Flavin-Dependent Enzymes in Cancer Prevention


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
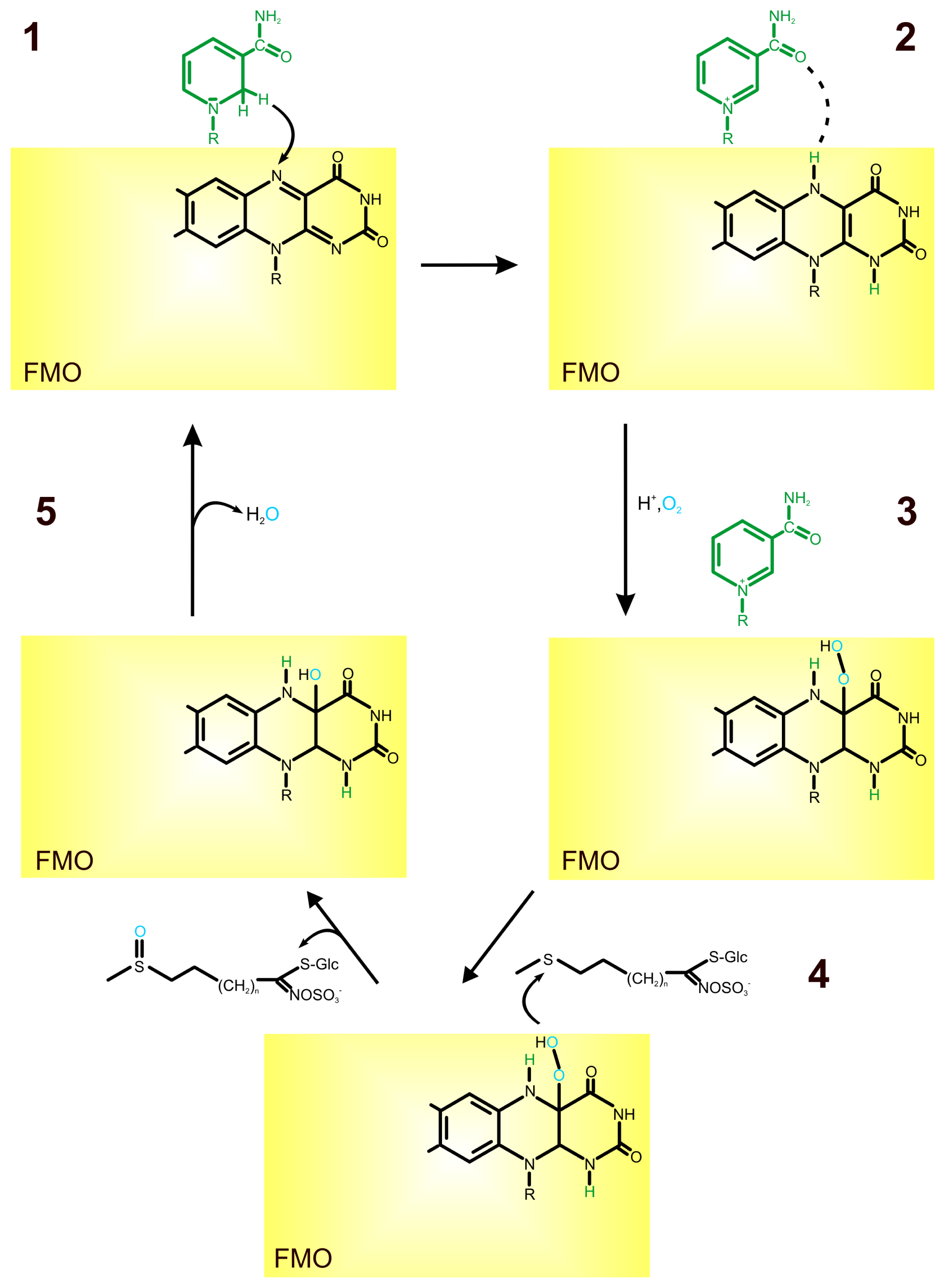
2. Role of Flavin-Containing Monooxygenase in Glucosinolate Synthesis
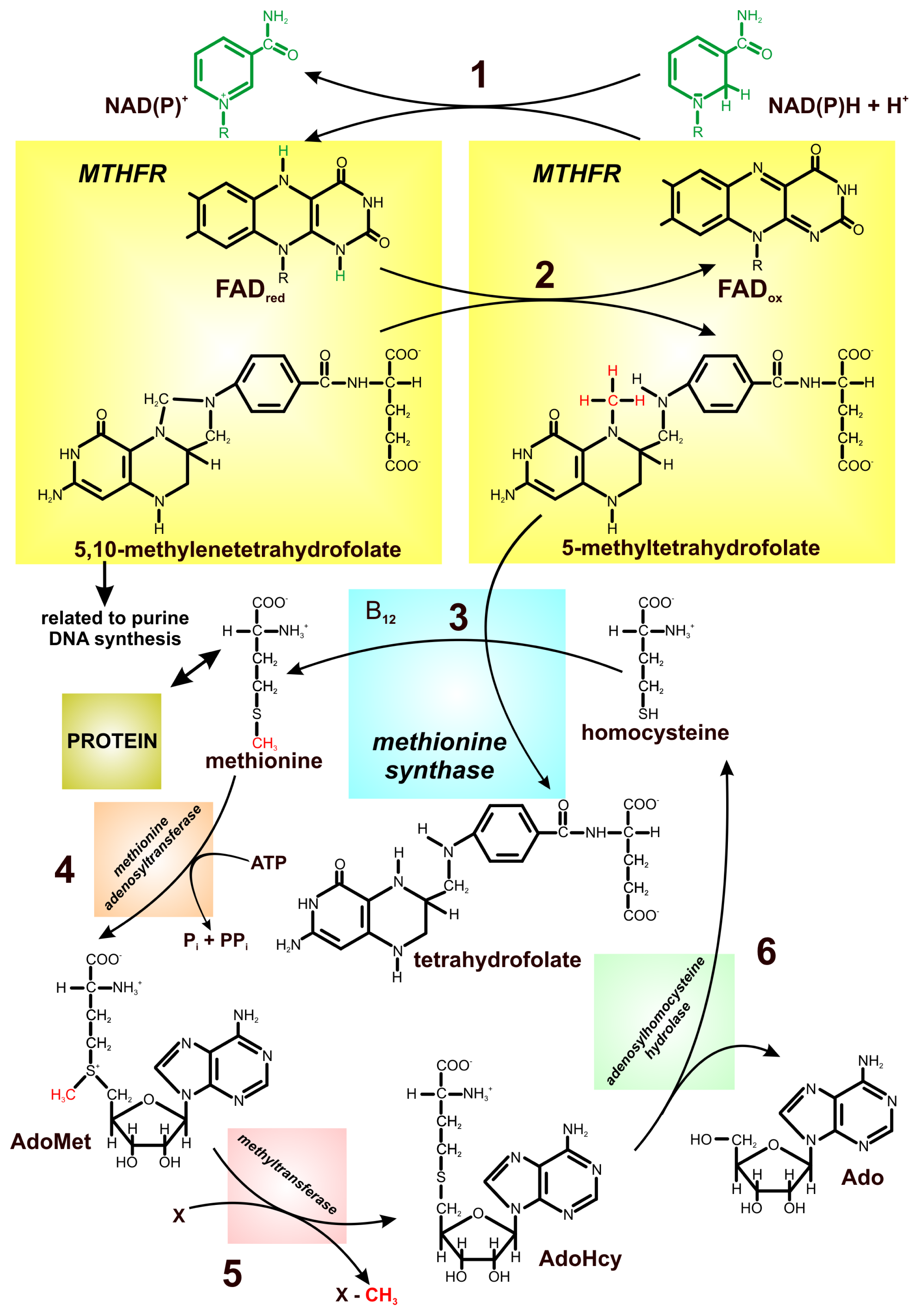
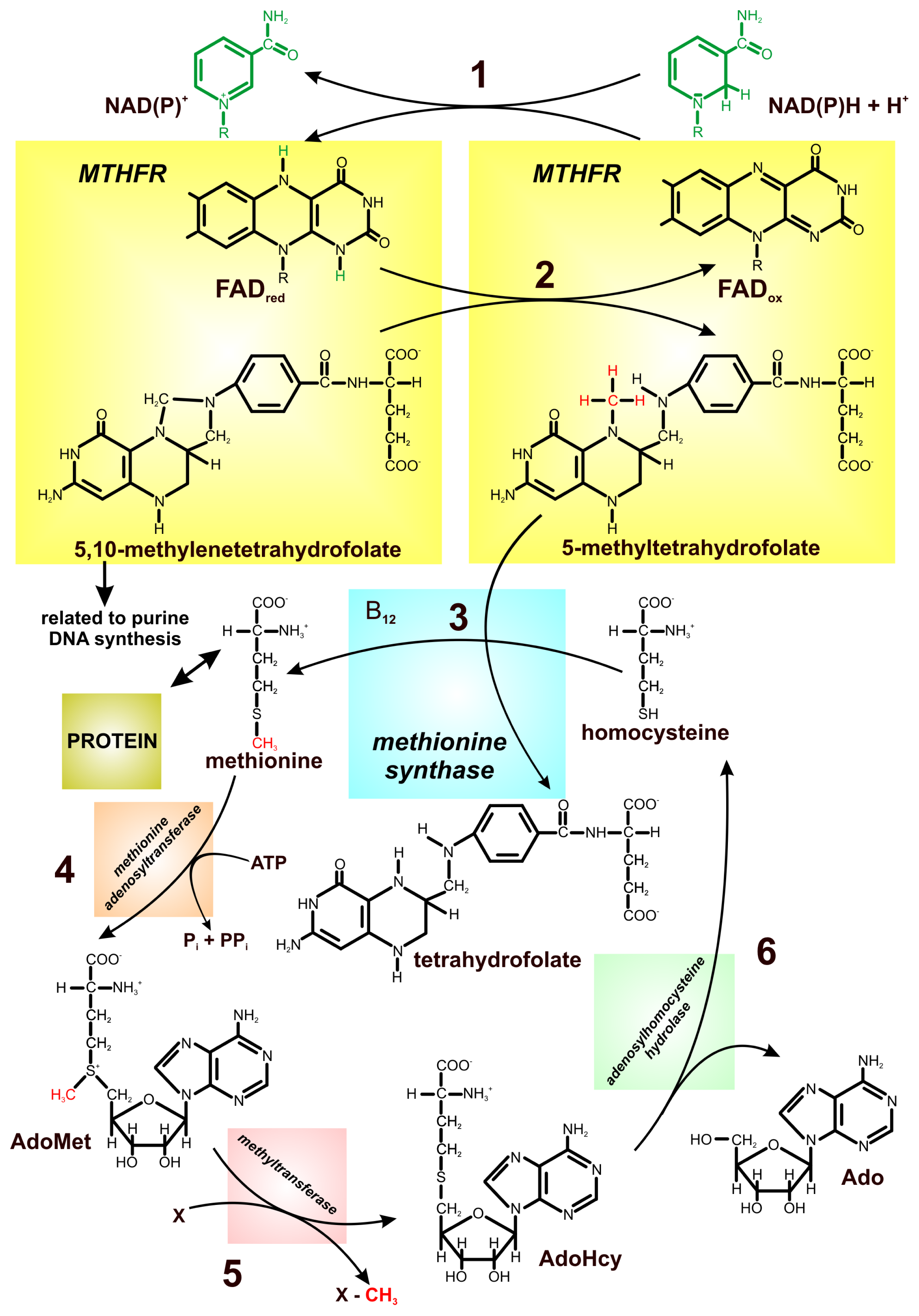
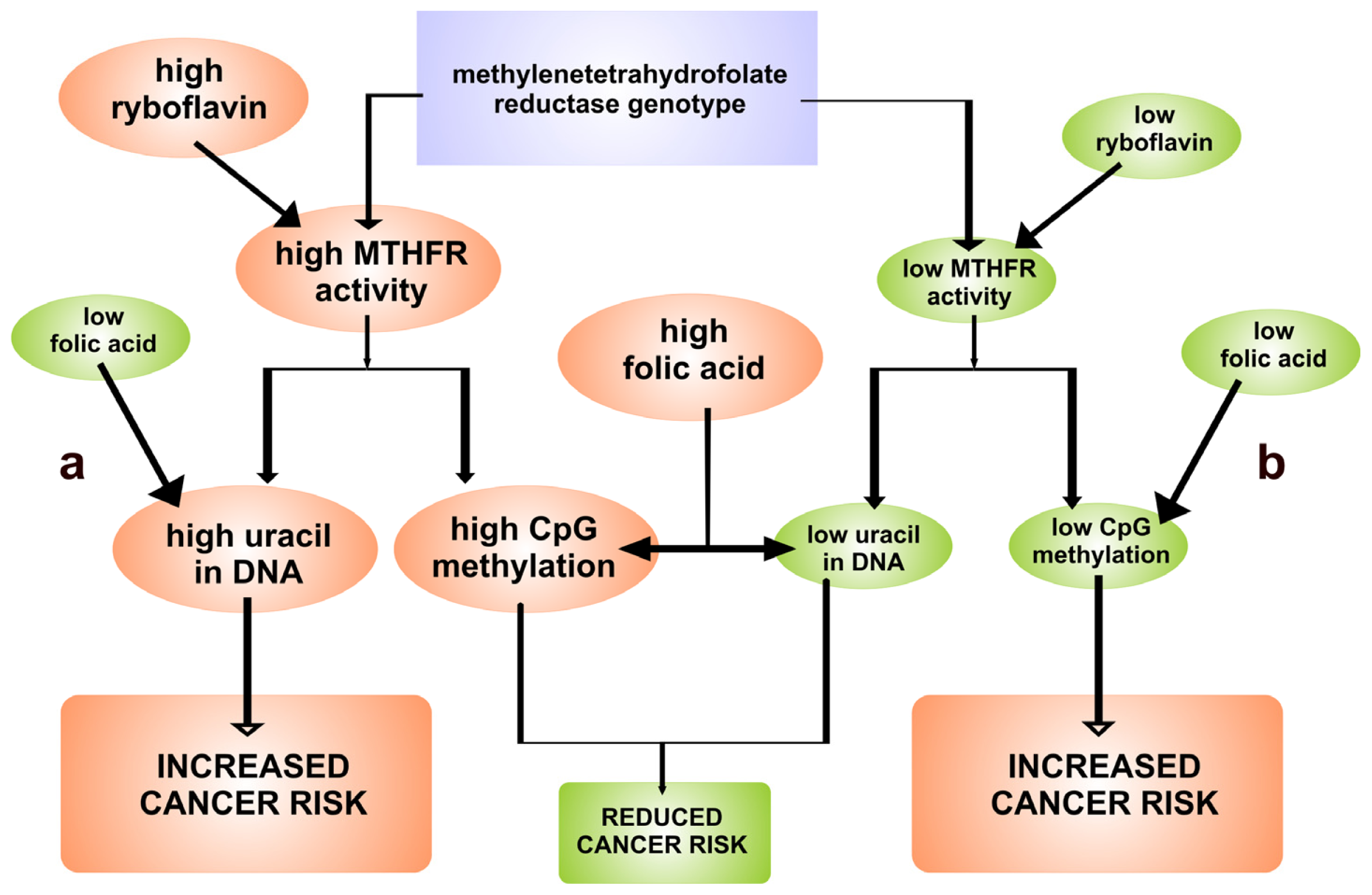
3. Relationship between FAD-Dependent 5,10-Methylenetetrahydrofolate Reductase and Cancer Risk
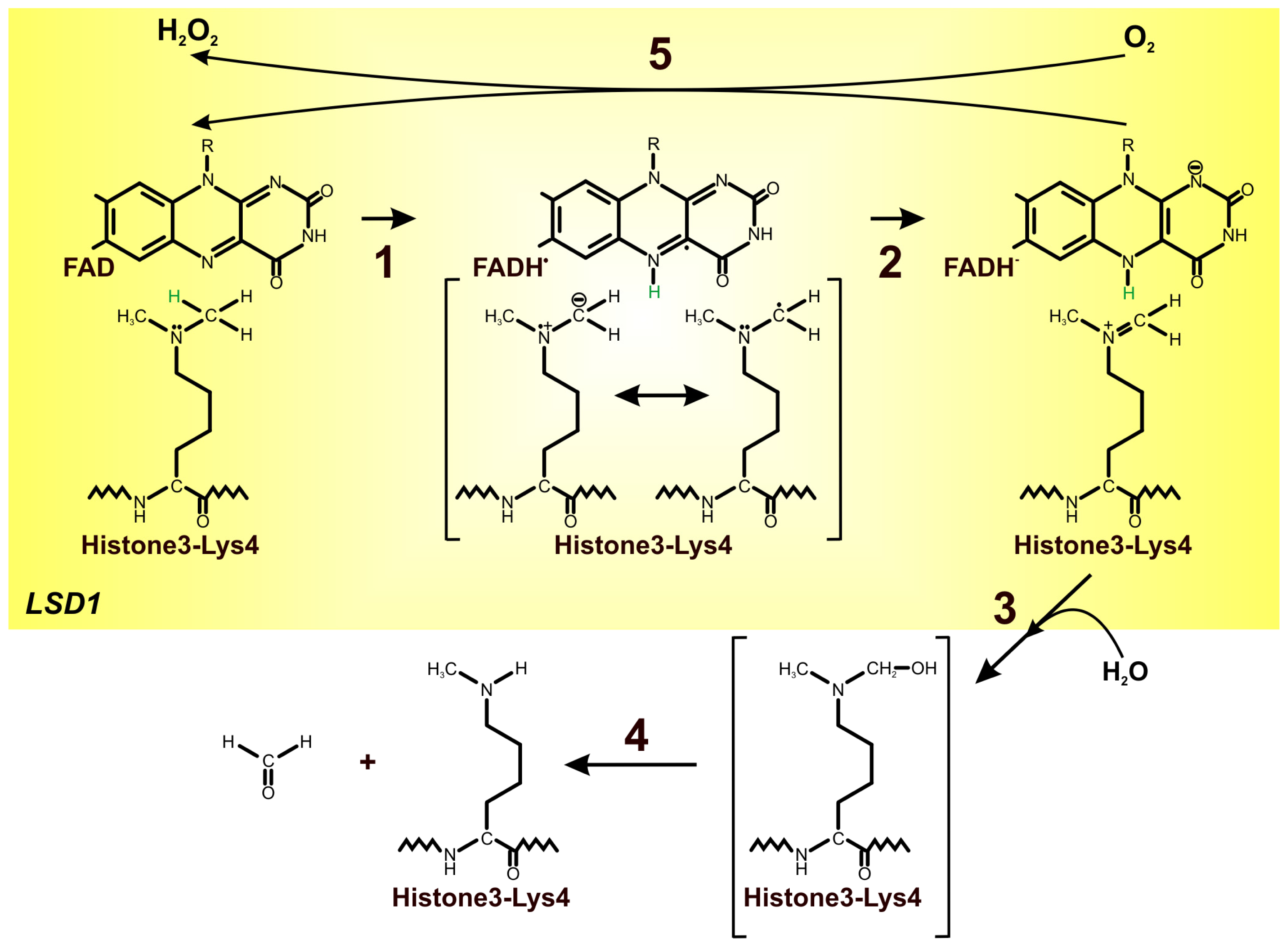
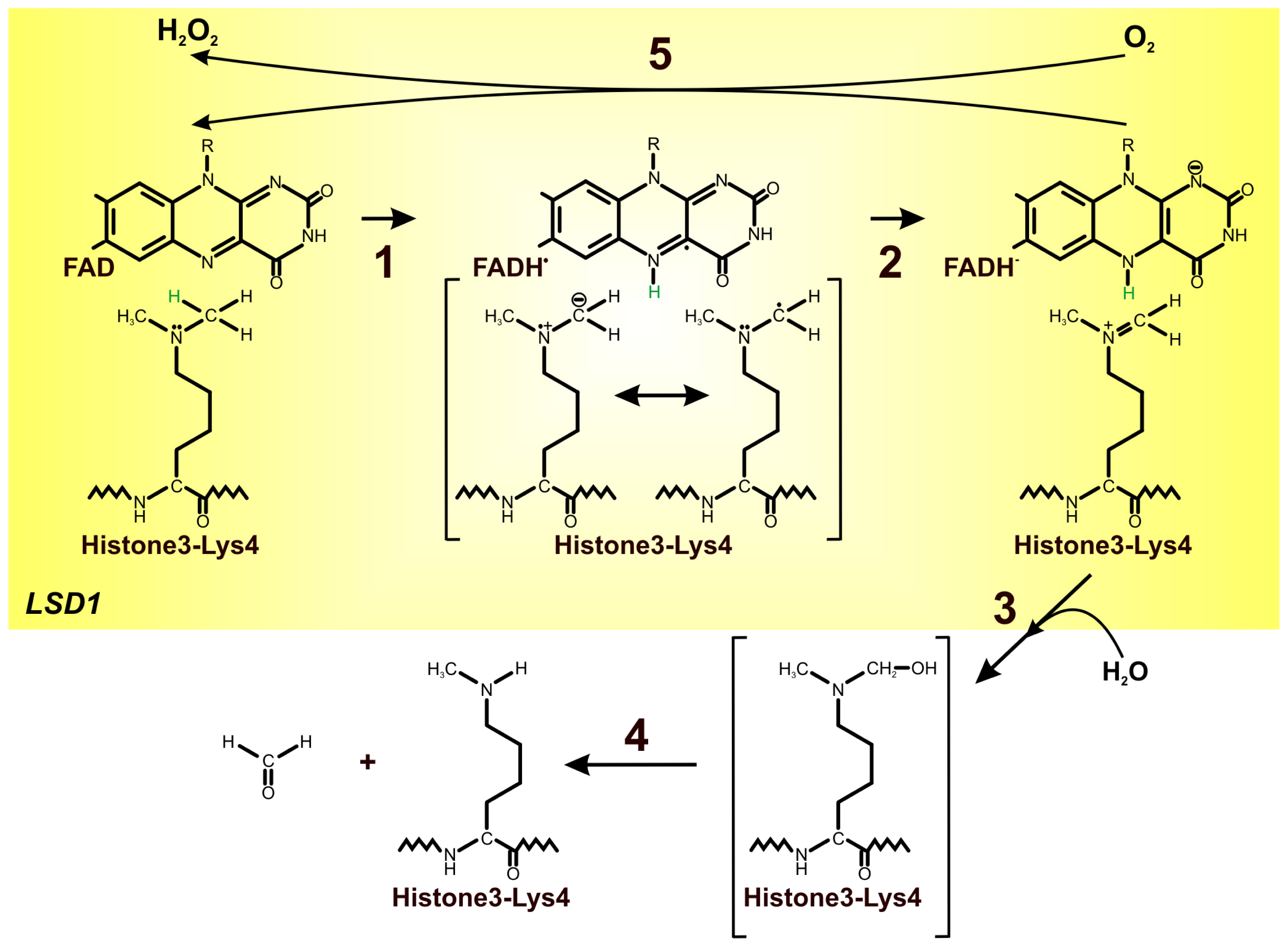
4. FAD as an Important Cofactor for the Demethylation of Histones
5. The Role of Riboflavin Supplements in the Prevention of Cancer
6. Conclusions
References
- Powers, H.J. Riboflavin (vitamin B-2) and health. Am. J. Clin. Nutr 2003, 77, 1352–1360. [Google Scholar]
- Basset, J.K.; Severi, G.; Hodge, A.M.; Baglietto, L.; Hopper, J.L.; English, D.R.; Giles, G.G. Dietary intake of B vitamins and methionine and prostate cancer incidence and mortality. Cancer Causes Control 2012, 23, 855–863. [Google Scholar]
- Kimura, M.; Umegaki, K.; Higuchi, M.; Thomas, P. Methylenetetrahydrofolate reductase C677T polymorphism, folic acid and riboflavin are important determinants of genome stability in culture human lymphocytes. J. Nutr 2004, 134, 48–56. [Google Scholar]
- Powers, H.J. Interaction among folate, riboflavin, genotype, and cancer, with reference to colorectal and cervical cancer. J. Nutr 2005, 135, 2960S–2966S. [Google Scholar]
- Powers, H.J.; Hill, M.H.; Welfare, M.; Spiers, A.; Bal, W.; Russell, J.; Duckworth, Y.; Gibney, E.; Williams, E.A.; Mathers, J.C. Responses of biomarkers of folate and riboflavin status to folate and riboflavin supplementation in healthy and colorectal polyp patients (The FAB2 study). Cancer Epidemiol. Biomarkers Prev 2007, 16, 2128–2135. [Google Scholar]
- Premkumar, V.G.; Yuvaraj, S.; Shanthi, P.; Sachdanandam, P. Co-enzyme Q10, riboflavin and niacin supplementation on alteration of DNA repair enzyme and DNA methylation in breast cancer patients undergoing tamoxifen therapy. Br. J. Nutr 2008, 100, 1179–1182. [Google Scholar]
- Vergara, F.; Wenzler, M.; Hansen, B.G.; Kliebenstein, D.J.; Halkier, B.A.; Gersgenzon, J.; Schneider, B. Determination of the absolute configuration of the glucosinolate methyl sulfoxide group reveals a stereospecific biosynthesis of the side chain. Phytochemistry 2008, 69, 2737–2742. [Google Scholar]
- Sønderby, I.E.; Geu-Flores, F.; Halkier, B.A. Biosynthesis of glucosinolates-gene discovery and beyond. Trends Plant Sci 2010, 15, 283–290. [Google Scholar]
- Wang, H.; Wu, J.; Sun, S.; Liu, B.; Cheng, F.; Sun, R.; Wang, X. Glucosinolate biosynthetic genes in Brassica rapa. Gene 2011, 487, 135–142. [Google Scholar]
- Katchamart, S.; Stresser, D.M.; Dehal, S.S.; Kupfer, D.; Williams, D.E. Concurrent flavin-containing monooxygenase down-regulation and cytochrome P-450 induction by dietary indoles in rat: implications for drug-drug interaction. Drug Metab. Dispos 2000, 28, 930–936. [Google Scholar]
- Hansen, B.G.; Kliebenstein, D.J.; Halkier, B.A. Identification of a flavin-monooxygenase as the S-oxygenating enzyme in aliphatic glucosinolate biosynthesis in Arabidopsis. Plant J 2007, 50, 902–910. [Google Scholar]
- Li, J.; Hansen, B.G.; Ober, J.A.; Kliebenstein, D.J.; Halkier, B.A. Subclade of flavin-monooxygenase involved in aliphatic glucosinolate biosynthesis. Plant Physiol 2008, 148, 1721–1733. [Google Scholar]
- Mikkelsen, M.D.; Olsen, C.E.; Halkier, B.A. Production of the cancer-preventive glucoraphanin in tobacco. Mol. Plant 2010, 4, 751–759. [Google Scholar]
- Li, J.; Kristiansen, K.A.; Hansen, B.G.; Halkier, B.A. Cellular and subcellular localization of flavin-monooxygenases involved in glucosinolate biosynthesis. J. Exp. Bot 2010, 62, 1337–1346. [Google Scholar]
- Verhoeven, D.T.H.; Goldbohm, R.A.; van Poppel, G.; Verhagen, H.; van den Brandt, P.A. Epidemiological studies on Brassica vegetables and cancer risk. Cancer Epidemiol. Biomark. Prev 1996, 5, 733–748. [Google Scholar]
- Fahey, J.W.; Zhang, Y.; Talalay, P. Broccoli sprouts: An exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc. Natl. Acad. Sci. USA 1997, 94, 10367–10372. [Google Scholar]
- Talalay, P.; Fahey, J.W. Phytochemicals from Cruciferous plants protect against cancer by modulating carcinogen metabolism. J. Nutr 2001, 131, 3027S–3033S. [Google Scholar]
- Schlaich, N.L. Flavin-containing monooxygenases in plants: Looking beyond detox. Trends Plant Sci 2007, 12, 412–418. [Google Scholar]
- Eswaramoorthy, S.; Bonanno, J.B.; Burley, S.K.; Swaminathan, S. Mechanism of action of a flavin-containing monooxygenase. Proc. Natl. Acad. Sci. USA 2006, 103, 9832–9837. [Google Scholar]
- Palfey, B.A.; McDonald, C.A. Control of catalysis in flavin-depndent monooxygenases. Arch. Biochem. Biophys 2010, 493, 26–36. [Google Scholar]
- Varalakshmi, K.; Savithri, H.S.; Rao, A. Purification and kinetic mechanism of 5,10-metliyienetetrahydrofolate reductase from sheep liver. J. Biosci 1983, 5, 287–299. [Google Scholar]
- Viel, A.; Dall’Agnese, L.; Simone, F.; Canzonieri, V.; Capozzi, E.; Visentin, M.C.; Valle, R.; Boiocchi, M. Loss of heterozygosity at the 5,10-methylenetetrahydrofolate reductase locus in human ovarian carcinomas. Br. J. Cancer 1997, 75, 1105–1110. [Google Scholar]
- Roje, S.; Wang, H.; McNeil, S.D.; Raymond, R.K.; Appling, D.R.; Shachar-Hill, Y.; Bohnert, H.J.; Hanson, A.D. Isolation, characterization, and functional expression of cDNAs encoding NADH-dependent methylenetetrahydrofolate reductase from higher plants. J. Biol. Chem 1999, 274, 36089–36096. [Google Scholar]
- Sheppard, C.A.; Trimmer, E.E.; Matthews, R.G. Purification and properties of NADH-depndent 5,10-methylenetetrahydrofolate reductase (MetF) from Escherichia coli. J. Bacteriol 1999, 181, 718–725. [Google Scholar]
- Chen, J.; Gammon, M.D.; Chan, W.; Palomeque, C.; Wetmur, J.G.; Kabat, G.C.; Teitelbaum, S.L.; Britton, J.A.; Terry, M.B.; Neugut, A.I.; et al. One-carbon metabolism, MTHFR polymorphisms, and risk of breast cancer. Cancer Res 2005, 65, 1606–1614. [Google Scholar]
- Pejchal, R.; Sargeant, R.; Ludwig, M.L. Structures of NADH and CH3-H4 folate complex of Escherichia coli methylenetetrahydrofolate reductase reveal a Spartan strategy for a ping-pong reaction. Biochemistry 2005, 44, 11447–11457. [Google Scholar]
- Igari, S.; Ohtaki, A.; Yamanaka, Y.; Sato, Y.; Yohda, M.; Odaka, M.; Noguchi, K.; Yamada, K. Properties and crystal structure of methylenetetrahydrofolate reductase from Thermus thermophilus HB8. PLoS One 2011, 6, e23716. [Google Scholar]
- Matthews, R.G.; Sheppard, S.; Goulding, C. Methylenetetrahydrofolate reductase and methionine synthase: Biochemistry and molecular biology. Eur. J. Pediatr 1998, 157, S54–S59. [Google Scholar]
- Ronien, K.; Ulrich, C.M. 5,10-Methylenetetrahydrofolate reductase polymorphisms and leukemia risk: A HuGE minireview. Am. J. Epidemiol 2003, 157, 571–582. [Google Scholar]
- Linnebank, M.; Semmler, A.; Moskau, S.; Smulders, Y.; Blom, H.; Simon, M. The methylenetetrahydrofolate reductase (MTHFR) variant c.677C > T (A222V) influences overall survival of patients with glioblastoma multifore. Neuro-Oncology 2008, 10, 548–552. [Google Scholar]
- Jacques, P.F.; Kalmbach, R.; Bagley, P.J.; Russo, G.T.; Rogers, G.; Wilson, P.W.F.; Rosenberg, I.H.; Selhub, J. The relationship between riboflavin and plasma total homocysteine in the Framingham Offspring Cohort is influenced by folate status and the C677T transition in the methylenetetrahydrofolate reductase gene. J. Nutr 2002, 132, 283–288. [Google Scholar]
- Forges, T.; Chery, C.; Audonnet, S.; Feillet, F.; Gueant, J.L. Life-treatening methylenetetrahydrofolate reductase (MTHFR) deficiency with extremely early onset: characterization of two novel mutations in compound heterozygous patients. Mol. Genet. Metab 2010, 100, 143–148. [Google Scholar]
- Hosseini, M.; Houshmand, M.; Ebrahimi, A. MTHFR polymorphisms and breast cancer risk. Arch. Med. Sci 2011, 7, 134–137. [Google Scholar]
- Chiuve, S.E.; Giovannucci, E.L.; Hankinson, S.E.; Hunter, D.J.; Stampfer, M.J.; Willett, W.C.; Rimm, E.B. Alcohol intake and methylenetetrahydrofolate reductase polymorphism modify the relation of folate intake to plasma homocysteine. Am. J. Clin. Nutr 2005, 82, 155–162. [Google Scholar]
- Kafadar, A.M.; Yilmaz, H.; Kafadar, D.; Ergen, A.; Zeybek, U.; Bozkurt, N.; Kuday, C.; Isbir, T. C677T gene polymorphism of methylenetetrahydrofolate reductase (MTHFR) in meningiomas and high-grade gliomas. Anticancer Res 2006, 26, 2445–2450. [Google Scholar]
- Hustad, S.; Nedrebø, B.G.; Ueland, P.M.; Schneede, J.; Vollset, S.E.; Ulvik, A.; Lien, E.A. Phenotypic expression of the methylenetetrahydrofolate reductase 677C→T polymorphism and flavin cofactor availability in thyroid dysfunction. Am. J. Clin. Nutr 2004, 80, 1050–1057. [Google Scholar]
- McNulty, H.; McKinley, M.C.; Wilson, B.; McPartlin, J.; Strain, J.J.; Weir, D.G.; Scott, J.M. Impaired functioning of thermolabile methylenetetrahydrofolate reductase is dependent on riboflavin status: implications for riboflavin requirements. Am. J. Clin. Nutr 2002, 76, 436–441. [Google Scholar]
- Hustad, S.; Midttun, Ø.; Schneede, J.; Vollset, S.E.; Grotmol, T.; Ueland, P.M. The methylenetetrahydrofolate reductase 677C→T polymorphism as a modulator of a B vitamin network with major effects on homocysteine metabolism. Am. J. Hum. Genet. 2007, 80, 846–855. [Google Scholar]
- Ferroni, P.; Palmirotta, R.; Martini, F.; Riondino, S.; Savonarola, A.; Spila, A.; Ciatti, S.; Sini, V.; Mariotti, S.; Del Monte, G.; et al. Determinants of homocysteine levels in colorectal and breast cancer patients. Anticancer Res 2009, 29, 4131–4138. [Google Scholar]
- Wu, L.L.; Wu, J.T. Hyperhomocysteinemia is a risk factor for cancer and a new potential tumor marker. Clin. Chim. Acta 2002, 322, 21–28. [Google Scholar]
- Lin, J.; Lee, I.M.; Song, Y.; Cook, N.R.; Selhub, J.; Manson, J.E.; B Uring, J.E.; Zhang, S.M. Plasma homocysteine and cysteine and risk of breast cancer in woman. Cancer Res 2010, 70, 2397–2405. [Google Scholar]
- Jakubowski, H. Metabolism of homocysteine thiolactone in human cell cultures. J. Biol. Chem 1997, 272, 1935–1942. [Google Scholar]
- Vinukonda, G. Plasma homocysteine and methylenetetrahydrofolate reductase gene polymorphism in human health and disease: An update. Int. J. Hum. Genet 2008, 8, 171–179. [Google Scholar]
- Ueland, P.M.; Hustad, S.; Schneede, J.; Refsum, H.; Vollset, S.E. Biological and clinical implications of the MTFR C677T polymorphism. Trends Pharmacol. Sci 2001, 22, 195–201. [Google Scholar]
- Moat, S.J.; Ashfield-Watt, P.A.L.; Powers, H.J.; Newcombe, R.G.; McDowell, I.F.W. Effect of riboflavin status on the homocysteine-lowering effect of folate in relation to the MTHFR (C677T) genotype. Clin. Chem 2003, 49, 295–302. [Google Scholar]
- Hustad, S.; Ueland, P.M.; Vollset, S.E.; Zhang, Y.; Bjørke-Monsen, A.L.; Schneede, J. Riboflavin as a determinant of plasma total homocysteine: Effect modification by the methylenetetrahydrofolate reductase C667T polymorphism. Clin. Chem 2000, 46, 1065–1071. [Google Scholar]
- Forneris, F.; Binda, C.; Vanoni, M.A.; Mattevi, A.; Battaglioli, E. Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process. FEBS Lett 2005, 579, 2203–2207. [Google Scholar]
- Cloos, P.A.C.; Christensen, J.; Agger, K.; Helin, K. Erasing the methyl mark: Histone demethylases at the center of cellular differentiation and disease. Genes Dev 2008, 22, 1115–1140. [Google Scholar]
- Forneris, F.; Battaglioli, E.; Mattevi, A.; Binda, C. New roles of flavoproteins in molecular cell biology: histone demethylase LSD1 and chromatin. FEBS J 2009, 276, 4304–4312. [Google Scholar]
- Karytinos, A.; Forneris, F.; Profumo, A.; Ciossani, G.; Battaglioli, E.; Binda, C.; Mattevi, A. A novel mammalian flavin-dependent histone demethylase. J. Biol. Chem 2009, 284, 17775–17782. [Google Scholar]
- Zhang, X.; Wen, H.; Shi, X. Lysine methylation: beyond histones. Acta Biochim. Biophys. Sin 2012, 44, 14–27. [Google Scholar]
- Culhane, J.C.; Cole, P.A. LSD1 and the chemistry of histone demethylation. Curr. Opin. Biol. Chem 2007, 11, 561–568. [Google Scholar]
- Schmidt, D.M.Z.; McCafferty, D.G. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry 2007, 46, 4408–4416. [Google Scholar]
- Wang, Y.; Zhang, H.; Chen, Y.; Sun, Y.; Yang, F.; Yu, W.; Liang, J.; Sun, L.; Yang, X.; Shi, L.; et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 2009, 138, 660–672. [Google Scholar]
- Binda, C.; Valente, S.; Romanenghi, M.; Pilotto, S.; Cirilli, R.; karytinos, A.; Ciossani, G.; Botrugno, O.A.; Forneris, F.; Tardugno, M.; et al. Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J. Am. Chem. Soc 2010, 132, 6827–6833. [Google Scholar]
- Upadhyay, A.K.; Horton, J.R.; Zhang, X.; Cheng, X. Coordinated methyl-lysine erasure: Structural and functional linkage of a Jumonji demethylase domain and a reader domain. Curr. Opin. Struct. Biol 2011, 21, 750–760. [Google Scholar]
- Heightman, T.D. Chemical biology of lysine demethylases. Curr. Chem. Genomics 2011, 5, 62–71. [Google Scholar]
- Baron, R.; Binda, C.; Tortorici, M.; McCammon, J.A.; Mattevi, A. Molecular mimicry and ligand recognition in binding and catalysis by the histone demethylase LSD1-CoREST complex. Structure 2011, 19, 212–220. [Google Scholar]
- Forneris, F.; Binda, C.; Battaglioli, E.; Mattevi, A. LSD1: Oxidative chemistry for multifaceted functions in chromatin regulation. Trends Biochem. Sci 2008, 33, 181–189. [Google Scholar]
- Chen, Y.; Yang, Y.; Wang, F.; Wan, K.; Yamane, K.; Zhang, Y.; Lei, M. Crystal structure of human histone lysine-specific demethylase 1 (LSD1). Proc. Natl. Acad. Sci. USA 2006, 103, 13956–13961. [Google Scholar]
- Fang, R.; Barbera, A.J.; Xu, Y.; Rutenberg, M.; Leonor, T.; Bi, Q.; Lan, F.; Mei, P.; Yuan, G.C.; Lian, C.; et al. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol. Cell 2010, 39, 222–233. [Google Scholar]
- Kong, X.; Ouyang, S.; Liang, Z.; Lu, J.; Chen, L.; Shen, B.; Li, D.; Zheng, M.; Li, K.K.; Luo, C.; et al. Catalytic mechanism investigation of lysine-specific demethylase 1 (LSD1): A computational study. PLoS One 2011, 6, e25444. [Google Scholar]
- Kurtz, K.A.; Rishavy, M.A.; Cleland, W.W.; Fitzpatrick, P.F. Nitrogen isotope effects as probes of the mechanism of D-amino acid oxidase. J. Am. Chem. Soc 2000, 122, 12896–12897. [Google Scholar]
- Edmondson, D.E.; Binda, C.; Mattevi, A. Structural insights into the mechanism of amine oxidation by monoamine oxidases A and B. Arch. Biochem. Biophys 2007, 464, 269–276. [Google Scholar]
- Shi, Y.; Whetstine, J.R. Dynamic regulation of histone lysine methylation by demethylases. Mol. Cell 2007, 25, 1–13. [Google Scholar]
- Yang, M.; Culhane, J.C.; Szewczuk, L.M.; Jalili, P.; Ball, H.L.; Machius, M.; Cole, P.A.; Yu, H. Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry 2007, 46, 8058–8065. [Google Scholar]
- Baron, R.; Vellore, N.A. LSD1/CoREST in an allosteric nanoscale clamp regulated by H3-histone-tail molecular recognition. Proc. Natl. Acad. Sci. USA 2012. [Google Scholar] [CrossRef]
- Huang, J.; Sengupta, R.; Espejo, A.B.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T.; et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007, 449, 105–109. [Google Scholar]
- He, Y.; Korboukh, I.; Jin, J.; Huang, J. Targeting protein lysine methylation and demethylation in cancer. Acta Biochim. Biophys. Sin 2012, 44, 70–79. [Google Scholar]
- Culhane, J.C.; Szewczuk, L.M.; Liu, X.; Da, G.; Marmorstein, R.; Cole, P.A. A mechanizm-based inactivator for histone demethylase LSD1. J. Am. Chem. Soc 2006, 128, 4536–4537. [Google Scholar]
- Culhane, J.C.; Wang, D.; Yen, P.M.; Cole, P.A. Comparative analysis of small molecules and histone substrate analogs as LSD1 lysine demethylase inhibitors. J. Am. Chem. Soc 2010, 132, 3164–3176. [Google Scholar]
- Schulte, J.H.; Lim, S.; Schramm, A.; Friedrichs, N.; Koster, J.; Versteeg, R.; Ora, I.; Pajtler, K.; Klein-Hitpass, L.; kuhfitting-Kulle, S.; et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: Implications for therapy. Cancer Res 2009, 69, 2065–2071. [Google Scholar]
- Lukaski, H.C. Vitamin and mineral status: Effects on physical performance. Nutrition 2004, 20, 632–644. [Google Scholar]
- Mannion, C.A.; Gray-Donald, K.; Koski, K.G. Association of low intake of milk and vitamin D during pregnancy with decreased birth weight. CMAJ 2006, 174, 1273–1277. [Google Scholar]
- He, Y.; Ye, L.; Shan, B.; Song, G.; Meng, F.; Wang, S. Effect of riboflavin-fortified salt nutrition intervention on esophaheal squamous call carcinoma in a high incidence area, China. Asian Pac. J. Cancer Prev 2009, 10, 619–622. [Google Scholar]
- Wang, G.Q.; Dawsey, S.M.; Li, J.Y.; Taylor, P.R.; Li, B.; Blot, W.J.; Weinstein, W.M.; Liu, F.S.; Lewin, K.J.; Wang, H.; et al. Effects of vitamin/mineral supplementation on the prevalence of histological dysplasia and early cancer of the esophagus and stomach: Results from the general population trial in Linxian, China. Cancer Epidemiol. Biomark. Prev 1994, 3, 161–166. [Google Scholar]
- de Vogel, S.; Dindore, V.; van Engeland, M.; Goldbohm, R.A.; van den Brandt, P.A.; Weijenberg, M.P. Dietary folate, methionine, riboflavin, and vitamin B-6 and risk of sporadic colorectal cancer. J. Nutr 2008, 138, 2372–2378. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wojcieszyńska, D.; Hupert-Kocurek, K.; Guzik, U. Flavin-Dependent Enzymes in Cancer Prevention. Int. J. Mol. Sci. 2012, 13, 16751-16768. https://doi.org/10.3390/ijms131216751
Wojcieszyńska D, Hupert-Kocurek K, Guzik U. Flavin-Dependent Enzymes in Cancer Prevention. International Journal of Molecular Sciences. 2012; 13(12):16751-16768. https://doi.org/10.3390/ijms131216751
Chicago/Turabian StyleWojcieszyńska, Danuta, Katarzyna Hupert-Kocurek, and Urszula Guzik. 2012. "Flavin-Dependent Enzymes in Cancer Prevention" International Journal of Molecular Sciences 13, no. 12: 16751-16768. https://doi.org/10.3390/ijms131216751