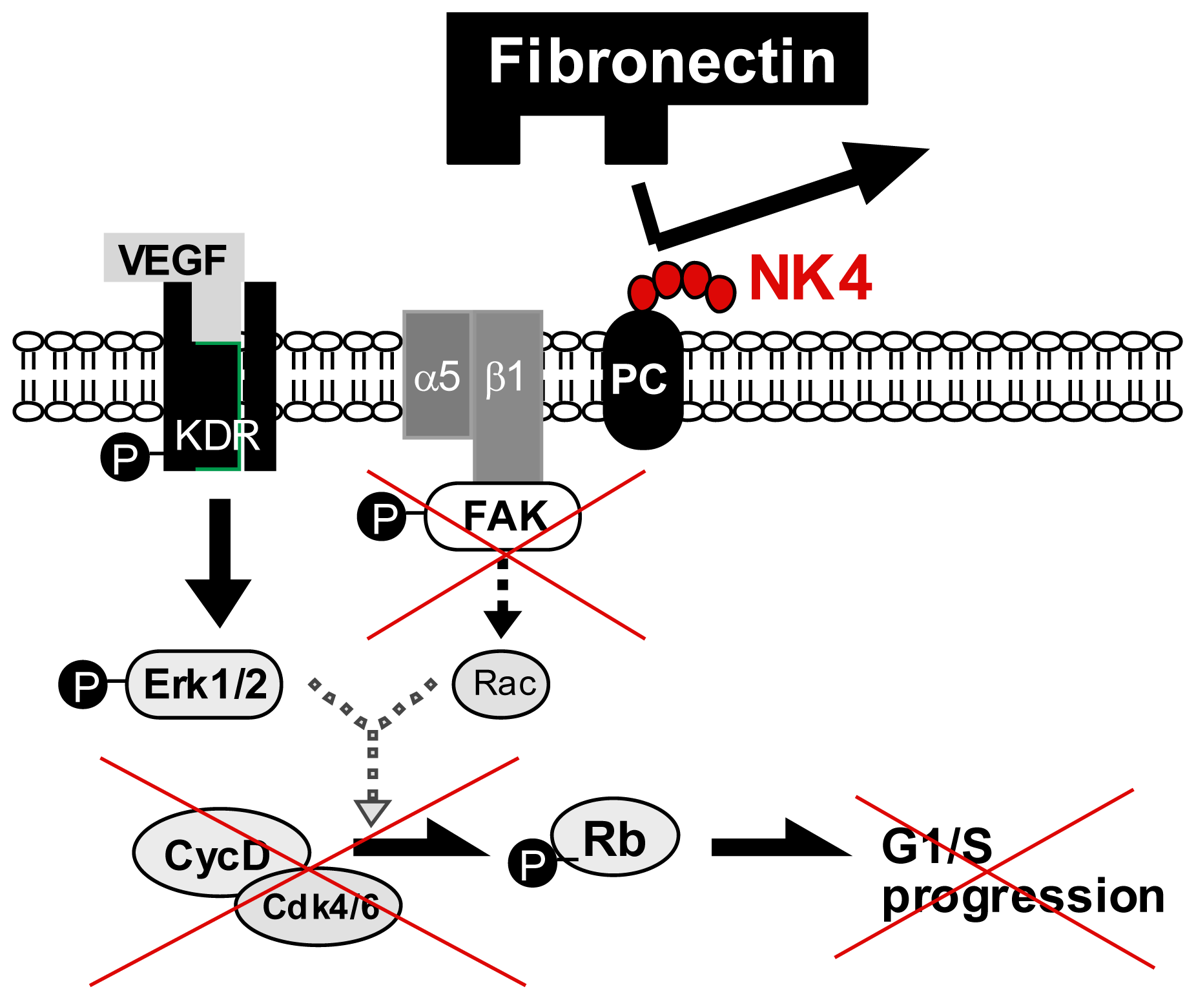
5.2. Perlecan-Dependent Anti-Angiogenic Mechanisms of NK4
5.3. Possible Cleavage of NK4-Like Fragment in Vivo
6. Anti-Cancer Strategy by NK4
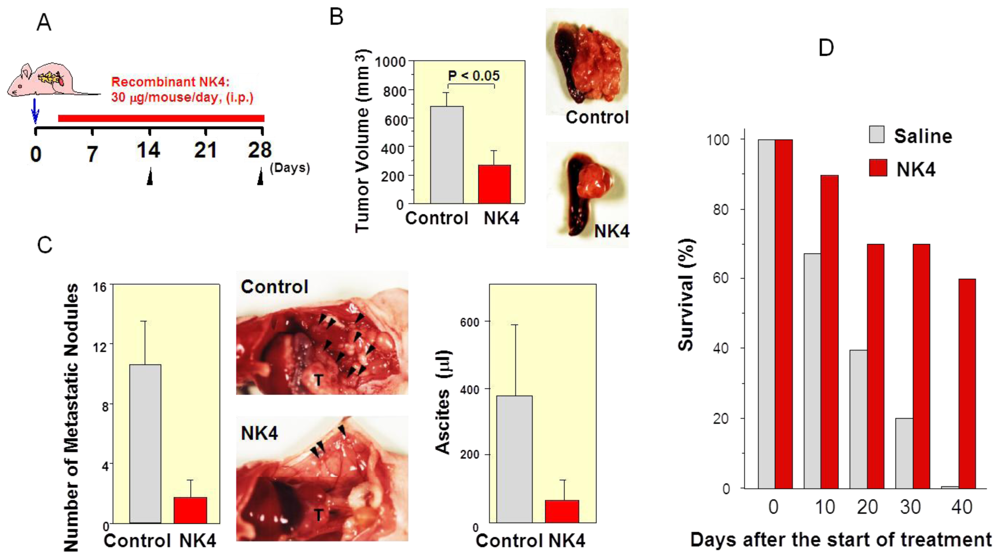
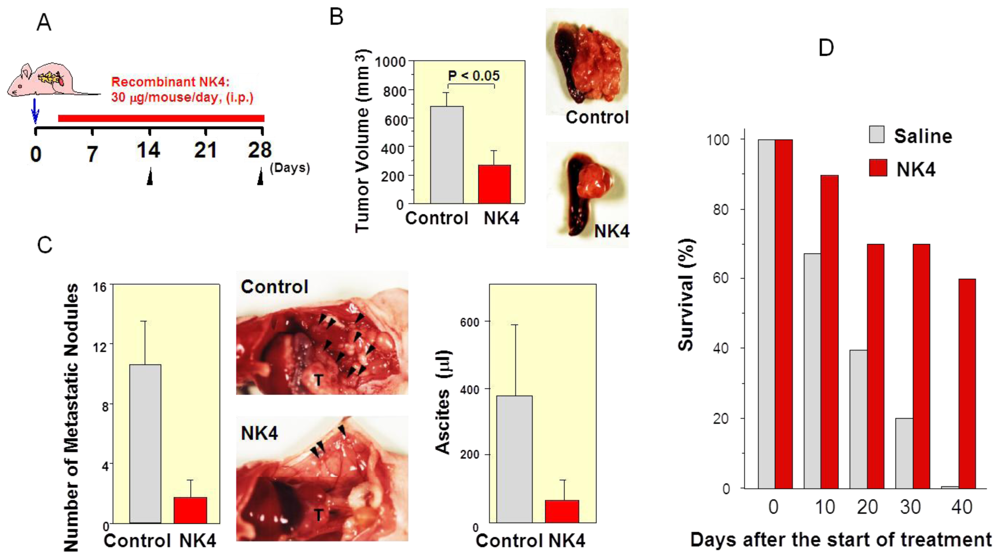
6.1. First Evidence of HGF-Antagonist for Inhibition of Tumor Progression in Vivo
6.2. Inhibition of Tumor Angiogenesis by NK4 Treatment
6.3. Delayed NK4 Therapy for Attenuation of End-Stage Carcinoma
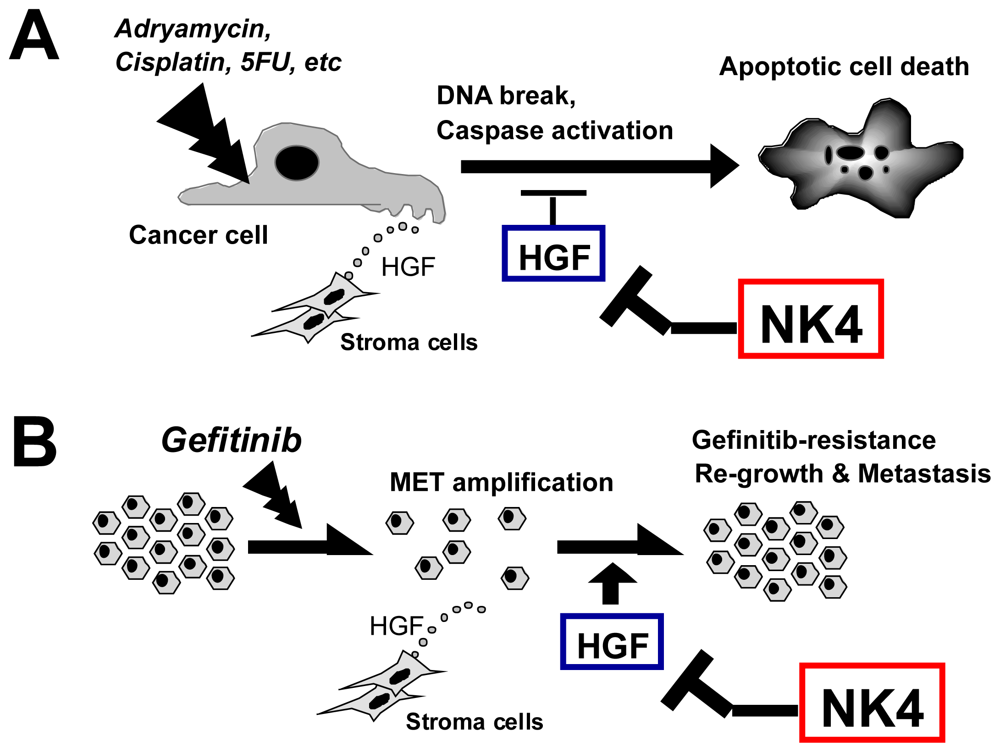
6.4. Therapy Combining NK4 with Other Treatments
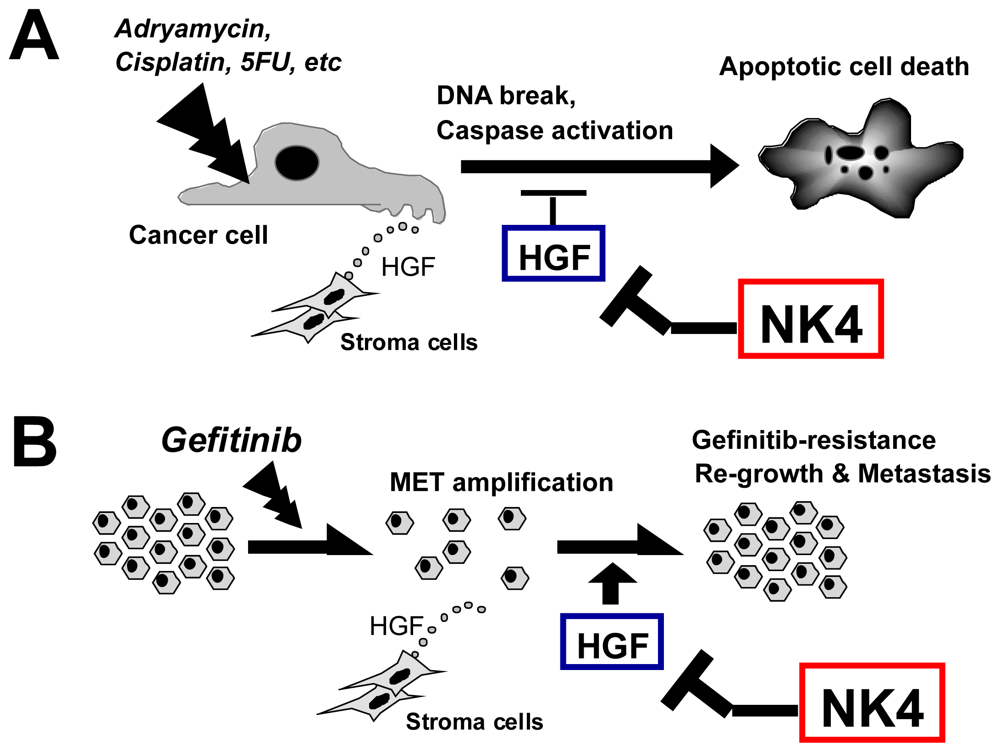
6.4.1. Chemotherapy
6.4.2. Radiation Therapy
6.4.3. Immune Therapy
6.4.4. Anti-Angiogenic Therapy
6.4.5. Anti-Cancer Stem Cell (CSC) Therapy
7. Other Anti-Cancer Strategies for Blocking HGF–MET Signaling
7.1. HGF-Mimic Molecules or HGF-Fragments
7.1.1. Pro-HGF
7.1.2. Engineering for HGF Mutation
7.1.3. NK1 and NK2
7.2. Antibodies to HGF or MET
7.2.1. Anti-HGF Antibodies
7.2.2. Anti-MET Antibody
7.3. Small-Sized MET TK Inhibitors
7.4. Decoys or MET Binders
8. Remarks and Perspective
Acknowledgements
References
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar]
- Nakamura, T.; Nawa, K.; Ichihara, A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem. Biophys. Res. Commun 1984, 122, 1450–1459. [Google Scholar]
- Nakamura, T.; Nishizawa, T.; Hagiya, M.; Seki, T.; Shimonishi, M.; Sugimura, A.; Tashiro, K.; Shimizu, S. Molecular cloning and expression of human hepatocyte growth factor. Nature 1989, 342, 440–443. [Google Scholar]
- Nakamura, T. Structure and function of hepatocyte growth factor. Prog. Growth Factor Res 1991, 3, 67–85. [Google Scholar]
- Boros, P.; Miller, C.M. Hepatocyte growth factor: A multifunctional cytokine. Lancet 1995, 345, 293–295. [Google Scholar]
- Rosário, M.; Birchmeier, W. How to make tubes: Signaling by the Met receptor tyrosine kinase. Trends Cell Biol 2003, 13, 328–335. [Google Scholar]
- Gherardi, E.; Sandin, S.; Petoukhov, M.V.; Finch, J.; Youles, M.E.; Ofverstedt, L.G.; Miguel, R.N.; Blundell, T.L.; Vande Woude, G.F.; Skoglund, U.; et al. Structural basis of hepatocyte growth factor/scatter factor and MET signaling. Proc. Natl. Acad. Sci 2006, 103, 4046–4051. [Google Scholar]
- Nakamura, T.; Mizuno, S. The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc. Jpn. Acad. Ser. B 2010, 86, 588–610. [Google Scholar]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.; Kmiecik, T.E.; Vande Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar]
- Higuchi, O.; Mizuno, K.; Vande Woude, G.F.; Nakamura, T. Expression of c-met proto-oncogene in COS cells induces the signal transducing high-affinity receptor for hepatocyte growth factor. FEBS Lett 1992, 301, 282–286. [Google Scholar]
- Matsumoto, K.; Matsumoto, K.; Nakamura, T.; Kramer, R.H. Hepatocyte growth factor/scatter factor induces tyrosine phosphorylation of focal adhesion kinase (p125FAK) and promotes migration and invasion by oral squamous cell carcinoma cells. J. Biol. Chem 1994, 269, 31807–31813. [Google Scholar]
- Nakamura, T.; Matsumoto, K.; Kiritoshi, A.; Tano, Y.; Nakamura, T. Induction of hepatocyte growth factor in fibroblasts by tumor-derived factors affects invasive growth of tumor cells: In vitro analysis of tumor-stromal interactions. Cancer Res 1997, 57, 3305–3313. [Google Scholar]
- Matsumoto, K.; Date, K.; Ohmichi, H.; Nakamura, T. Hepatocyte growth factor in lung morphogenesis and tumor invasion: role as a mediator in epithelium-mesenchyme and tumor-stroma interactions. Cancer Chemother. Pharmacol 1996, 38, S42–S47. [Google Scholar]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nature Genet 1997, 16, 68–73. [Google Scholar]
- Date, K.; Matsumoto, K.; Shimura, H.; Tanaka, M.; Nakamura, T. HGF/NK4 is a specific antagonist for pleiotrophic actions of hepatocyte growth factor. FEBS Lett 1997, 420, 1–6. [Google Scholar]
- Date, K.; Matsumoto, K.; Kuba, K.; Shimura, H.; Tanaka, M.; Nakamura, T. Inhibition of tumor growth and invasion by a four-kringle antagonist (HGF/NK4) for hepatocyte growth factor. Oncogene 1998, 17, 3045–3054. [Google Scholar]
- Dean, M.; Park, M.; Le Beau, M.M.; Robins, T.S.; Diaz, M.O.; Rowley, J.D.; Blair, D.G.; Vande Woude, G.F. The human met oncogene is related to the tyrosine kinase oncogenes. Nature 1985, 318, 385–388. [Google Scholar]
- Nakamura, T.; Nawa, K.; Ichihara, A.; Kaise, N.; Nishino, T. Purification and subunit structure of hepatocyte growth factor from rat platelets. FEBS Lett 1987, 224, 311–316. [Google Scholar]
- Stoker, M.; Gherardi, E.; Perryman, M.; Gray, J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature 1987, 327, 239–242. [Google Scholar]
- Konishi, T.; Takehara, T.; Tsuji, T.; Ohsato, K.; Matsumoto, K.; Nakamura, T. Scatter factor from human embryonic lung fibroblasts is probably identical to hepatocyte growth factor. Biochem. Biophys. Res. Commun 1991, 180, 765–773. [Google Scholar]
- Weidner, K.M.; Arakaki, N.; Hartmann, G.; Vandekerckhove, J.; Weingart, S.; Rieder, H.; Fonatsch, C.; Tsubouchi, H.; Hishida, T.; Daikuhara, Y.; et al. Evidence for the identity of human scatter factor and human hepatocyte growth factor. Proc. Natl. Acad. Sci 1991, 88, 7001–7005. [Google Scholar]
- Ohmichi, H.; Koshimizu, U.; Matsumoto, K.; Nakamura, T. Hepatocyte growth factor (HGF) acts as a mesenchyme-derived morphogenic factor during fetal lung development. Development 1998, 125, 1315–1324. [Google Scholar]
- Ishiki, Y.; Ohnishi, H.; Muto, Y.; Matsumoto, K.; Nakamura, T. Direct evidence that hepatocyte growth factor is a hepatotrophic factor for liver regeneration and has a potent antihepatitis effect in vivo. Hepatology 1992, 16, 1227–1235. [Google Scholar]
- Nakamura, T.; Mizuno, S.; Matsumoto, K.; Sawa, Y.; Matsuda, H.; Nakamura, T. Myocardial protection from ischemia/reperfusion injury by endogenous and exogenous HGF. J. Clin. Invest 2000, 106, 1511–1519. [Google Scholar]
- Mizuno, S.; Ohnishi, H.; Nakamura, T. Hepatocyte growth factor (HGF), an endogenous pulmotrophic regulator, for the rescue of acute and chronic lung diseases. Curr. Signal Transduct. Ther 2011, 6, 210–220. [Google Scholar]
- Kanda, H.; Tajima, H.; Lee, G.H.; Nomura, K.; Ohtake, K.; Matsumoto, K.; Nakamura, T.; Kitagawa, T. Hepatocyte growth factor transforms immortalized mouse liver epithelial cells. Oncogene 1993, 8, 3047–3053. [Google Scholar]
- Takayama, H.; LaRochelle, W.J.; Sharp, R.; Otsuka, T.; Kriebel, P.; Anver, M.; Aaronson, S.A.; Merlino, G. Diverse tumorigenesis associated with aberrant development in mice overexpressing hepatocyte growth factor/scatter factor. Proc. Natl. Acad. Sci 1997, 94, 701–706. [Google Scholar]
- Stabile, L.P.; Lyker, J.S.; Land, S.R.; Dacic, S.; Zamboni, B.A.; Siegfried, J.M. Transgenic mice overexpressing hepatocyte growth factor in the airways show increased susceptibility to lung cancer. Carcinogenesis 2006, 27, 1547–1555. [Google Scholar]
- Di Renzo, M.F.; Olivero, M.; Martone, T.; Maffe, A.; Maggiora, P.; Stefani, A.D.; Valente, G.; Giordano, S.; Cortesina, G.; Comoglio, P.M. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinoma. Oncogene 2000, 19, 1547–1555. [Google Scholar]
- Ma, P.C.; Kijima, T.; Maulik, G.; Fox, E.A.; Sattler, M.; Griffin, J.D.; Johnson, B.E.; Salgia, R. c-MET mutational analysis in small cell lung cancer: Novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003, 63, 6272–6281. [Google Scholar]
- Krishnaswamy, S.; Kanteti, R.; Duke-Cohan, J.S.; Loganathan, S.; Liu, W.; Ma, P.C.; Sattler, M.; Singleton, P.A.; Ramnath, N.; Innocenti, F.; et al. Ethnic differences and functional analysis of MET mutations in lung cancer. Clin. Cancer Res 2009, 15, 5714–5723. [Google Scholar]
- Giordano, S.; Maffe, A.; Williams, T.A.; Artigiani, S.; Gual, P.; Bardelli, A.; Basilico, C.; Michieli, P.; Comoglio, P.M. Different point mutations in the met oncogene elicit distinct biological properties. FASEB J 2000, 14, 399–406. [Google Scholar]
- Danilkovitch-Miagkova, A.; Miagkov, A.; Skeel, A.; Nakaigawa, N.; Zbar, B.; Leonard, E.J. Oncogenic mutants of RON and MET receptor tyrosine kinases cause activation of the beta-catenin pathway. Mol. Cell Biol 2001, 21, 5857–5868. [Google Scholar]
- Nakaigawa, N.; Weirich, G.; Schmidt, L.; Zbar, B. Tumorigenesis mediated by MET mutant M1268T is inhibited by dominant-negative Src. Oncogene 2000, 19, 2996–3002. [Google Scholar]
- Lin, H.C.; Lai, P.Y.; Lin, Y.P.; Huang, J.Y.; Yang, B.C. Fas ligand enhances malignant behavior of tumor cells through interaction with Met, hepatocyte growth factor receptor, in lipid rafts. J. Biol. Chem 2012, 287, 20664–20673. [Google Scholar]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar]
- Karamouzis, M.V.; Konstantinopoulos, P.A.; Papavassiliou, A.G. Targeting MET as a strategy to overcome crosstalk-related resistance to EGFR inhibitors. Lancet Oncol 2009, 10, 709–717. [Google Scholar]
- Shattuck, D.L.; Miller, J.K.; Carraway, K.L.; Sweeney, C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res 2008, 68, 1471–1477. [Google Scholar]
- Guo, A.; Villén, J.; Kornhauser, J.; Lee, K.A.; Stokes, M.P.; Rikova, K.; Possemato, A.; Nardone, J.; Innocenti, G.; Wetzel, R.; et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc. Natl. Acad. Sci 2008, 105, 692–697. [Google Scholar]
- Tanizaki, J.; Okamoto, I.; Sakai, K.; Nakagawa, K. Differential roles of trans-phosphorylated EGFR, HER2, HER3, and RET as heterodimerisation partners of MET in lung cancer with MET amplification. Br. J. Cancer 2011, 105, 807–813. [Google Scholar]
- Miura, H.; Nishimura, K.; Tsujimura, A.; Matsumiya, K.; Matsumoto, K.; Nakamura, T.; Okuyama, A. Effects of hepatocyte growth factor on E-cadherin-mediated cell-cell adhesion in DU145 prostate cancer cells. Urology 2001, 58, 1064–1069. [Google Scholar]
- Kimura, T.; Sakisaka, T.; Baba, T.; Yamada, T.; Takai, Y. Involvement of the Ras-Ras-activated Rab5 guanine nucleotide exchange factor RIN2-Rab5 pathway in the hepatocyte growth factor-induced endocytosis of E-cadherin. J. Biol. Chem 2006, 281, 10598–10609. [Google Scholar]
- Hiscox, S.; Jiang, W.G. Hepatocyte growth factor/scatter factor disrupts epithelial tumour cell-cell adhesion: Involvement of beta-catenin. Anticancer Res 1999, 19, 509–517. [Google Scholar]
- Royal, I.; Lamarche-Vane, N.; Lamorte, L.; Kaibuchi, K.; Park, M. Activation of cdc42, rac, PAK, and rho-kinase in response to hepatocyte growth factor differentially regulates epithelial cell colony spreading and dissociation. Mol. Biol. Cell 2000, 11, 1709–1725. [Google Scholar]
- Parr, C.; Davies, G.; Nakamura, T.; Matsumoto, K.; Mason, M.D.; Jiang, W.G. The HGF/SF-induced phosphorylation of paxillin, matrix adhesion, and invasion of prostate cancer cells were suppressed by NK4, an HGF/SF variant. Biochem. Biophys. Res. Commun 2001, 285, 1330–1337. [Google Scholar]
- Li, H.; Shimura, H.; Aoki, Y.; Date, K.; Matsumoto, K.; Nakamura, T.; Tanaka, M. Hepatocyte growth factor stimulates the invasion of gallbladder carcinoma cell lines in vitro. Clin. Exp. Metastasis 1998, 16, 74–82. [Google Scholar]
- Nagakawa, O.; Murakami, K.; Yamaura, T.; Fujiuchi, Y.; Murata, J.; Fuse, H.; Saiki, I. Expression of membrane-type 1 matrix metalloproteinase (MT1-MMP) on prostate cancer cell lines. Cancer Lett 2000, 155, 173–179. [Google Scholar]
- Jiang, Y.; Xu, W.; Lu, J.; He, F.; Yang, X. Invasiveness of hepatocellular carcinoma cell lines: Contribution of hepatocyte growth factor, c-met, and transcription factor Ets-1. Biochem. Biophys. Res. Commun 2001, 286, 1123–1130. [Google Scholar]
- Zeng, Q.; Chen, S.; You, Z.; Yang, F.; Carey, T.E.; Saims, D.; Wang, C.Y. Hepatocyte growth factor inhibits anoikis in head and neck squamous cell carcinoma cells by activation of ERK and Akt signaling independent of NFkappa B. J. Biol. Chem 2002, 277, 25203–25208. [Google Scholar]
- Franco, M.; Muratori, C.; Corso, S.; Tenaglia, E.; Bertotti, A.; Capparuccia, L.; Trusolino, L.; Comoglio, P.M.; Tamagnone, L. The tetraspanin CD151 is required for Met-dependent signaling and tumor cell growth. J. Biol. Chem 2010, 285, 38756–38764. [Google Scholar]
- Huang, S.; Ouyang, N.; Lin, L.; Chen, L.; Wu, W.; Su, F.; Yao, Y.; Yao, H. HGF-induced PKCζ activation increases functional CXCR4 expression in human breast cancer cells. PLoS One 2012, 7, e29124. [Google Scholar]
- Tu, H.; Zhou, Z.; Liang, Q.; Li, Z.; Li, D.; Qing, J.; Wang, H.; Zhang, L. CXCR4 and SDF-1 production are stimulated by hepatocyte growth factor and promote glioma cell invasion. Onkologie 2009, 32, 331–336. [Google Scholar]
- Bussolino, F.; Di Renzo, M.F.; Ziche, M.; Bocchietto, E.; Olivero, M.; Naldini, L.; Gaudino, G.; Tamagnone, L.; Coffer, A.; Comoglio, P.M. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J. Cell Biol 1992, 119, 629–641. [Google Scholar]
- Kuba, K.; Matsumoto, K.; Date, K.; Shimura, H.; Tanaka, M.; Nakamura, T. HGF/NK4, a four-kringle antagonist of hepatocyte growth factor, is an angiogenesis inhibitor that suppresses tumor growth and metastasis in mice. Cancer Res 2000, 60, 6737–6743. [Google Scholar]
- Kubota, T.; Taiyoh, H.; Matsumura, A.; Murayama, Y.; Ichikawa, D.; Okamoto, K.; Fujiwara, H.; Ikoma, H.; Nakanishi, M.; Kikuchi, S.; et al. NK4, an HGF antagonist, prevents hematogenous pulmonary metastasis by inhibiting adhesion of CT26 cells to endothelial cells. Clin. Exp. Metastasis 2009, 26, 447–456. [Google Scholar]
- Jiang, W.G.; Martin, T.A.; Matsumoto, K.; Nakamura, T.; Mansel, R.E. Hepatocyte growth factor/scatter factor decreases the expression of occludin and transendothelial resistance (TER) and increases paracellular permeability in human vascular endothelial cells. J. Cell Physiol 1999, 181, 319–329. [Google Scholar]
- Rutella, S.; Danese, S.; Leone, G. Tolerogenic dendritic cells: cytokine modulation comes of age. Blood 2006, 108, 1435–1440. [Google Scholar]
- Michieli, P.; Basilico, C.; Pennacchietti, S.; Maffè, A.; Tamagnone, L.; Giordano, S.; Bardelli, A.; Comoglio, P.M. Mutant Met-mediated transformation is ligand-dependent and can be inhibited by HGF antagonists. Oncogene 1999, 18, 5221–5231. [Google Scholar]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar]
- Kitajima, Y.; Ide, T.; Ohtsuka, T.; Miyazaki, K. Induction of hepatocyte growth factor activator gene expression under hypoxia activates the hepatocyte growth factor/c-Met system via hypoxia inducible factor-1 in pancreatic cancer. Cancer Sci 2008, 99, 1341–1347. [Google Scholar]
- Nakamura, Y.; Morishita, R.; Higaki, J.; Kida, I.; Aoki, M.; Moriguchi, A.; Yamada, K.; Hayashi, S.; Yo, Y.; Nakano, H.; et al. Hepatocyte growth factor is a novel member of the endothelium-specific growth factors: Additive stimulatory effect of hepatocyte growth factor with basic fibroblast growth factor but not with vascular endothelial growth factor. J. Hypertens 1996, 14, 1067–1072. [Google Scholar]
- Laterra, J.; Nam, M.; Rosen, E.; Rao, J.S.; Lamszus, K.; Goldberg, I.D.; Johnston, P. Scatter factor/hepatocyte growth factor gene transfer enhances glioma growth and angiogenesis in vivo. Lab. Invest 1997, 76, 565–577. [Google Scholar]
- Kubota, T.; Taiyoh, H.; Matsumura, A.; Murayama, Y.; Ichikawa, D.; Okamoto, K.; Fujiwara, H.; Ikoma, H.; Nakanishi, M.; Kikuchi, S.; et al. Gene transfer of NK4, an angiogenesis inhibitor, induces CT26 tumor regression via tumor-specific T lymphocyte activation. Int. J. Cancer 2009, 125, 2879–2886. [Google Scholar]
- Matsumoto-Taniura, N.; Matsumoto, K.; Nakamura, T. Prostaglandin production in mouse mammary tumour cells confers invasive growth potential by inducing hepatocyte growth factor in stromal fibroblasts. Br. J. Cancer 1999, 81, 194–202. [Google Scholar]
- Hasina, R.; Matsumoto, K.; Matsumoto-Taniura, N.; Kato, I.; Sakuda, M.; Nakamura, T. Autocrine and paracrine motility factors and their involvement in invasiveness in a human oral carcinoma cell line. Br. J. Cancer 1999, 80, 1708–1717. [Google Scholar]
- Matsumoto, K.; Nakamura, T. NK4 (HGF-antagonist/angiogenesis inhibitor) in cancer biology and therapeutics. Cancer Sci 2003, 94, 321–327. [Google Scholar]
- Wislez, M.; Rabbe, N.; Marchal, J.; Milleron, B.; Crestani, B.; Mayaud, C.; Antoine, M.; Soler, P.; Cadranel, J. Hepatocyte growth factor production by neutrophils infiltrating bronchioloalveolar subtype pulmonary adenocarcinoma: role in tumor progression and death. Cancer Res 2003, 63, 1405–1412. [Google Scholar]
- Osada, S.; Kanematsu, M.; Imai, H.; Goshima, S. Clinical significance of serum HGF and c-Met expression in tumor tissue for evaluation of properties and treatment of hepatocellular carcinoma. Hepatogastroenterology 2008, 55, 544–549. [Google Scholar]
- Beppu, K.; Uchiyama, A.; Morisaki, T.; Matsumoto, K.; Nakamura, T.; Tanaka, M.; Katano, M. Hepatocyte growth factor production by peripheral blood mononuclear cells of recurrent cancer patients. Anticancer Res 2001, 21, 2195–2200. [Google Scholar]
- Matsumoto, K.; Kataoka, H.; Date, K.; Nakamura, T. Cooperative interaction between alpha- and beta-chains of hepatocyte growth factor on c-Met receptor confers ligand-induced receptor tyrosine phosphorylation and multiple biological responses. J. Biol. Chem 1998, 273, 22913–22920. [Google Scholar]
- Otsuka, T.; Jakubczak, J.; Vieira, W.; Bottaro, D.P.; Breckenridge, D.; Larochelle, W.J.; Merlino, G. Disassociation of met-mediated biological responses in vivo: The natural hepatocyte growth factor/scatter factor splice variant NK2 antagonizes growth but facilitates metastasis. Mol. Cell Biol 2000, 20, 2055–2065. [Google Scholar]
- Jiang, W.G.; Hiscox, S.E.; Parr, C.; Martin, T.A.; Matsumoto, K.; Nakamura, T.; Mansel, R.E. Antagonistic effect of NK4, a novel hepatocyte growth factor variant, on in vitro angiogenesis of human vascular endothelial cells. Clin. Cancer Res 1999, 5, 3695–3703. [Google Scholar]
- Sakai, K.; Nakamura, T.; Matsumoto, K.; Nakamura, T. Angioinhibitory action of NK4 involves impaired extracellular assembly of fibronectin mediated by perlecan-NK4 association. J. Biol. Chem 2009, 284, 22491–22499. [Google Scholar]
- Ohnishi, H.; Oka, K.; Mizuno, S.; Nakamura, T. Identification of mannose receptor as receptor for hepatocyte growth factor beta-chain: novel ligand-receptor pathway for enhancing macrophage phagocytosis. J. Biol. Chem 2012, 287, 13371–13381. [Google Scholar]
- Buchstein, N.; Hoffmann, D.; Smola, H.; Lang, S.; Paulsson, M.; Niemann, C.; Krieg, T.; Eming, S.A. Alternative proteolytic processing of hepatocyte growth factor during wound repair. Am. J. Pathol 2009, 174, 2116–21128. [Google Scholar]
- Wright, T.G.; Singh, V.K.; Li, J.J.; Foley, J.H.; Miller, F.; Jia, Z.; Elliott, B.E. Increased production and secretion of HGF alpha-chain and an antagonistic HGF fragment in a human breast cancer progression model. Int. J. Cancer 2009, 125, 1004–1015. [Google Scholar]
- Dai, Y.; Siemann, D.W. Constitutively active c-Met kinase in PC-3 cells is autocrine- independent and can be blocked by the Met kinase inhibitor BMS-777607. BMC Cancer 2012, 12, e198. [Google Scholar]
- Yancopoulos, G.D.; Klagsbrun, M.; Folkman, J. Vasculogenesis, angiogenesis, and growth factors: Ephrins enter the fray at the border. Cell 1998, 93, 661–664. [Google Scholar]
- Tomioka, D.; Maehara, N.; Kuba, K.; Mizumoto, K.; Tanaka, M.; Matsumoto, K.; Nakamura, T. Inhibition of growth, invasion, and metastasis of human pancreatic carcinoma cells by NK4 in an orthotopic mouse model. Cancer Res 2001, 61, 7518–7524. [Google Scholar]
- Saimura, M.; Nagai, E.; Mizumoto, K.; Maehara, N.; Minamishima, Y.A.; Katano, M.; Matsumoto, K.; Nakamura, T.; Tanaka, M. Tumor suppression through angiogenesis inhibition by SUIT-2 pancreatic cancer cells genetically engineered to secrete NK4. Clin. Cancer Res 2002, 8, 3243–3249. [Google Scholar]
- Kishi, Y.; Kuba, K.; Nakamura, T.; Wen, J.; Suzuki, Y.; Mizuno, S.; Nukiwa, T.; Matsumoto, K.; Nakamura, T. Systemic NK4 gene therapy inhibits tumor growth and metastasis of melanoma and lung carcinoma in syngeneic mouse tumor models. Cancer Sci 2009, 100, 1351–1358. [Google Scholar]
- Fan, S.; Ma, Y.X.; Wang, J.A.; Yuan, R.Q.; Meng, Q.; Cao, Y.; Laterra, J.J.; Goldberg, I.D.; Rosen, E.M. The cytokine hepatocyte growth factor/scatter factor inhibits apoptosis and enhances DNA repair by a common mechanism involving signaling through phosphatidyl inositol 3′ kinase. Oncogene 2000, 19, 2212–2223. [Google Scholar]
- Bowers, D.C.; Fan, S.; Walter, K.A.; Abounader, R.; Williams, J.A.; Rosen, EM.; Laterra, J. Scatter factor/hepatocyte growth factor protects against cytotoxic death in human glioblastoma via phosphatidylinositol 3-kinase-AKT-dependent pathways. Cancer Res. 2000, 60, 4277–4283. [Google Scholar]
- Matsumoto, G.; Omi, Y.; Lee, U.; Kubota, E.; Tabata, Y. NK4 gene therapy combined with cisplatin inhibits tumour growth and metastasis of squamous cell carcinoma. Anticancer Res 2011, 31, 105–111. [Google Scholar]
- Yano, S.; Wang, W.; Li, Q.; Matsumoto, K.; Sakurama, H.; Nakamura, T.; Ogino, H.; Kakiuchi, S.; Hanibuchi, M.; Nishioka, Y.; et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res 2008, 68, 9479–9487. [Google Scholar]
- Ohuchida, K.; Mizumoto, K.; Murakami, M.; Qian, L.W.; Sato, N.; Nagai, E.; Matsumoto, K.; Nakamura, T.; Tanaka, M. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res 2004, 64, 3215–3222. [Google Scholar]
- Sheng-Hua, C.; Yan-Bin, M.; Zhi-An, Z.; Hong, Z.; Dong-Fu, F.; Zhi-Qiang, L.; Xian-Hou, Y. Radiation-enhanced hepatocyte growth factor secretion in malignant glioma cell lines. Surg. Neurol 2007, 68, 610–613. [Google Scholar]
- Hu, S.Y.; Duan, H.F.; Li, Q.F.; Yang, Y.F.; Chen, J.L.; Wang, L.S.; Wang, H. Hepatocyte growth factor protects endothelial cells against gamma ray irradiation-induced damage. Acta Pharmacol. Sin 2009, 30, 1415–1420. [Google Scholar]
- Lynn, K.D.; Brekken, R.A. Anti-VEGF therapy revived by c-Met inhibition, but is c-Met the answer? Cancer Discov 2012, 2, 211–213. [Google Scholar]
- Li, Y.; Li, A.; Glas, M.; Lal, B.; Ying, M.; Sang, Y.; Xia, S.; Trageser, D.; Guerrero-Cázares, H.; Eberhart, C.G.; et al. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc. Natl. Acad. Sci 2011, 108, 9951–9956. [Google Scholar]
- Sun, S.; Wang, Z. Head neck squamous cell carcinoma c-Met+ cells display cancer stem cell properties and are responsible for cisplatin-resistance and metastasis. Int. J. Cancer 2011, 129, 2337–2348. [Google Scholar]
- Matsumoto, K.; Nakamura, T. Mechanisms and significance of bifunctional NK4 in cancer treatment. Biochem. Biophys. Res. Commun 2005, 333, 316–327. [Google Scholar]
- Ueda, K.; Iwahashi, M.; Matsuura, I.; Nakamori, M.; Nakamura, M.; Ojima, T.; Naka, T.; Ishida, K.; Matsumoto, K.; Nakamura, T.; et al. Adenoviral-mediated gene transduction of the hepatocyte growth factor (HGF) antagonist, NK4, suppresses peritoneal metastases of gastric cancer in nude mice. Eur. J. Cancer 2004, 40, 2135–2142. [Google Scholar]
- Son, G.; Hirano, T.; Seki, E.; Iimuro, Y.; Nukiwa, T.; Matsumoto, K.; Nakamura, T.; Fujimoto, J. Blockage of HGF/c-Met system by gene therapy (adenovirus-mediated NK4 gene) suppresses hepatocellular carcinoma in mice. J. Hepatol 2006, 45, 688–695. [Google Scholar]
- Wen, J.; Matsumoto, K.; Taniura, N.; Tomioka, D.; Nakamura, T. Hepatic gene expression of NK4, an HGF-antagonist/angiogenesis inhibitor, suppresses liver metastasis and invasive growth of colon cancer in mice. Cancer Gene Ther 2004, 11, 419–430. [Google Scholar]
- Maemondo, M.; Narumi, K.; Saijo, Y.; Usui, K.; Tahara, M.; Tazawa, R.; Hagiwara, K.; Matsumoto, K.; Nakamura, T.; Nukiwa, T. Targeting angiogenesis and HGF function using an adenoviral vector expressing the HGF antagonist NK4 for cancer therapy. Mol. Therapy 2002, 5, 177–185. [Google Scholar]
- Suzuki, Y.; Sakai, K.; Ueki, J.; Xu, Q.; Nakamura, T.; Shimada, H.; Nakamura, T.; Matsumoto, K. Inhibition of Met/HGF receptor and angiogenesis by NK4 leads to suppression of tumor growth and migration in malignant pleural mesothelioma. Int. J. Cancer 2010, 127, 1948–1957. [Google Scholar]
- Davies, G.; Mason, M.D.; Martin, T.A.; Parr, C.; Watkins, G.; Lane, J.; Matsumoto, K.; Nakamura, T.; Jiang, W.G. The HGF/SF antagonist NK4 reverses fibroblast- and HGF-induced prostate tumor growth and angiogenesis in vivo. Int. J. Cancer 2003, 106, 348–354. [Google Scholar]
- Saga, Y.; Mizukami, H.; Suzuki, M.; Urabe, M.; Kume, A.; Nakamura, T.; Sato, I.; Ozawa, K. Expression of HGF/NK4 in ovarian cancer cells suppresses intraperitoneal dissemination and extends host survival. Gene Ther 2001, 8, 1450–1455. [Google Scholar]
- Kikuchi, T.; Maemondo, M.; Narumi, K.; Matsumoto, K.; Nakamura, T.; Nukiwa, T. Tumor suppression induced by intratumor administration of adenovirus vector expressing NK4, a 4-kringle antagonist of hepatocyte growth factor, and naive dendritic cells. Blood 2002, 100, 3950–3959. [Google Scholar]
- Du, W.; Hattori, Y.; Yamada, T.; Matsumoto, K.; Nakamura, T.; Sagawa, M.; Otsuki, T.; Niikura, T.; Nukiwa, T.; Ikeda, Y. NK4, an antagonist of hepatocyte growth factor (HGF), inhibits growth of multiple myeloma cells: Molecular targeting of angiogenic growth factor. Blood 2007, 109, 3042–3049. [Google Scholar]
- Brockmann, M.A.; Papadimitriou, A.; Brandt, M.; Fillbrandt, R.; Westphal, M.; Lamszus, K. Inhibition of intracerebral glioblastoma growth by local treatment with the scatter factor/hepatocyte growth factor-antagonist NK4. Clin. Cancer Res 2003, 9, 4578–4585. [Google Scholar]
- Martin, T.A.; Parr, C.; Davies, G.; Watkins, G.; Lane, J.; Matsumoto, K.; Nakamura, T.; Mansel, R.E.; Jiang, W.G. Growth and angiogenesis of human breast cancer in a nude mouse tumour model is reduced by NK4, a HGF/SF antagonist. Carcinogenesis 2003, 24, 1317–1323. [Google Scholar]
- Abounader, R.; Ranganathan, S.; Lal, B.; Fielding, K.; Book, A.; Dietz, H.; Burger, P.; Laterra, J. Reversion of human glioblastoma malignancy by U1 small nuclear RNA/ribozyme targeting of scatter factor/hepatocyte growth factor and c-met expression. J. Natl. Cancer Inst 1999, 91, 1548–1556. [Google Scholar]
- Cao, B.; Su, Y.; Oskarsson, M.; Zhao, P.; Kort, E.J.; Fisher, R.J.; Wang, L.M.; Vande Woude, G.F. Neutralizing monoclonal antibodies to hepatocyte growth factor/scatter factor (HGF/SF) display antitumor activity in animal models. Proc. Natl. Acad. Sci 2001, 98, 7443–7448. [Google Scholar]
- Mazzone, M.; Basilico, C.; Cavassa, S.; Pennacchietti, S.; Risio, M.; Naldini, L.; Comoglio, P.M.; Michieli, P. An uncleavable form of pro-scatter factor suppresses tumor growth and dissemination in mice. J. Clin. Invest 2004, 114, 1418–1432. [Google Scholar]
- Kirchhofer, D.; Lipari, M.T.; Santell, L.; Billeci, K.L.; Maun, H.R.; Sandoval, W.N.; Moran, P.; Ridgway, J.; Eigenbrot, C.; Lazarus, R.A. Utilizing the activation mechanism of serine proteases to engineer hepatocyte growth factor into a Met antagonist. Proc. Natl. Acad. Sci 2007, 104, 5306–5311. [Google Scholar]
- Jones, D.S.; Tsai, P.C.; Cochran, J.R. Engineering hepatocyte growth factor fragments with high stability and activity as Met receptor agonists and antagonists. Proc. Natl. Acad. Sci 2011, 108, 13035–13040. [Google Scholar]
- Otsuka, T.; Takagi, H.; Horiguchi, N.; Toyoda, M.; Sato, K.; Takayama, H.; Mori, M. CCl4-induced acute liver injury in mice is inhibited by hepatocyte growth factor overexpression but stimulated by NK2 overexpression. FEBS Lett. 2002, 532, 391–395. [Google Scholar]
- Burgess, T.; Coxon, A.; Meyer, S.; Sun, J.; Rex, K.; Tsuruda, T.; Chen, Q.; Ho, S.Y.; Li, L.; Kaufman, S.; et al. Fully human monoclonal antibodies to hepatocyte growth factor with therapeutic potential against hepatocyte growth factor/c-Met-dependent human tumors. Cancer Res 2006, 66, 1721–1729. [Google Scholar]
- Martens, T.; Schmidt, N.O.; Eckerich, C.; Fillbrandt, R.; Merchant, M.; Schwall, R.; Westphal, M.; Lamszus, K. A novel one-armed anti-c-Met antibody inhibits glioblastoma growth in vivo. Clin. Cancer Res 2006, 12, 6144–6152. [Google Scholar]
- Petrelli, A.; Circosta, P.; Granziero, L.; Mazzone, M.; Pisacane, A.; Fenoglio, S.; Comoglio, P.M.; Giordano, S. Ab-induced ectodomain shedding mediates hepatocyte growth factor receptor down-regulation and hampers biological activity. Proc. Natl. Acad. Sci 2006, 103, 5090–5095. [Google Scholar]
- Greenall, S.A.; Gherardi, E.; Liu, Z.; Donoghue, J.F.; Vitali, A.A.; Li, Q.; Murphy, R.; Iamele, L.; Scott, A.M.; Johns, T.G. Non-agonistic bivalent antibodies that promote c-MET degradation and inhibit tumor growth and others specific for tumor related c-MET. PLoS One 2012, 7, e34658. [Google Scholar]
- Bardelli, A.; Longati, P.; Gramaglia, D.; Stella, M.C.; Comoglio, P.M. Gab1 coupling to the HGF/Met receptor multifunctional docking site requires binding of Grb2 and correlates with the transforming potential. Oncogene 1997, 15, 3103–3111. [Google Scholar]
- Puri, N.; Khramtsov, A.; Ahmed, S.; Nallasura, V.; Hetzel, J.T.; Jagadeeswaran, R.; Karczmar, G.; Salgia, R. A selective small molecule inhibitor of c-Met, PHA665752, inhibits tumorigenicity and angiogenesis in mouse lung cancer xenografts. Cancer Res 2007, 67, 3529–3534. [Google Scholar]
- Pan, B.S.; Chan, G.K.; Chenard, M.; Chi, A.; Davis, L.J.; Deshmukh, S.V.; Gibbs, J.B.; Gil, S.; Hang, G.; Hatch, H.; et al. MK-2461, a novel multitargeted kinase inhibitor, preferentially inhibits the activated c-Met receptor. Cancer Res 2010, 70, 1524–1533. [Google Scholar]
- Cecchi, F.; Rabe, D.C.; Bottaro, D.P. Targeting the HGF/Met signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 553–572. [Google Scholar]
- Buraschi, S.; Pal, N.; Tyler-Rubinstein, N.; Owens, R.T.; Neill, T.; Iozzo, R.V. Decorin antagonizes Met receptor activity and down-regulates β-catenin and Myc levels. J. Biol. Chem 2010, 285, 42075–42085. [Google Scholar]
- Yamamoto, B.J.; Elias, P.D.; Masino, J.A.; Hudson, B.D.; McCoy, A.T.; Anderson, Z.J.; Varnum, M.D.; Sardinia, M.F.; Wright, J.W.; Harding, J.W. The angiotensin IV analog Nle-Tyr-Leu-psi-(CH2-NH2)3–4-His-Pro-Phe (norleual) can act as a hepatocyte growth factor/c-Met inhibitor. J. Pharmacol. Exp. Ther 2010, 333, 161–173. [Google Scholar]
- Kong-Beltran, M.; Stamos, J.; Wickramasinghe, D. The Sema domain of Met is necessary for receptor dimerization and activation. Cancer Cell 2004, 6, 75–84. [Google Scholar]
- Zhao, P.; Grabinski, T.; Gao, C.; Skinner, R.S.; Giambernardi, T.; Su, Y.; Hudson, E.; Resau, J.; Gross, M.; Vande Woude, G.F.; et al. Identification of a met-binding peptide from a phage display library. Clin. Cancer Res 2007, 13, 6049–6055. [Google Scholar]
- Tuynman, J.B.; Vermeulen, L.; Boon, E.M.; Kemper, K.; Zwinderman, A.H.; Peppelenbosch, M.P.; Richel, D.J. Cyclooxygenase-2 inhibition inhibits c-Met kinase activity and Wnt activity in colon cancer. Cancer Res 2008, 68, 1213–1220. [Google Scholar]
- Lee, W.J.; Chen, W.K.; Wang, C.J.; Lin, W.L.; Tseng, T.H. Apigenin inhibits HGF-promoted invasive growth and metastasis involving blocking PI3K/Akt pathway and beta 4 integrin function in MDA-MB-231 breast cancer cells. Toxicol. Appl. Pharmacol 2008, 226, 178–191. [Google Scholar]
- Koh, Y.W.; Choi, E.C.; Kang, S.U.; Hwang, H.S.; Lee, M.H.; Pyun, J.; Park, R.; Lee, Y.; Kim, C.H. Green tea (−)-epigallocatechin-3-gallate inhibits HGF-induced progression in oral cavity cancer through suppression of HGF/c-Met. J. Nutr. Biochem 2011, 22, 1074–1083. [Google Scholar]
- Hung, C.M.; Kuo, D.H.; Chou, C.H.; Su, Y.C.; Ho, C.T.; Way, T.D. Osthole suppresses hepatocyte growth factor (HGF)-induced epithelial-mesenchymal transition via repression of the c-Met/Akt/mTOR pathway in human breast cancer cells. J. Agric. Food Chem 2011, 59, 9683–9690. [Google Scholar]
- Ma, J.; DeFrances, M.C.; Zou, C.; Johnson, C.; Ferrell, R.; Zarnegar, R. Somatic mutation and functional polymorphism of a novel regulatory element in the HGF gene promoter causes its aberrant expression in human breast cancer. J. Clin. Invest 2009, 119, 478–491. [Google Scholar]
- Chen, J.T.; Lin, T.S.; Chow, K.C.; Huang, H.H.; Chiou, S.H.; Chiang, S.F.; Chen, H.C.; Chuang, T.L.; Lin, T.Y.; Chen, C.Y. Cigarette smoking induces overexpression of hepatocyte growth factor in type II pneumocytes and lung cancer cells. Am. J. Respir. Cell Mol. Biol 2006, 34, 264–273. [Google Scholar]
- Kanehira, M.; Xin, H.; Hoshino, K.; Maemondo, M.; Mizuguchi, H.; Hayakawa, T.; Matsumoto, K.; Nakamura, T.; Nukiwa, T.; Saijo, Y. Targeted delivery of NK4 to multiple lung tumors by bone marrow-derived mesenchymal stem cells. Cancer Gene Ther 2007, 14, 894–903. [Google Scholar]
- Okasora, T.; Jo, J.I.; Tabata, Y. Augmented anti-tumor therapy through natural targetability of macrophages genetically engineered by NK4 plasmid DNA. Gene Ther 2008, 15, 524–530. [Google Scholar]
- Kushibiki, T.; Matsumoto, K.; Nakamura, T.; Tabata, Y. Suppression of tumor metastasis by NK4 plasmid DNA released from cationized gelatin. Gene Ther. 2004, 11, 1205–1214. [Google Scholar]
- Hay, R.V.; Cao, B.; Skinner, R.S.; Su, Y.; Zhao, P.; Gustafson, M.F.; Qian, C.N.; The, B.T.; Knudsen, B.S.; Resau, J.H.; et al. Nuclear imaging of Met-expressing human and canine cancer xenografts with radiolabeled monoclonal antibodies (MetSeek). Clin. Cancer Res 2005, 11, 7064s–7069s. [Google Scholar]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar]
- Eder, J.P.; Vande Woude, G.F.; Boerner, S.A.; LoRusso, P.M. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin. Cancer Res 2009, 15, 2207–2214. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target cells | Effect | Involved mechanism | Reference |
|---|---|---|---|
| Cancer cells | Growth | β-catenin, Src, RAS activations | [32–34] |
| FasL-Stat3 activation | [35] | ||
| Dissociation | Cadherin endocytosis | [41–43] | |
| β-catenin activation | |||
| Migration ECM breakdown Anti-anoikis | cdc42-rac-PAK activation Induction of MMP Activations of PI3K-AKT Integrin-CD151 pathways | [44,45] [46–48] [49,50] | |
| Homing | Increased CXCR4 | [51,52] | |
| Enhanced response to SDF1 | |||
| Vascular cells | |||
| Endothelium | Mitogenesis | ERK1/2 activation | [53,54] |
| Cancer-adhesion | Integrin-β4 involvement | [45,55] | |
| Permeability | Occludin downregulation | [56] | |
| Pericytes | Migration | PI3K-AKT activations | [8] |
| Immune cells | |||
| DC | Tolerogenic effects | TH1 << TH2 balance | [57] |
| T-lymphocytes | Anti-proliferation | Reduced IFN-γ | [8] |
| Tumor diseases | Animal model | Approach | Outcome | Reference |
|---|---|---|---|---|
| Digestive system | ||||
| Gastric carcinoma | TMK1 cells, ip (Mouse) | Adeno-NK4, ip | Inhibition of growth, Anti-metastasis, Anti-angiogenesis, Reduced ascites | [93] |
| Hepatic carcinoma | HUH7 cells, portal vein (Mouse) | Adeno-NK4, iv | Inhibition of growth, Anti-angiogenesis, Prolonged survival | [94] |
| Gallbladder carcinoma | GB-d1, sc (Mouse) | r-NK4, sc | Inhibition of growth, Anti-invasion | [16] |
| Pancreatic cancer | SUIT-2 cells, intra-pancreas (Mouse) | r-NK4, ip | Inhibition of growth, Anti-metastasis, Anti-angiogenesis, Reduced ascites, Prolonged survival | [79] |
| Colon carcinoma | MC-38 cells, intra-spleen (Mouse) | NK4 cDNA, bolus iv (hydrodynamics) | Inhibition of growth, Anti-metastasis, Anti-angiogenesis, Prolonged survival | [95] |
| Respiratory tissue | ||||
| Lung carcinoma | Lewis lung cancer, sc (Mouse) | r-NK4, sc | Inhibition of growth, Anti-metastasis, Anti-angiogenesis | [54] |
| Lung carcinoma | A549 cells, sc (Mouse) | Adeno-NK4, intra-tumor or ip | Inhibition of growth, Anti-angiogenesis | [96] |
| Mesothelioma | EHMES-10 cells, sc (Mouse) | Adeno-NK4, intra-tumor | Inhibition of growth, Enhanced apoptosis, Anti-angiogenesis | [97] |
| Reproductive organ | ||||
| Prostate carcinoma | PC-3 cells, sc (Mouse) | r-NK4, sc (osmotic pump) | Inhibitions of growth, Anti-angiogenesis | [98] |
| Ovarian carcinoma | HRA cells, ip (Mouse) | NK4 gene, Stable transfection | Anti-metastasis, Prolonged survival | [99] |
| Blood system | ||||
| Lymphoma | E.G7-OVA cells, sc (Mouse) | Adeno-NK4, intra-tumor (with DC) | Inhibition of growth, Anti-angiogenesis, Induction of CTL | [100] |
| Multiple myeloma | KMS11/34 cells, sc (Mouse) | Adeno-NK4, im | Inhibition of growth, Anti-angiogenesis, Enhanced apoptosis | [101] |
| Others | ||||
| Melanoma | B16F10 cells, sc (Mouse) | Adeno-NK4, iv | Inhibition of growth, Anti-metastasis, Anti-angiogenesis | [81] |
| Glioblastoma | U-87 MG cells, intra-brain (Mouse) | r-NK4, intra-tumor | Inhibition of growth, Anti-angiogenesis, Enhanced apoptosis | [102] |
| Breast carcinoma | MDAMB231 cells, sc (Mouse) | r-NK4, sc | Inhibition of growth, Anti-angiogenesis | [103] |
| Tumor diseases | Animal model | Treatment | Outcome | Reference |
|---|---|---|---|---|
| Anti-HGF approaches | ||||
| HGF knock-down | U87 glioblastoma, brain (Mouse) | HGF ribozyme, cell implant (brain) | Reduced mass size, Anti-proliferation | [104] |
| Uncleavable pro-HGF | MDA-MB435 breast cancer, sc (Mouse) | Pro-HGF cDNA, lentivirus vector, intra-tumor, 18 days | Anti-proliferation, Anti-angiogenesis, Enhanced apoptosis | [106] |
| Anti-HGF antibody | U118 glioblastoma, sc (Nude mice) | Anti-HGF IgG, sc, 2 times/week × 10 | Reduced mass size | [105] |
| Anti-HGF antibody (AMG102) * | U87 glioblastoma, sc (Mouse) | Anti-HGF IgG, sc, 2 times/week × 5 | Anti-proliferation, Enhanced caspase-3 | [110] |
| MET-inhibitions | ||||
| Anti-MET antibody (MetMab) ** | U87 glioblastoma, brain (Mouse) | Antibody, intra-brain, pump, 4 weeks | Anti-proliferation, Enhanced apoptosis | [111] |
| Anti-MET antibody (DN30) | GTL16 gastric cancer, sc (Mouse) | Antibody, sc, 2 times/week × 4 | Anti-proliferation, MET shedding | [112] |
| Anti-pro MET antibody (LMH-80) | U87 glioblastoma, brain (Mouse) | Antibody, 3 times, sc | Anti-proliferation, Binding to pro-MET | [113] |
| Decorin | A431 epidermoid cancer, sc (Mouse) | 5 mg/kg/48 hr, ip, 12 times | Growth arrest, β-catenin inactivation | [118] |
| Angiotensin-IV (Norleual) | B16F10 melanoma, iv (Mouse) | 50 μg/kg/day, ip, 14 days | Anti-metastasis, Gab1 inactivation | [119] |
| PHA665752 | NCI-H69 lung cancer, sc (Mouse) | 16.5 μg/day, intra-tumor, 8 days | Anti-angiogenesis, Increased TSP-1 | [115] |
| MK-2461 *** | GTL16 gastric cancer, sc (Mouse) | 200 mg/kg/day, po, 20 days | MET Y-1349 inhibition, Growth arrest | [116] |
| Apigenin (Flavonoids) | MDA-MB231 breast cancer, iv (Mouse) | 40 μM, iv, with cancer cells + HGF | Anti-metastasis | [123] |
| EGCG (Green tea) | SCC-VII/SF, sc (Mouse) | 75 mg/kg/day, ip, 21 days | Anti-proliferation, Enhanced apoptosis | [124] |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mizuno, S.; Nakamura, T. HGF–MET Cascade, a Key Target for Inhibiting Cancer Metastasis: The Impact of NK4 Discovery on Cancer Biology and Therapeutics. Int. J. Mol. Sci. 2013, 14, 888-919. https://doi.org/10.3390/ijms14010888
Mizuno S, Nakamura T. HGF–MET Cascade, a Key Target for Inhibiting Cancer Metastasis: The Impact of NK4 Discovery on Cancer Biology and Therapeutics. International Journal of Molecular Sciences. 2013; 14(1):888-919. https://doi.org/10.3390/ijms14010888
Chicago/Turabian StyleMizuno, Shinya, and Toshikazu Nakamura. 2013. "HGF–MET Cascade, a Key Target for Inhibiting Cancer Metastasis: The Impact of NK4 Discovery on Cancer Biology and Therapeutics" International Journal of Molecular Sciences 14, no. 1: 888-919. https://doi.org/10.3390/ijms14010888