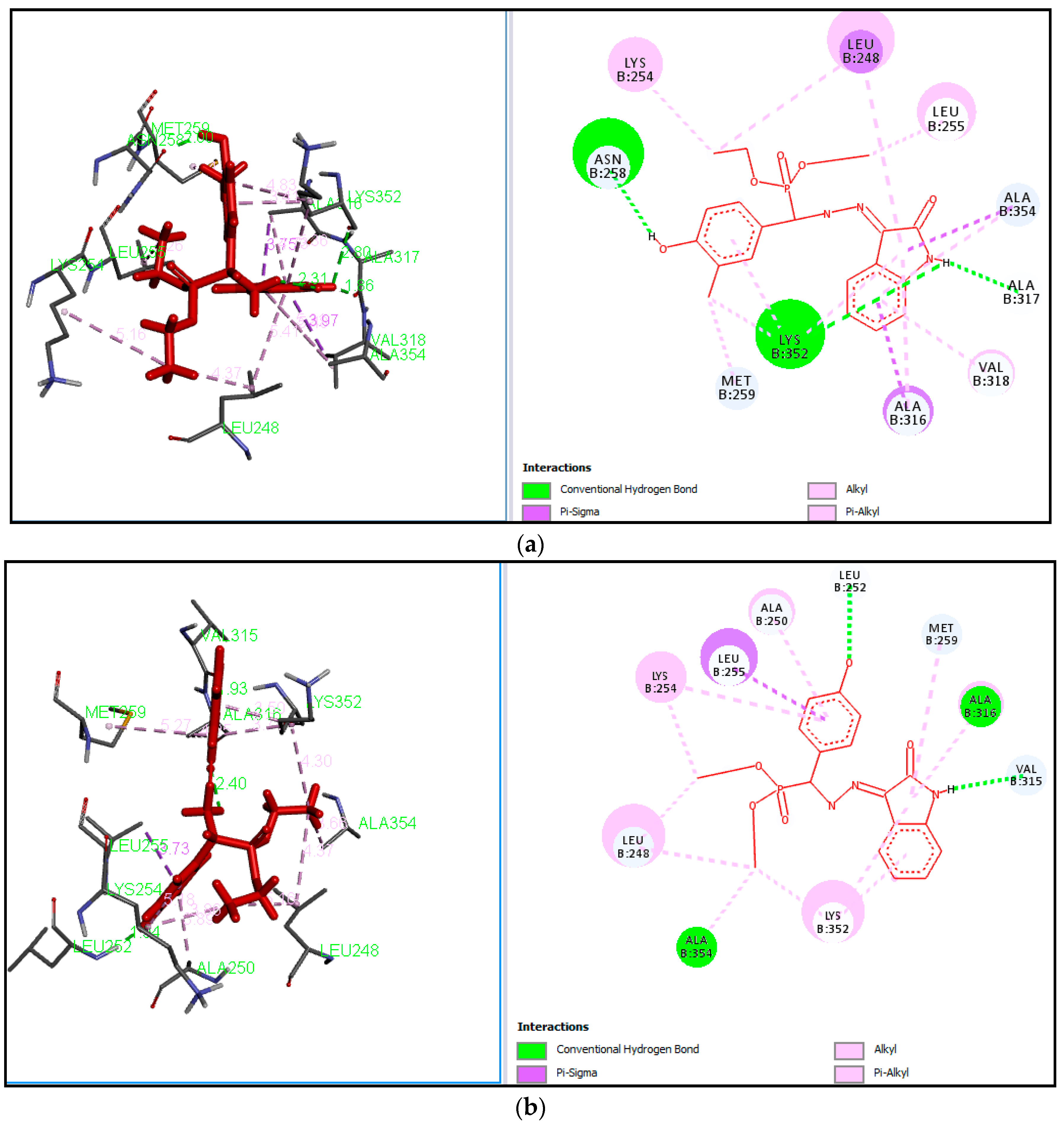
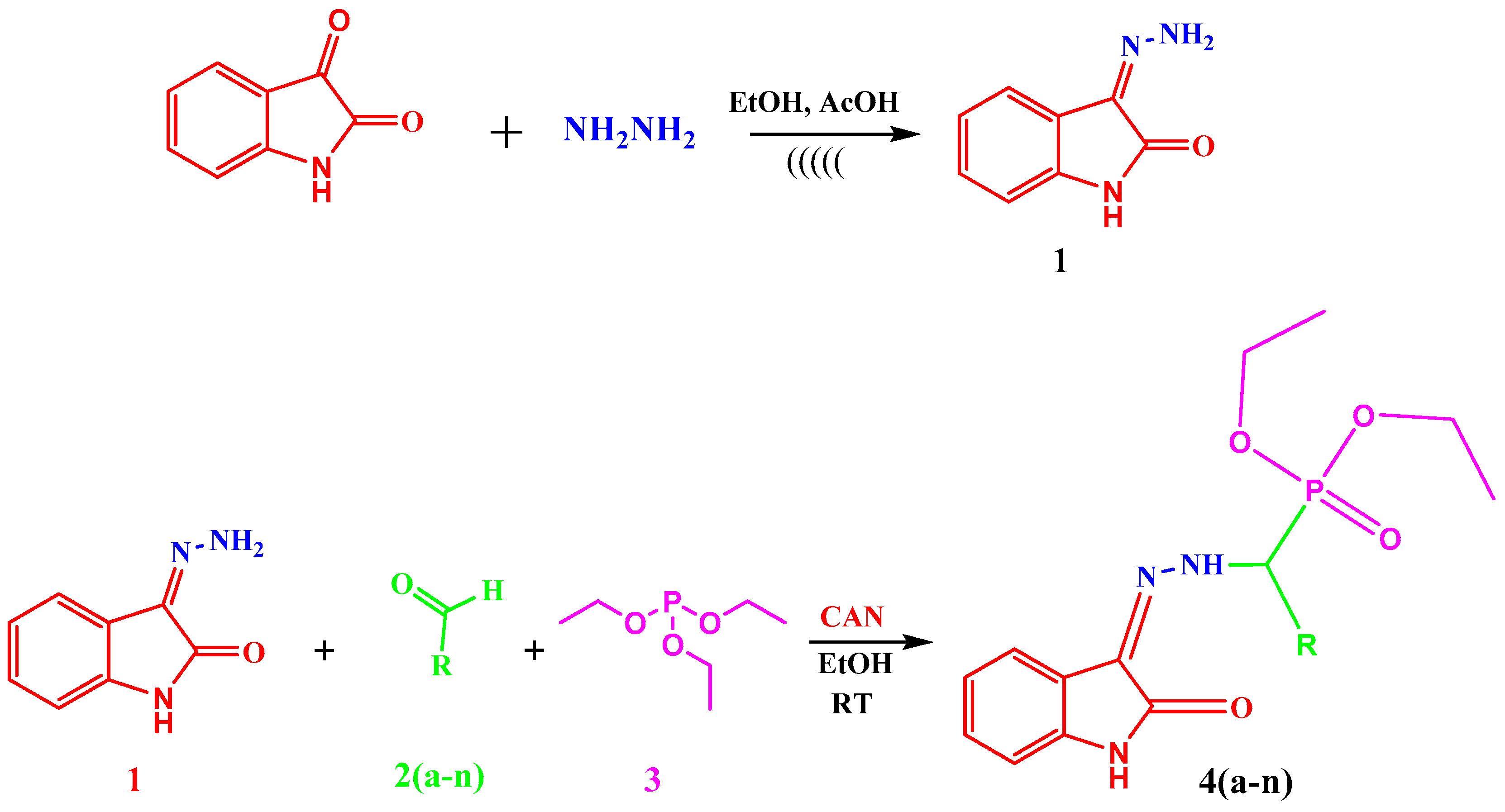
3.3.2. General Procedure for the Synthesis of Diethyl(substituted phenyl/heteroaryl)(2-(2-oxoindolin-3-ylidene)hydrazinyl)methylphosphonates
Equimolar quantities of 3-hydrazonoindolin-2-one (
1, 1 mmol), a substituted aromatic aldehyde/heteroaldehyde
2(
a–
n) (1 mmol) and triethylphosphite (
3, 1 mmol) were stirred at room temperature in absolute ethanol, in the presence of ceric ammonium nitrate (CAN, 0.003 mmol) as a catalyst. The TLC method was used to verify the completion of the reaction. After completion of the reaction, the reaction mixture was cooled and poured into water, filtered and the solid obtained was dried and recrystallized using ethanol. The time required for completion of reaction varies from 70 min to 89 min. The details are shown in
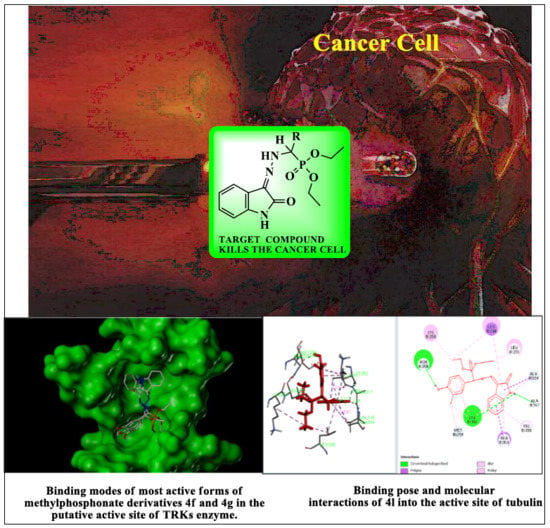
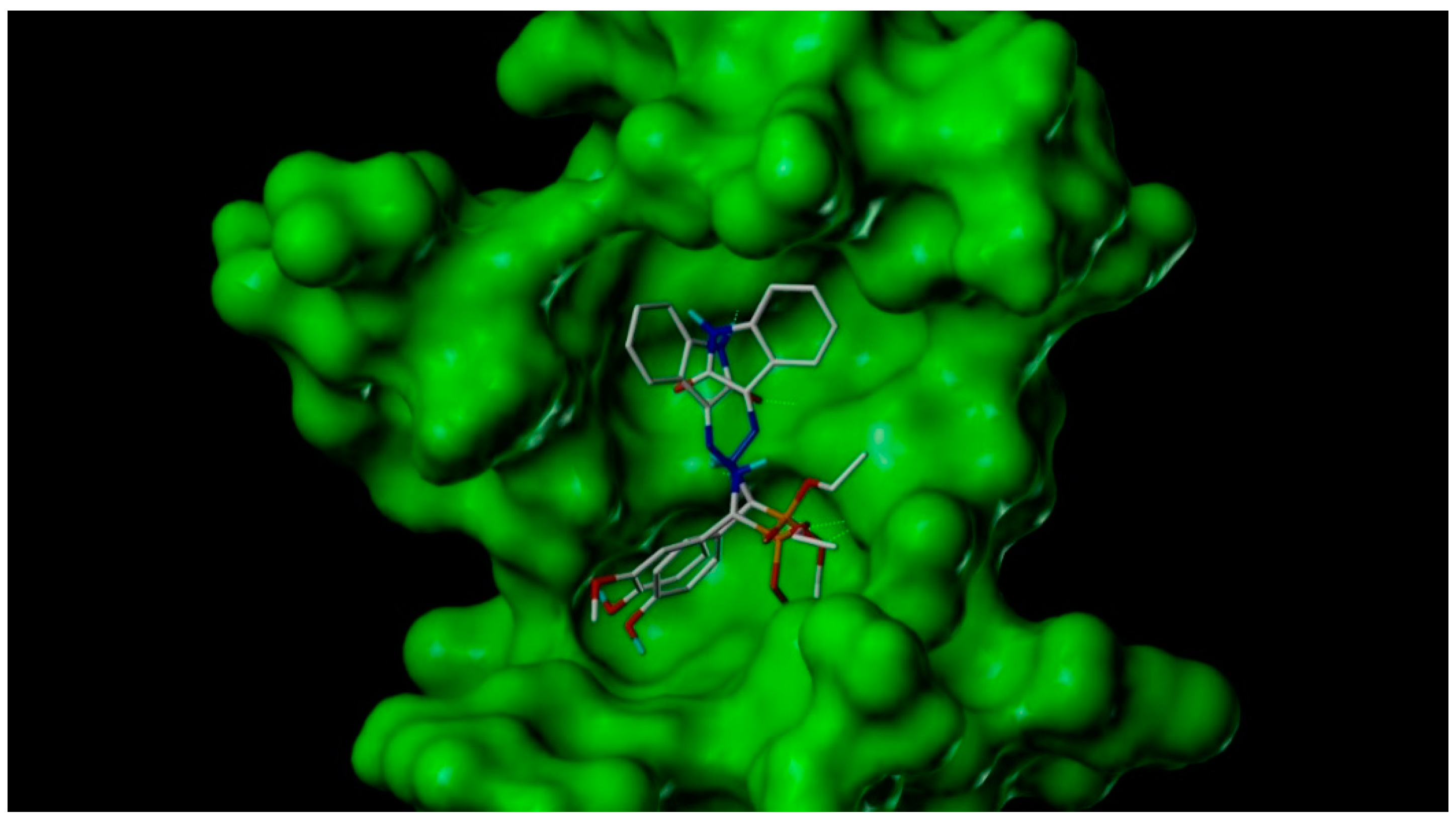
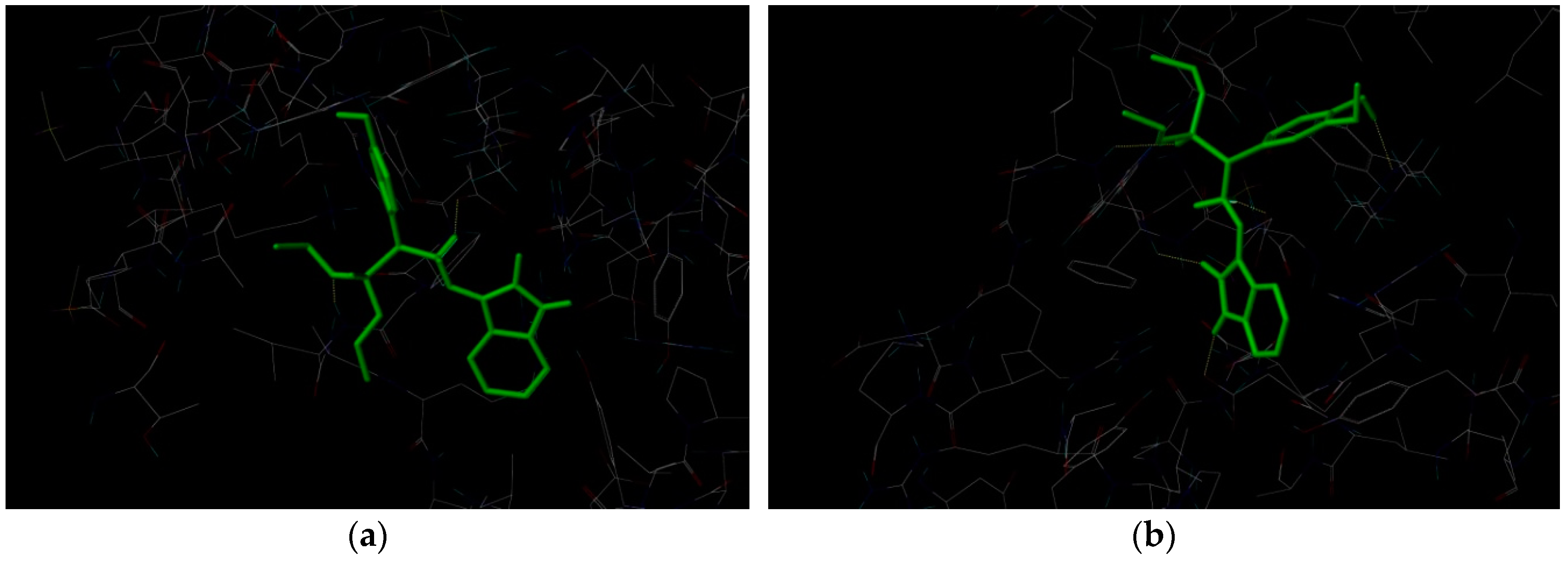
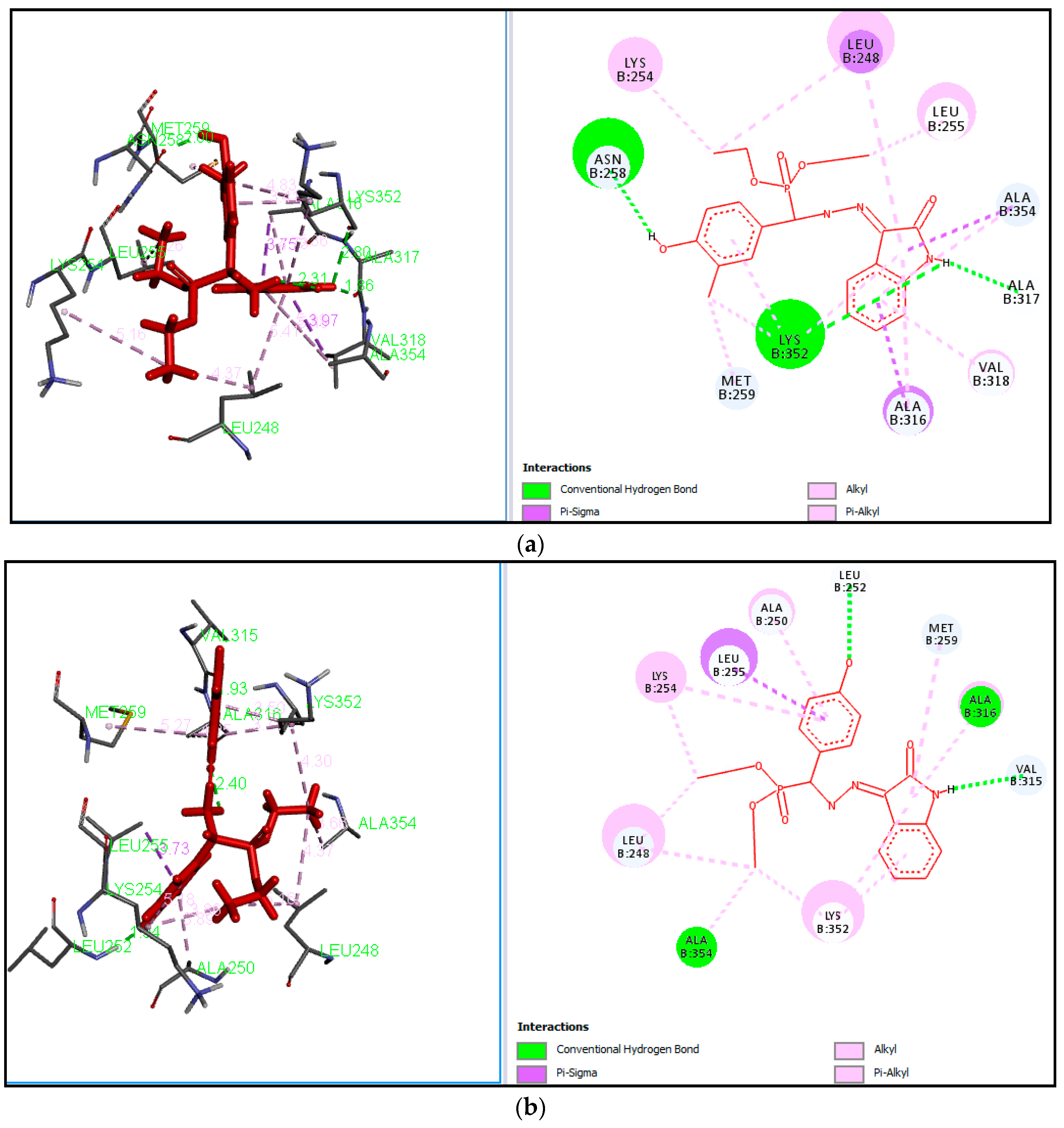
Table 1. Our work represents a one pot Kabachnik-Fields synthesis of diethyl (substitutedphenyl/heteroaryl)(2-(2-oxoindolin-3-ylidene)hydrazinyl)-methylphosphonate derivatives from 3-hydrazonoindolin-2-one and substituted aromatic aldehyde/heteroaldehydes using CAN as a green catalyst at room temperature with better yield 84–95%.
Diethyl(phenyl)(2-(2-oxoindolin-3-ylidene)hydrazinyl)methylphosphonate (4a): Yield: 90%; M.P. 195–196 °C; 1H-NMR (CDCl3) δ: 1.2 (t, J = 7.11 Hz, 6H, 2×OCH2CH3),3.17 (d, J = 8.51 Hz, 1H, -CH), 4.70 (q, J = 7.11 Hz, 4H, 2×OCH2CH3), 7.10 (m, 9H, -CH), 8.61 (s, 1H, -NH), 10.90 (s, 1H, -NH of indole); 13C-NMR (CDCl3) δ: 16.31, 60.11, 63.32, 110.32, 119.25, 124.32, 126.25, 126.52, 128.12, 128.32, 129.22, 130.32, 162.11; 31P-NMR (CDCl3) δ: 19.90; ESI-MS: m/z calculated for C19H22N3O4P (M + H+): 388.84; found: 389.88 (M+1); IR (KBr) cm−1: 3340.31 (N-H stretching), 2960.41 (CH stretching of aromatic), 2837.21 (CH stretching of alkyl), 2300.23 (N-H stretching), 1620.33 (C=O stretching of amide), 1466.55 (CH Bending of CH2); Elemental analysis calculated for C19H22N3O4P: C, 58.91; H, 5.72; N, 10.85; P, 7.58; found; C, 58.88; H, 5.75; N, 10.87; P, 7.60.
Diethyl(4-chlorophenyl)(2-(2-oxoindolin-3-ylidene)hydrazinyl)methylphosphonate (4b): Yield 92%; M.P. 150–152 °C; 1H-NMR (CDCl3) δ: 1.20 (t, J = 7.11 Hz, 6H, 2×OCH2CH3), 2.53 (d, J = 8.11 Hz, 1H, -CH), 4.10 (q, J = 7.11 Hz, 4H, 2×OCH2CH3), 7.40 (m, 8H, -CH), 8.58 (s, 1H, -NH), 11.55 (s, 1H, -NH of indole); 13C-NMR (CDCl3) δ: 16.15, 40.17, 60.25, 78.07, 110.26, 117.96,127.79, 128.21, 128.83, 129.54, 130.19, 158.78, 164.52; 31P-NMR (CDCl3) δ: 18.84; ESI-MS: m/z calculated for C19H21ClN3O4P (M+1): 421.09; found: 422.33 (M+1); IR (KBr) cm−1: 3350.41 (N-H stretching), 2970.06 (CH stretching of aromatic), 2800.22 (CH stretching of alkyl), 2350.36 (N-H stretching), 1710.01 (C=O stretching of amide), 1454.75 (CH Bending of CH2); Elemental analysis calculated for C19H21ClN3O4P: C, 54.10; H, 5.02; N, 9.96; P, 7.34; found; C, 54.12; H, 5.04; N, 9.99; P, 7.39
Diethyl(4-flurophenyl)(2-(2-oxoindolin-3-ylidene)hydrazinyl)methylphosphonate (4c): Yield 95%; M.P. 176–180 °C; 1H-NMR (CDCl3) δ: 1.31 (t, J = 7.11 Hz, 6H, 2×OCH2CH3), 3.64 (d, J=8.45 Hz, 1H, -CH), 4.41 (q, J = 7.11 Hz, 4H, 2×OCH2CH3), 8.79 (m, 8H, -CH), 8.84 (s, 1H, -NH), 10.14 (s, 1H, -NH of indole); 13C-NMR (CDCl3) δ: 18.12, 65.21, 68.21, 123.32, 114.21, 117.14, 120.85, 127.55, 128.85, 131.36, 144.74, 146.96, 161.11, 164.85; 31P-NMR (CDCl3) δ: 18.54; ESI-MS: m/z calculated for C19H21FN3O4P (M + H+): 406.13; found: 407.20 (M + H+); IR (KBr) cm−1:3340.11 (N-H stretching), 2910.16 (CH stretching of aromatic), 2800.48 (CH stretching of alkyl), 2200.50 (N-H stretching), 1620.17 (C=O stretching of amide), 1464.47 (CH Bending of CH2); Elemental analysis calculated for C19H21FN3O4P: C, 56.30; H, 5.22; N, 10.37; P, 7.64 found; C, 56.33; H, 5.23; N, 10.40; P, 7.67
(Z)-Diethyl(4-methoxyphenyl)(2-(2-oxoindolin-3-ylidene)hydrazinyl)methylphosphonate (4d): Yield 89%; M.P. 178–179 °C; 1H-NMR (CDCl3) δ: 1.25 (t, J = 7.11 Hz, 6H, 2×OCH2CH3), 2.54 (s, 3H, O CH3), 3.42 (d, J = 8.41 Hz, 1H, -CH), 4.11 (q, J = 7.11 Hz, 4H, 2×OCH2CH3), 7.00 (m, 8H, -CH), 8.60 (s, 1H, -NH), 10.94 (s, 1H, -NH of indole); 13C-NMR (CDCl3) δ: 14.23, 55.13, 78.34, 79.88, 99.49, 110.93, 113.77, 119.25, 122.98, 126.47, 128.23, 129.85, 133.99, 144.85, 160.22, 168.98;31P-NMR (CDCl3) δ: 19.84; ESI-MS: m/z calculated for C20H24N3O5P (M + H+): 417.15; found: 418.42 (M + H+); IR (KBr) cm−1: 3350.11 (N-H stretching), 3070.76 (CH stretching of aromatic), 2800.96 (CH stretching of alkyl), 2300.11 (N-H stretching), 1610.47 (C=O stretching of amide), 1025.74 (-O- stretching); Elemental analysis calculated for C20H24N3O5P: C, 57.55; H, 5.80; N, 10.07; P, 7.42 found; C, 57.58; H, 5.82; N, 10.10; P, 7.45.
Diethyl(3,4-dimethoxyphenyl)(2-(2-oxoindolin-3-ylidene)hydrazinyl)methylphosphonate (4e): Yield 90%; M.P. 189–190 °C; 1H-NMR (CDCl3) δ: 1.21 (t, J = 7.11 Hz, 6H, 2×OCH2CH3), 2.55 (s, 6H, OCH3), 3.67 (d, J = 8.45 Hz, 1H, -CH), 4.21 (q, J = 7.11 Hz, 4H, 2×OCH2CH3), 7.37 (m, 7H, -CH), 8.42 (s, 1H, -NH), 10.03 (s, 1H, -NH of indole); 13C-NMR: (CDCl3) δ: 20.22, 58.69, 60.21, 61.12, 66.32, 111.78, 120.78, 121.36, 131.85, 132.11, 133.25, 141.74, 148.23, 150.41, 151.12, 167.47; 31P-NMR (CDCl3) δ: 18.94; ESI-MS: m/z calculated for C21H26N3O6P (M + H+): 448.16; found: 449.44 (M + H+); IR (KBr)cm−1: 3250.01 (N-H stretching), 2890.76 (CH stretching of aromatic), 2800.57 (CH stretching of alkyl), 2350.78 (N-H stretching), 1650.23 (C=O stretching of amide), 1002.44 (-O- stretching); Elemental analysis calculated for C21H26N3O6P: C, 56.37; H, 5.86; N, 9.39; P, 6.92 found; C, 56.40; H, 5.89; N, 9.41; P, 6.94.
Diethyl(4-hydroxyphenyl)(2-(2-oxoindolin-3-ylidene)hydrazinyl)methylphosphonate (4f): Yield 88%; M.P. 140–142 °C; 1H-NMR: (CDCl3) δ: 1.31 (t, J = 7.11 Hz, 6H, 2×OCH2CH3), 3.71 (d, J = 8.41 Hz, 1H, -CH), 4.54 (q, J = 7.11 Hz, 4H, 2×OCH2CH3), 5.61 (s, 1H, OH), 7.20 (m, 8H, -CH), 8.53 (s, 1H, -NH), 10.44 (s, 1H, -NH of indole); 13C-NMR (, CDCl3) δ: 17.26, 61.25, 69.74, 115.47,116.23, 117.63, 123.52, 129.85, 130.47, 134.52, 136.12, 145.32, 156.54, 164.41; 31P-NMR (CDCl3) δ:19.64; ESI-MS: m/z calculated for C19H22N3O5P (M + H+): 404.13; found: 405.37 (M + H+); IR (KBr) cm−1: 3600.10 (aromatic OH), 3440.44 (N-H stretching), 2980.88 (CH stretching of aromatic), 2800.01 (CH stretching of alkyl), 2280.21 (N-H stretching), 1710.22 (C=O stretching of amide); Elemental analysis calculated for C19H22N3O5P: C, 56.57; H, 5.50; N, 10.42; P, 7.68 found; C, 56.60; H, 5.54; N, 10.44; P, 7.70.
Diethyl(4-hydroxy-3-methoxyphenyl)(2-(2-oxoindolin-3-ylidene)hydrazinyl)methylphosphonates (4g): Yield: 94%; M.P. 112–114 °C; 1H-NMR (CDCl3) δ: 1.37 (t, J = 7.11 Hz, 6H, 2×OCH2CH3), 2.58 (s, 3H, OCH3), 3.36 (d, J = 8.41 Hz, 1H, CH), 4.14 (q, J = 7.11 Hz, 4H, 2×OCH2CH3), 6.84 (s, 1H, OH), 7.73 (m, 7H, CH), 9.77 (s, 1H, -NH), 10.55 (s, 1H, -NH of indole); 13C-NMR (CDCl3) δ: 16.05, 40.17, 55.40, 62.57, 110.36, 116.74, 120.48, 125.29, 132.66,134.25, 136.24, 138.59, 138.90, 144.46, 150.94, 190.22; 31P-NMR (CDCl3) δ: 19.94; ESI-MS: m/z calculated for C20H24N3O6P (M + H+): 434.14; found: 435.39 (M + H+); IR (KBr) cm−1:3610.45 (aromatic OH), 3450.64 (N-H stretching), 2996.74 (CH stretching of aromatic), 2830.74 (CH stretching of alkyl), 2310.21 (N-H stretching), 1680.12 (C=O stretching of amide), 1030.14 (-O-stretching); Elemental analysis calculated for C20H24N3O6P: C, 55.43; H, 5.51; N, 9.70; found; C, 55.49; H, 5.60; N, 9.75.
(Z)-Diethyl(3-ethoxy-4-hydroxyphenyl)(2-(2-oxoindolin-3-ylidene)hydrazinyl)methylphosphonate (4h): Yield 92%; M.P. 160–162 °C; 1H-NMR (CDCl3) δ: 1.40 (t, J = 7.11 Hz, 9H, 3×OCH2CH3), 3.90 (d, J = 8.41 Hz, 1H, CH), 4.51 (q, J = 7.11 Hz, 6H, 3×OCH2CH3), 5.42 (s, 1H, OH), 7.71 (m, 7H, CH), 8.8 (s, 1H, -NH),10.72 (s, 1H, -NH of indole); 13C-NMR (CDCl3) δ: 16.21, 16.52,18.21, 61.52, 64.85, 68.74, 117.12, 116.41, 119.32, 121.92, 122.74, 130.96, 133.12, 136.24, 141.23, 142.74, 167.74; 31P-NMR (CDCl3) δ: 18.65; ESI-MS: m/z calculated for C21H26N3O6P (M + H+): 448.16; found: 449.40 (M + H+); IR (KBr) cm−1: 3550.50 (-OH),3420.32 (N-H stretching), 2999.45 (CH stretching of aromatic), 2813.87 (CH stretching of alkyl), 2350.52 (N-H stretching), 1710.72 (C=O stretching of amide), 1020.42 (-O- stretching); Elemental analysis calculated for C21H26N3O6P: C, 56.37; H, 5.86; N, 9.39; P, 6.92 found; C, 56.40; H, 5.88; N, 9.41; P, 6.94.
(Z)-Diethyl(2-(2-oxoindolin-3-ylidene)hydrazinyl)(thiophen-2-yl)methylphosphonate (4i): Yield 87%; M.P. 179–182 °C; 1H-NMR (CDCl3) δ: 1.01 (t, J = 7.11 Hz, 6H, 2×OCH2CH3), 3.64 (d, J = 8.44 Hz, 1H, CH), 3.82 (s, 1H, CH), 4.51 (q, J = 7.11 Hz, 4H, 2×OCH2CH3), 7.81 (m, 6H, CH), 8.57 (s, 1H, -NH), 10.77 (s, 1H, -NH of indole); 13C-NMR: (CDCl3) δ: 17.12, 63.52, 69.96, 119.12, 120.35, 124.18, 128.47, 129.96, 130.18, 150.52, 152.74, 155.54, 165.65, 170.65,174.96; 31P-NMR (CDCl3) δ: 18.45; ESI-MS: m/z calculated for C17H20N3O4PS (M + H+): 394.09; found: 395.48 (M + H+);IR (KBr) cm−1: 3520.72 (N-H stretching), 2912.88 (CH stretching of aromatic), 2800.32 (CH stretching of alkyl), 2340.54 (N-H stretching), 1710.72 (C=O stretching of amide); Elemental analysis calculated for C17H20N3O4PS: C, 51.90; H, 5.12; N, 10.68; P, 7.87; S, 8.15 found; C, 51.91; H, 5.14; N, 10.70; P, 7.89; S, 8.17.
(Z)-Diethylfuran-2-yl(2-(2-oxoindolin-3-ylidene)hydrazinyl)methylphosphonate (4j): Yield 84%; M.P. 176–178 °C; 1H-NMR (CDCl3) δ: 1.20 (t, J=7.11 Hz, 6H, 2×OCH2CH3), 3.81 (s, 1H, CH), 3.42 (d, J = 8.44 Hz, 1H, CH), 4.62 (q, J = 7.11 Hz, 4H, 2×OCH2CH3), 7.98 (m, 6H, CH), 8.44 (s, 1H, -NH), 10.45 (s, 1H, -NH of indole); 13C-NMR (CDCl3) δ: 22.21, 50.11, 65.52, 112.31, 119.95, 121.36, 128.63, 130.36, 131.21, 133.65, 147.33, 151.25, 153.23, 155.39, 164.21; 31P-NMR (CDCl3) δ: 18.56; ESI-MS: m/z calculated for C17H20N3O5P (M + H+): 378.12; found: 379.33 (M + H+);IR (KBr)cm−1: 3280.32 (NH stretching), 2945.46 (CH aromatic), 2850.63 (CH stretching of alkyl), 2440.85 (C=N stretching), 1710.33 (C=O stretching), 1070.10 (-O- stretching); Elemental analysis calculated for C17H20N3O5P: C, 54.11; H, 5.34; N, 11.14; P, 8.21 found; C, 54.14; H, 5.36; N, 11.16; P, 8.24.
(Z)-Diethyl (2-hydroxyphenyl)(2-(2-oxoindolin-3-ylidene)hydrazinyl)methylphosphonate (4k): Yield 88%; M.P.152–154 °C; 1H-NMR (DMSO-d6) δ: 1.29 (t, J = 7.11 Hz, 6H, 2×OCH2CH3), 4.07 (q, J = 7.12 Hz, 4H, 2×OCH2CH3),5. 35 (s, 1H, OH), 6.80–8.08 (m, 9H, aromatic), 8.0 (s, 1H, -NH), 10.46 (s, 1H, -NH of indole); 13C-NMR: (DMSO-d6) δ ppm: 16.31, 61.33,62.24, 115.41, 117.45, 119.05, 121.33, 121.56, 124.42, 128.46, 128.73, 129.11, 131.91, 133.22, 134.59, 141.44, 155.81, 168.44; 31P-NMR (CDCl3) δ: 19.56; ESI-MS: m/z calculated for C19H22N3O5P (M + H+): 403.13; found: 404.37 (M + H+); IR (KBr) cm−1: NH 3313.45 (N-H stretching), 3033.42 (C-H stretching of aromatic), 2653.64 (C-H stretching of alkyl), 2133.63 (C=N Stretching), 1723.05 (C-O stretching), 1690.80 (C=O stretching), 1554.65 (C-N Stretching), 1034.65 (O- stretching),Elemental Analysis calculated for C19H22N3O5P: C, 56.57; H, 5.50; N, 10.42; P, 7.68 found; C, 56.59; H, 5.52; N, 10.44; P, 7.69.
(Z)-Diethyl (4-hydroxy-3-methylphenyl)(2-(2-oxoindolin-3-ylidene)hydrazinyl)methylphosphonate (4l): Yield 75%; M.P.190–192 °C; 1H-NMR (DMSO-d6) δ: 1.29 (t, J = 7.11 Hz, 6H, 2×OCH2CH3), 2.15 (s, 1H, CH3), 4.07.(q, J = 7.12 Hz, 4H, 2×OCH2CH3), 5. 35 (s, 1H, OH), 6.80–8.08 (m, 9H, aromatic), 8.0 (s, 1H, -NH), 10.45 (s, 1H, -NH of indole); 13C-NMR: (DMSO-d6) δ ppm: 15.32, 16.33, 62.22, 67.72, 115.4, 118.44, 119.93, 124.49, 124.88, 125.94, 128.32, 129.81, 130.59, 131.72, 133.49, 141.45, 152.21, 168.41;31P-NMR (CDCl3) δ: 19.56; ESI-MS: m/z calculated for C20H24N3O5P (M + H+): 417.15; found: 417.40; IR (KBr) cm−1: NH 3303.34 (N-H stretching), 3013.32 (C-H stretching of aromatic), 2653.64 (C-H stretching of alkyl), 2133.63 (C=N Stretching), 1723.05 (C-O stretching), 1690.80 (C=O stretching), 1554.65 (C-N Stretching), 1034.65 (O- stretching),Elemental Analysis calculated for C20H24N3O5P: C, 57.55; H, 5.80; N, 10.07; P, 7.42 found; C, 57.59; H, 5.82; N, 10.09; P, 7.43.
(Z)-Diethyl (4-nitrophenyl)(2-(2-oxoindolin-3-ylidene)hydrazinyl)methylphosphonate (4m): Yield 90%; M.P.172–174 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 1.29 (t, J = 7.11 Hz, 6H, 2×OCH2CH3), 4.07 (q, J = 7.11 Hz, 4H, 2×OCH2CH3), 5. 35 (s, 1H, OH), 6.80–8.08 (m, 9H, aromatic), 8.0 (s, 1H, -NH), 10.45 (s, 1H, -NH of indole);13C-NMR: (DMSO-d6) δ ppm: 16.37, 62.24, 68.88, 117.76, 119,82, 123.37, 124.43, 127.94, 129.53, 131.22, 134.54, 141.14, 142.39, 145.45, 168.76; 31P-NMR (,CDCl3) δ: 19.56; ESI-MS: m/z calculated for C19H21N4O6P: (M + H+): 432.12, found: 433.37; IR (KBr) cm−1: NH 3303.34 (N-H stretching), 3013.32 (C-H stretching of aromatic), 2653.64 (C-H stretching of alkyl), 2133.63 (C=N Stretching), 1723.05 (C-O stretching), 1690.80 (C=O stretching), 1554.65 (C-N Stretching), 1034.65 (O- stretching); Elemental Analysis calculated for C19H21N4O6P: C, 52.78; H, 4.90; N, 12.96; P, 7.16 found; C, 52.79; H, 4.93; N, 12.98; P, 7.17.
(Z)-Diethyl (4-methylthiazole-5-yl)(2-(2-oxoindolin-3-ylidene)hydrazinyl)methylphosphonate(4n): Yield 70%; M.P. 188–190 °C; 1H-NMR (DMSO-d6) δ: 1.29 (t, J = 7.11 Hz, 6H, 2×OCH2CH3), 2.46 (s, 1H, CH3), 4.07.(q, J = 7.12 Hz, 4H, 2×OCH2CH3), 5. 35 (s, 1H, OH), 6.80–8.08 (m, 9H, aromatic), 8.0 (s, 1H, -NH), 10.45 (s, 1H, -NH of indole); 13C-NMR: (DMSO-d6) δ ppm: 14.73, 16.39, 62.28, 65.77, 117.78, 121.3, 124.48, 129.47, 130.13, 131.1 (C), 133,34, 141.22, 148.77, 151.34, 168.79; 31P-NMR (,CDCl3) δ: 19.54; ESI-MS: m/z calculated for C17H21N4O4PS: (M + H+): 408.10, found: 408.41; IR (KBr) cm−1: NH 3303.34 (N-H stretching), 3013.32 (C-H stretching of aromatic), 2653.64 (C-H stretching of alkyl), 2133.63 (C=N Stretching), 1723.05 (C-O stretching), 1690.80 (C=O stretching), 1554.65 (C-N Stretching), 1034.65 (O-stretching),2723.65 (COOH); Elemental Analysis calculated for C17H21N4O4PS: C, 47.57; H, 5.10; N, 12.33; P, 6.82 found; C, 47.59; H, 5.13; N, 12.38; P, 6.80.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}