ANRIL: Molecular Mechanisms and Implications in Human Health

Abstract
:1. Introduction
1.1. The Chr9p21 Locus
1.2. The Long Non Coding RNA in the Chromosome 9p21: ANRIL
2. ANRIL, Polycomb Proteins and Epigenetic Regulation of the CDKN2A/B Locus
3. ANRIL Expression in Disease
4. Conclusions and Discussion
Acknowledgements
References
- The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678.
- Broadbent, H.M.; Peden, J.F.; Lorkowski, S.; Goel, A.; Ongen, H.; Green, F.; Clarke, R.; Collins, R.; Franzosi, M.G.; Tognoni, G.; et al. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked, SNPs in the ANRIL locus on chromosome 9p. Hum. Mol. Genet 2008, 17, 806–814. [Google Scholar]
- Cugino, D.; Gianfagna, F.; Santimone, I.; de Gaetano, G.; Donati, M.B.; Iacoviello, L.; di Castelnuovo, A. Type 2 diabetes and polymorphisms on chromosome 9p21: A meta-analysis. Nutr. Metab. Cardiovasc. Dis 2012, 22, 619–625. [Google Scholar]
- Emanuele, E.; Lista, S.; Ghidoni, R.; Binetti, G.; Cereda, C.; Benussi, L.; Maletta, R.; Bruni, A.C.; Politi, P. Chromosome 9p21.3 genotype is associated with vascular dementia and Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1231–1235. [Google Scholar]
- Burdon, K.P.; Macgregor, S.; Hewitt, A.W.; Sharma, S.; Chidlow, G.; Mills, R.A.; Danoy, P.; Casson, R.; Viswanathan, A.C.; Liu, J.Z. Genome-wide association study identifies susceptibility loci for open angle glaucoma at TMCO1 and CDKN2BAS1. Nat. Genet 2011, 43, 574–578. [Google Scholar]
- Ramdas, W.D.; van Koolwijk, L.M.; Lemij, H.G.; Pasutto, F.; Cree, A.J.; Thorleifsson, G.; Janssen, S.F.; Jacoline, T.B.; Amin, N.; Rivadeneira, F. Common genetic variants associated with open-angle glaucoma. Hum. Mol. Genet 2011, 20, 2464–2471. [Google Scholar]
- Uno, S.; Zembutsu, H.; Hirasawa, A.; Takahashi, A.; Kubo, M.; Akahane, T.; Aoki, D.; Kamatani, N.; Hirata, K.; Nakamura, Y. A genome-wide association study identifies genetic variants in the CDKN2BAS locus associated with endometriosis in Japanese. Nat. Genet 2010, 42, 707–710. [Google Scholar]
- Schaefer, A.S.; Richter, G.M.; Groessner-Schreiber, B.; Noack, B.; Nothnagel, M.; el Mokhtari, N.E.; Loos, B.G.; Jepsen, S.; Schreiber, S. Identification of a shared genetic susceptibility locus for coronary heart disease and periodontitis. PLoS Genet 2009, 5, e1000378. [Google Scholar]
- Sherborne, A.L.; Hosking, F.J.; Prasad, R.B.; Kumar, R.; Koehler, R.; Vijayakrishnan, J.; Papaemmanuil, E.; Bartram, C.R.; Stanulla, M.; Schrappe, M.; et al. Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat. Genet 2010, 42, 492–494. [Google Scholar]
- Turnbull, C.; Ahmed, S.; Morrison, J.; Pernet, D.; Renwick, A.; Maranian, M.; Seal, S.; Ghoussaini, M.; Hines, S.; Healey, C.S.; et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat. Genet 2010, 42, 504–507. [Google Scholar]
- Stacey, S.N.; Sulem, P.; Masson, G.; Gudjonsson, S.A.; Thorleifsson, G.; Jakobsdottir, M.; Sigurdsson, A.; Gudbjartsson, D.F.; Sigurgeirsson, B.; Benediktsdottir, K.R.; et al. New common variants affecting susceptibility to basal cell carcinoma. Nat. Genet 2009, 41, 909–914. [Google Scholar]
- Kumar, R.; Smeds, J.; Berggren, P.; Straume, O.; Rozell, B.L.; Akslen, L.A.; Hemminki, K.A. A single nucleotide polymorphism in the 3′ untranslated region of the CDNK2A gene is common in sporadic primary melanomas but mutations in the CDKN2B, CDKN2C, CDK4 and P53 genes are rare. Int. J. Cancer 2001, 95, 388–393. [Google Scholar]
- Yeh, I.; Bastian, B.C. Genome-wide associations studies for melanoma and nevi. Pigment Cell Melanoma Res 2009, 22, 527–528. [Google Scholar]
- Chen, J.; Li, D.; Wei, C.; Sen, S.; Killary, A.M.; Amos, C.I.; Evans, D.B.; Abbruzzese, J.L.; Frazier, M.L. Aurora-A and p16 polymorphisms contribute to an earlier age at diagnosis of pancreatic cancer in Caucasians. Clin. Cancer Res 2007, 13, 3100–3104. [Google Scholar]
- Gayther, S.A.; Song, H.; Ramus, S.J.; Kjaer, S.K.; Whittemore, A.S.; Quaye, L.; Tyrer, J.; Shadforth, D.; Hogdall, E.; Hogdall, C.; et al. Tagging single nucleotide polymorphisms in cell cycle control genes and susceptibility to invasive epithelial ovarian cancer. Cancer Res 2007, 67, 3027–3035. [Google Scholar]
- Rajaraman, P.; Melin, B.S.; Wang, Z.; McKean-Cowdin, R.; Michaud, D.S.; Wang, S.S.; Bondy, M.; Houlston, R.; Jenkins, R.B.; Wrensch, M.; et al. Genome-wide association study of glioma and meta-analysis. Hum. Genet 2010, 131, 1877–1888. [Google Scholar]
- Matheu, A.; Maraver, A.; Collado, M.; Garcia-Cao, I.; Cañamero, M.; Borras, C.; Flores, J.M.; Klatt, P.; Viña, J.; Serrano, M. Anti-aging activity of the Ink4/Arf locus. Aging Cell 2009, 8, 152–161. [Google Scholar]
- Cánepa, E.T.; Scassa, M.E.; Ceruti, J.M.; Marazita, M.C.; Carcagno, A.L.; Sirkin, P.F.; Ogara, M.F. INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions. IUBMB Life 2007, 59, 419–426. [Google Scholar]
- Schmid, M.; Malicki, D.; Nobori, T.; Rosenbach, M.D.; Campbell, K.; Carson, D.A.; Carrera, C.J. Homozygous deletions of methylthioadenosine phosphorylase (MTAP) are more frequent than p16 non-small cell lung cancers (NSCLC). Oncogene 1998, 17, 2669–2675. [Google Scholar]
- Hellerbrand, C.; Mühlbauer, M.; Wallner, S.; Schuierer, M.; Behrmann, I.; Bataille, F.; Weiss, T.; Schölmerich, J.; Bosserhoff, A.K. Promoter-hypermethylation is causing functional relevant downregulation of methylthioadenosine phosphorylase (MTAP) expression in hepatocellular carcinoma. Carcinogenesis 2006, 27, 64–72. [Google Scholar]
- Congrains, A.; Kamide, K.; Oguro, R.; Yasuda, O.; Miyata, K.; Yamamoto, E.; Kawai, T.; Kusunoki, H.; Yamamoto, H.; Takeya, Y.; et al. Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B. Atherosclerosis 2012, 220, 449–455. [Google Scholar]
- Holdt, L.M.; Beutner, F.; Scholz, M.; Gielen, S.; Gäbel, G.; Bergert, H.; Schuler, G.; Thiery, J.; Teupser, D. ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscler. Thromb. Vasc. Biol 2010, 30, 620–627. [Google Scholar]
- Cunnington, M.S.; Santibanez Koref, M.; Mayosi, B.M.; Burn, J.; Keavney, B. Chromosome 9p21 SNPs associated with multiple disease phenotypes correlate with ANRIL expression. PLoS Genet 2010, 6, e1000899. [Google Scholar]
- Harismendy, O.; Notani, D.; Song, X.; Rahim, N.G.; Tanasa, B.; Heintzman, N.; Ren, B.; Fu, X.D.; Topol, E.J.; Rosenfeld, M.G.; et al. 9p21 DNA variants associated with coronary artery disease impair interferon-γ signalling response. Nature 2011, 470, 264–268. [Google Scholar]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15INK4B tumor suppressor gene. Oncogene 2011, 30, 1956–1962. [Google Scholar]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.M. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar]
- Pasmant, E.; Laurendeau, I.; Héron, D.; Vidaud, M.; Vidaud, D.; Bièche, I. Characterization of a germ-line deletion. Including the entire INK4/ARF locus, in a melanoma-neural system tumor family identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res 2007, 67, 3963–3969. [Google Scholar]
- Folkersen, L.; Kyriakou, T.; Goel, A.; Peden, J.; Mälarstig, A.; Paulsson-Berne, G.; Hamsten, A.; Watkins, H.; Franco-Cereceda, A.; Gabrielsen, A.; et al. PROCARDIS consortia. Relationship between CAD risk genotype in the chromosome 9p21 locus and gene expression. Identification of eight new ANRIL splice variants. PLoS One 2009, 4, e7677. [Google Scholar]
- Burd, C.E.; Jeck, W.R.; Liu, Y.; Sanoff, H.K.; Wang, Z.; Sharpless, N.E. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet 2010, 6, e1001233. [Google Scholar]
- Kanhere, A.; Viiri, K.; Araújo, C.C.; Rasaiyaah, J.; Bouwman, R.D.; Whyte, W.A.; Pereira, C.F.; Brookes, E.; Walker, K.; Bell, G.W.; et al. Short RNAs are transcribed from repressed polycomb target genes and interact with polycomb repressive complex-2. Mol. Cell 2010, 38, 675–688. [Google Scholar]
- Glazko, G.V.; Zybailov, B.L.; Rogozin, I.B. Computational prediction of polycomb-associated long non-coding RNAs. PLoS One 2012, 7, e44878. [Google Scholar]
- Guil, S.; Soler, M.; Portela, A.; Carrère, J.; Fonalleras, E.; Gómez, A.; Villanueva, A.; Esteller, M. Intronic RNAs mediate EZH2 regulation of epigenetic targets. Nat. Struct. Mol. Biol 2012, 19, 664–670. [Google Scholar]
- Duszczyk, M.M.; Zanier, K.; Sattler, M. A NMR strategy to unambiguously distinguish nucleic acid hairpin and duplex conformations applied to a Xist RNA A-repeat. Nucleic Acids Res 2008, 36, 7068–7077. [Google Scholar]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.J.; Lee, J.T. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 2008, 322, 750–756. [Google Scholar]
- Hansen, T.B.; Wiklund, E.D.; Bramsen, J.B.; Villadsen, S.B.; Statham, A.L.; Clark, S.J.; Kjems, J. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J 2011, 30, 4414–4422. [Google Scholar]
- Orlando, V. Polycomb, epigenomes, and control of cell identity. Cell 2003, 112, 599–606. [Google Scholar]
- Mohammad, H.P.; Cai, Y.; McGarvey, K.M.; Easwaran, H.; van Neste, L.; Ohm, J.E.; O’Hagan, H.M.; Baylin, S.B. Polycomb CBX7 promotes initiation of heritable repression of genes frequently silenced with cancer-specific DNA hypermethylation. Cancer Res 2009, 69, 6322–6330. [Google Scholar]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar]
- Ordovás, J.M.; Smith, C.E. Epigenetics and cardiovascular disease. Nat. Rev. Cardiol 2010, 7, 510–519. [Google Scholar]
- Koleganova, N.; Benz, K.; Piecha, G.; Ritz, E.; Amann, K. Renal, cardiovascular and metabolic effects of fetal programming. Nephrol. Dial. Transplant 2012, 27, 3003–3007. [Google Scholar]
- Okamura, M.; Inagaki, T.; Tanaka, T.; Sakai, J. Role of histone methylation and demethylation in adipogenesis and obesity. Organogenesis 2010, 6, 24–32. [Google Scholar]
- Aguilo, F.; Zhou, M.M.; Walsh, M.J. Long noncoding RNA, polycomb, and the ghosts haunting INK4b-ARF-INK4a expression. Cancer Res 2011, 71, 5365–5369. [Google Scholar]
- Vincenz, C.; Kerppola, T.K. Different polycomb group CBX family proteins associate with distinct regions of chromatin using nonhomologous protein sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 16572–16577. [Google Scholar]
- Gil, J.; Bernard, D.; Martínez, D.; Beach, D. Polycomb CBX7 has a unifying role in cellular lifespan. Nat. Cell Biol 2004, 6, 67–72. [Google Scholar]
- Popov, N.; Gil, J. Epigenetic regulation of the INK4b-ARF-INK4a locus: In sickness and in health. Epigenetics 2010, 5, 685–690. [Google Scholar]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar]
- Pandey, R.R.; Mondal, T.; Mohammad, F.; Enroth, S.; Redrup, L.; Komorowski, J.; Nagano, T.; Mancini-Dinardo, D.; Kanduri, C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 2008, 32, 232–246. [Google Scholar]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Morales, D.R.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar]
- Sato, K.; Nakagawa, H.; Tajima, A.; Yoshida, K.; Inoue, I. ANRIL is implicated in the regulation of nucleus and potential transcriptional target of E2F1. Oncol. Rep 2010, 24, 701–707. [Google Scholar]
- Ghosh, A.K.; Varga, J. The transcriptional coactivator and acetyltransferase p300 in fibroblast biology and fibrosis. J. Cell Physiol 2007, 213, 663–671. [Google Scholar]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [Green Version]
- Bandyopadhyay, D.; Okan, N.A.; Bales, E.; Nascimento, L.; Cole, P.A.; Medrano, E.E. Down-regulation of p300/CBP histone acetyltransferase activates a senescence checkpoint in human melanocytes. Cancer Res 2002, 62, 6231–6239. [Google Scholar]
- Li, Q.; Xiao, H.; Isobe, K. Histone acetyltransferase activities of cAMP-regulated enhancer-binding protein p300 in tissues of fetal, young and old mice. J. Gerontol. A 2002, 57, B93–B98. [Google Scholar]
- Ooi, L.; Wood, I.C. Chromatin crosstalk in development and disease: Lessons from REST. Nat. Rev. Genet 2007, 8, 544–554. [Google Scholar]
- Wolf, S.S.; Patchev, V.K.; Obendorf, M. A novel variant of the putative demethylase gene, s-JMJD1C, is a coactivator of the AR. Arch. Biochem. Biophys 2007, 460, 56–66. [Google Scholar]
- Lluis, F.; Ombrato, L.; Pedone, E.; Pepe, S.; Merrill, B.J.; Cosma, M.P. T-cell factor 3 (Tcf3) deletion increases somatic cell reprogramming by inducing epigenome modifications. Proc. Natl. Acad. Sci. USA 2011, 108, 11912–11917. [Google Scholar]
- Visel, A.; Zhu, Y.; May, D.; Afzal, V.; Gong, E.; Attanasio, C.; Blow, M.J.; Cohen, J.C.; Rubin, E.M.; Pennacchio, L.A. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature 2010, 464, 409–412. [Google Scholar]
- Pasmant, E.; Sabbagh, A.; Vidaud, M.; Bièche, I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J 2011, 25, 444–448. [Google Scholar]
- Rangarajan, A.; Weinberg, R.A. Comparative biology of mouse versus human cells: Modeling human cancer in mice. Nat. Rev. Cancer 2003, 3, 952–959. [Google Scholar]
- Wei, W.; Hemmer, R.M.; Sedivy, J.M. Role of p14ARF in replicative and induced senescence of human fibroblasts. Mol. Cell. Biol 2001, 21, 6748–6757. [Google Scholar]
- Yu, W.; Gius, D.; Onyango, P.; Muldoon-Jacobs, K.; Karp, J.; Feinberg, A.P.; Cui, H. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature 2008, 451, 202–206. [Google Scholar]
- Guil, S.; Esteller, M. Cis-acting noncoding RNAs: Friends and foes. Nat. Struct. Mol. Biol 2012, 19, 1068–1075. [Google Scholar]
- Congrains, A.; Kamide, K.; Katsuya, T.; Yasuda, O.; Oguro, R.; Yamamoto, K.; Ohishi, M.; Rakugi, H. CVD-associated non-coding RNA, ANRIL, modulates expression of atherogenic pathways in VSMC. Biochem. Biophys. Res. Commun. 2012, 419, 612–616. [Google Scholar]
- Pasmant, E.; Sabbagh, A.; Masliah-Planchon, J.; Ortonne, N.; Laurendeau, I.; Melin, L.; Ferkal, S.; Hernandez, L.; Leroy, K.; Valeyrie-Allanore, L.; et al. Role of noncoding RNA ANRIL in genesis of plexiform neurofibromas in neurofibromatosis type 1. J. Natl. Cancer Inst 2011, 103, 1713–1722. [Google Scholar]
- Gschwendtner, A.; Bevan, S.; Cole, J.W.; Plourde, A.; Matarin, M.; Ross-Adams, H.; Meitinger, T.; Wichmann, E.; Mitchell, B.D.; Furie, K.; et al. Sequence variants on chromosome 9p21.3 confer risk for atherosclerotic stroke. Ann. Neurol 2009, 65, 531–539. [Google Scholar]
- Helgadottir, A.; Thorleifsson, G.; Manolescu, A.; Gretarsdottir, S.; Blondal, T.; Jonasdottir, A.; Jonasdottir, A.; Sigurdsson, A.; Baker, A.; Palsson, A.; et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 2007, 316, 1491–1493. [Google Scholar]
- Bos, J.L. Ras oncogenes in human cancer: A review. Cancer Res 1989, 49, 4682. [Google Scholar]
- Minamino, T.; Yoshida, T.; Tateno, K.; Miyauchi, H.; Zou, Y.; Toko, H.; Komuro, I. Ras induces vascular smooth muscle cell senescence and inflammation in human atherosclerosis. Circulation 2003, 108, 2264–2269. [Google Scholar]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis—Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar]
- Hirosue, A.; Ishihara, K.; Tokunaga, K.; Watanabe, T.; Saitoh, N.; Nakamoto, M.; Chandra, T.; Narita, M.; Shinohara, M.; Nakao, M. Quantitative assessment of higher-order chromatin structure of the INK4/ARF locus in human senescent cells. Aging Cell 2012, 11, 553–556. [Google Scholar]
- Kim, W.Y.; Sharpless, N.E. The regulation of INK4/ARF in cancer and aging. Cell 2006, 127, 265–275. [Google Scholar]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Invest 2004, 114, 1299–1307. [Google Scholar]
- Li, H.; Collado, M.; Villasante, A.; Strati, K.; Ortega, S.; Cañamero, M.; Blasco, M.A.; Serrano, M. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 2009, 460, 1136–1139. [Google Scholar]
- Krishnamurthy, J.; Ramsey, M.R.; Ligon, K.L.; Torrice, C.; Koh, A.; Bonner-Weir, S.; Sharpless, N.E. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006, 443, 453–457. [Google Scholar]
- Molofsky, A.V.; Slutsky, S.G.; Joseph, N.M.; He, S.; Pardal, R.; Krishnamurthy, J.; Sharpless, N.E.; Morrison, S.J. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 2006, 443, 448–452. [Google Scholar]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet 2006, 15, R17–R29. [Google Scholar]
- Bracken, A.P.; Dietrich, N.; Pasini, D.; Hansen, K.H.; Helin, K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev 2006, 20, 1123–1136. [Google Scholar]
- Ku, M.; Koche, R.P.; Rheinbay, E.; Mendenhall, E.M.; Endoh, M.; Mikkelsen, T.S.; Presser, A.; Nusbaum, C.; Xie, X.; Chi, A.S.; et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 2008, 4, e1000242. [Google Scholar]
- Kim, J.B.; Deluna, A.; Mungrue, I.N.; Vu, C.; Pouldar, D.; Civelek, M.; Orozco, L.; Wu, J.; Wang, X.; Charugundla, S.; et al. The effect of 9p21.3 coronary artery disease locus neighboring genes on atherosclerosis in mice. Circulation 2012. [Google Scholar] [CrossRef]
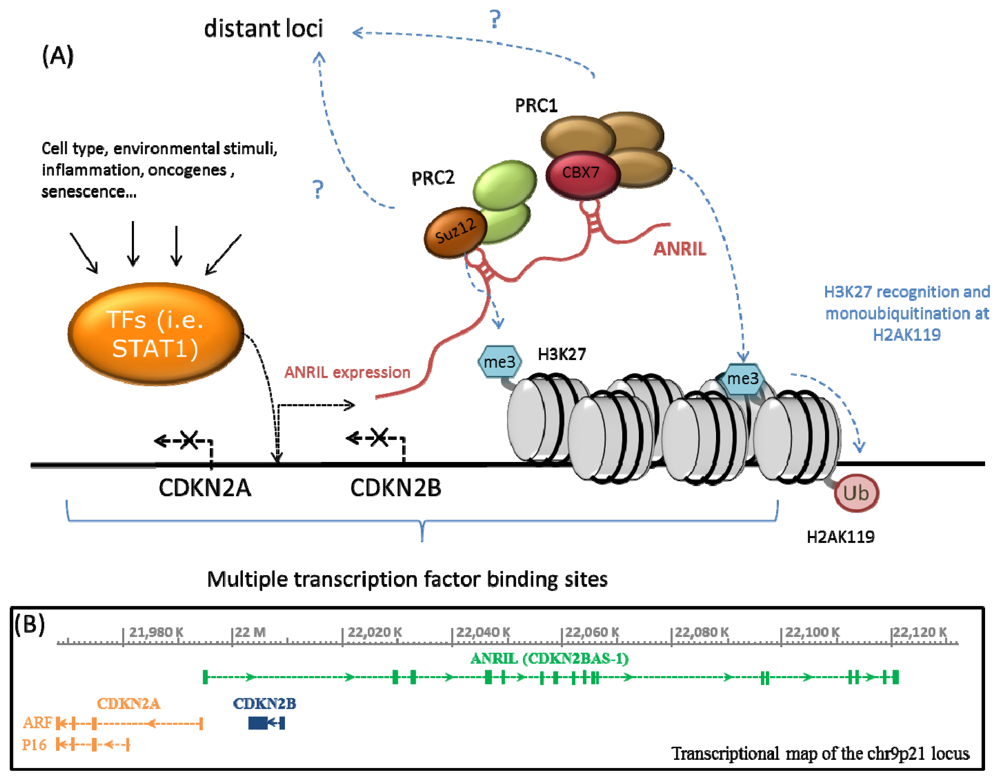

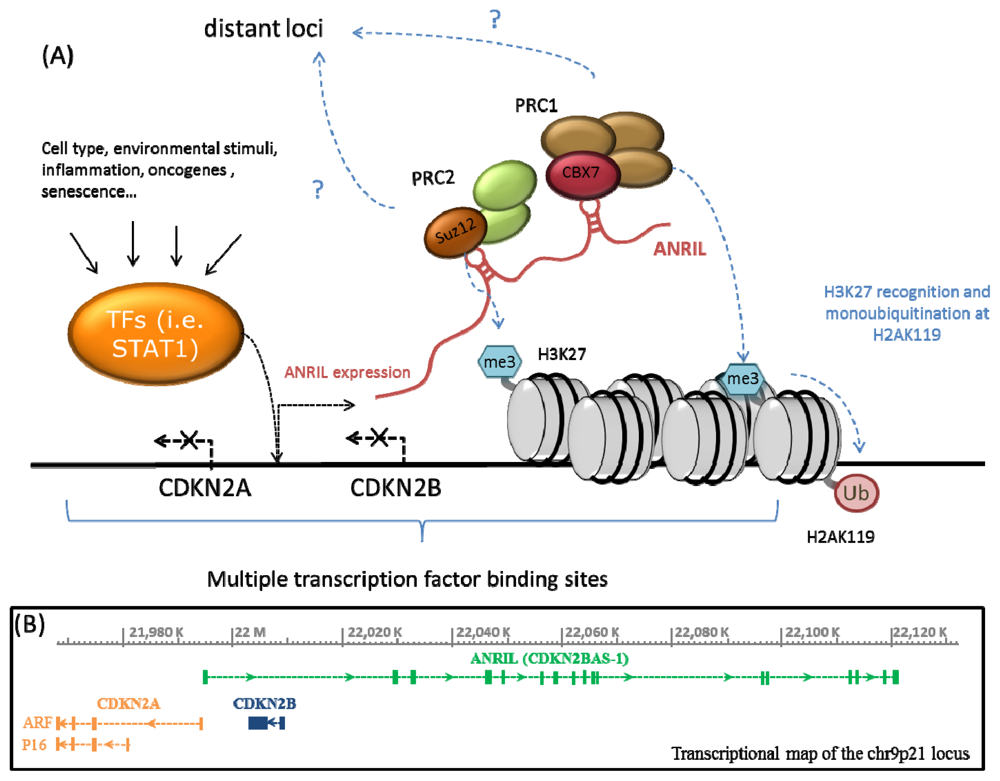{kind=link}
| SNP | Disease association | Reported effect on ANRIL expression |
|---|---|---|
| rs564398-A | Risk allele for diabetes and atherosclerotic stroke [65] | Reduced ANRIL expression (exons 1–2). Twice reported to exert the strongest influence in ANRIL expression in peripheral blood [21,23] |
| rs1063192-C | Risk for glioma [16] and open angle glaucoma [6] | Increased ANRIL expression (exons 1–2) [21,23] |
| rs1011970-T | Melanoma [13] | Reduced expression of ANRIL (exons 1–2) [23] |
| rs2151280-T | Risk allele for plexiform neurofibroma development [64] and protective allele for BCC [11] | Reduced expression of ANRIL (exons 15–16) [64] |
| rs3731217-G | Risk allele for Acute lymphoblastic leukemia [9] | Reduced expression of ANRIL (exons 17–18) [21] |
| rs496892-G | Risk for atherosclerotic stroke [65] and periodontitis | Reduced ANRIL expression (exons 1–2) [21,23] |
| rs10757278-G | Lead SNP for CAD risk [66] | Increased ANRIL variant EU741058 expression (exons 1–5 of the long transcript) [22], but reduced expression of ANRIL exons 1–2 [23]. |
| rs3731257-G | Risk allele for ovarian cancer [15] | Increased expression of ANRIL (exons 1–2) [23] |
| rs10811661-T | Risk allele for diabetes [2] | Reduced ANRIL expression (exons 1–2) [23] |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Congrains, A.; Kamide, K.; Ohishi, M.; Rakugi, H. ANRIL: Molecular Mechanisms and Implications in Human Health. Int. J. Mol. Sci. 2013, 14, 1278-1292. https://doi.org/10.3390/ijms14011278
Congrains A, Kamide K, Ohishi M, Rakugi H. ANRIL: Molecular Mechanisms and Implications in Human Health. International Journal of Molecular Sciences. 2013; 14(1):1278-1292. https://doi.org/10.3390/ijms14011278
Chicago/Turabian StyleCongrains, Ada, Kei Kamide, Mitsuru Ohishi, and Hiromi Rakugi. 2013. "ANRIL: Molecular Mechanisms and Implications in Human Health" International Journal of Molecular Sciences 14, no. 1: 1278-1292. https://doi.org/10.3390/ijms14011278