
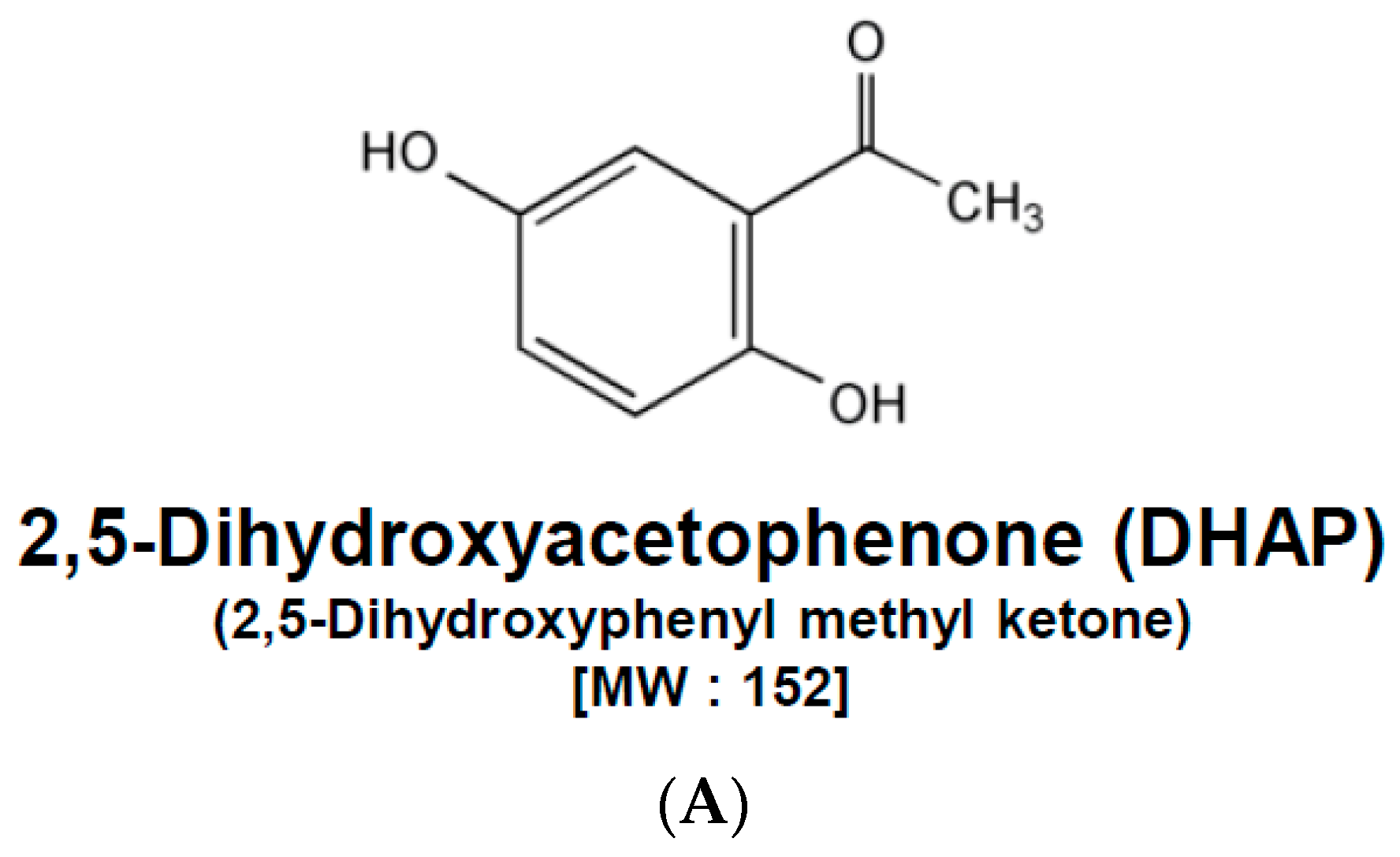
2,5-Dihydroxyacetophenone Induces Apoptosis of Multiple Myeloma Cells by Regulating the MAPK Activation Pathway


 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
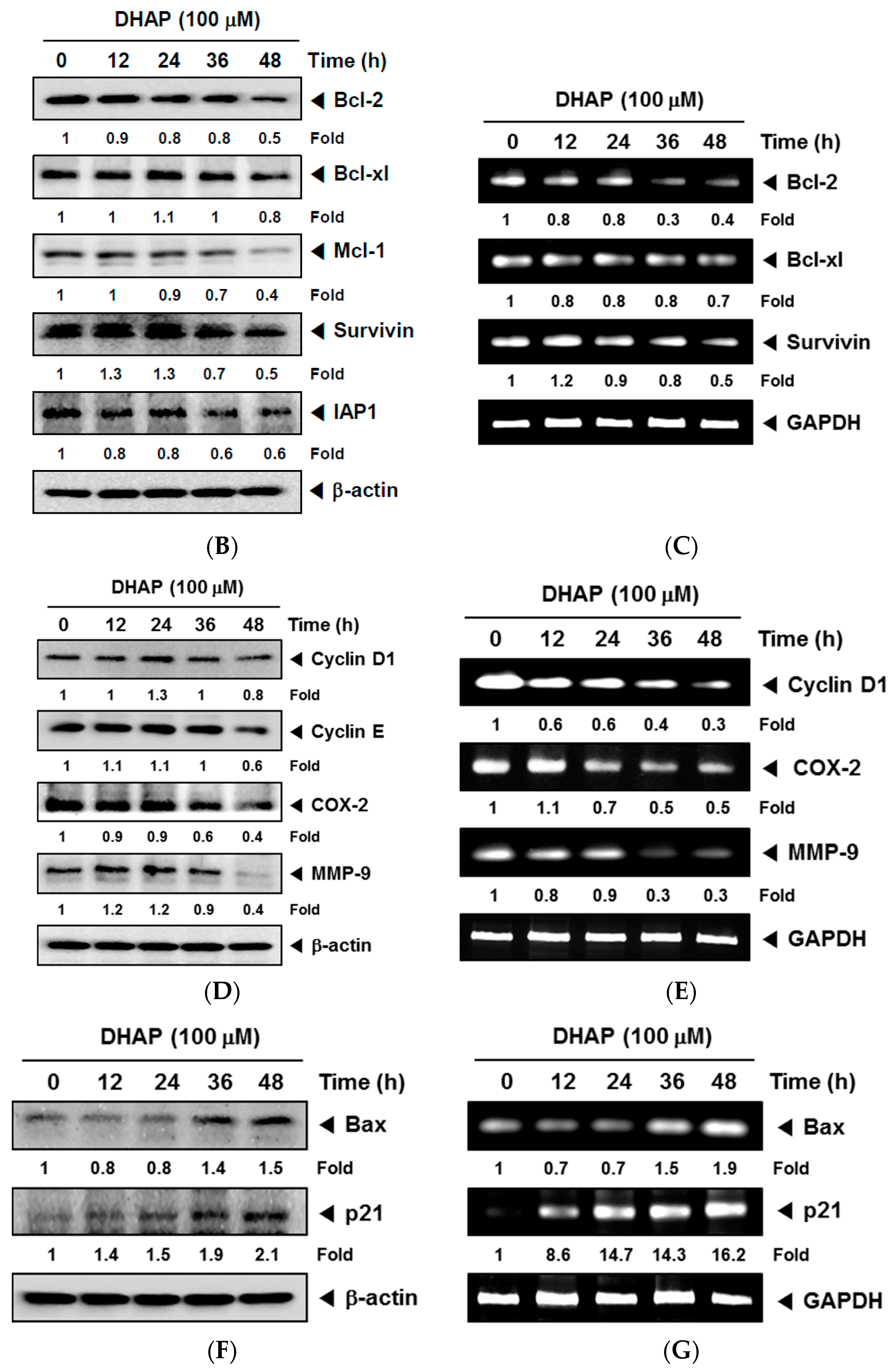
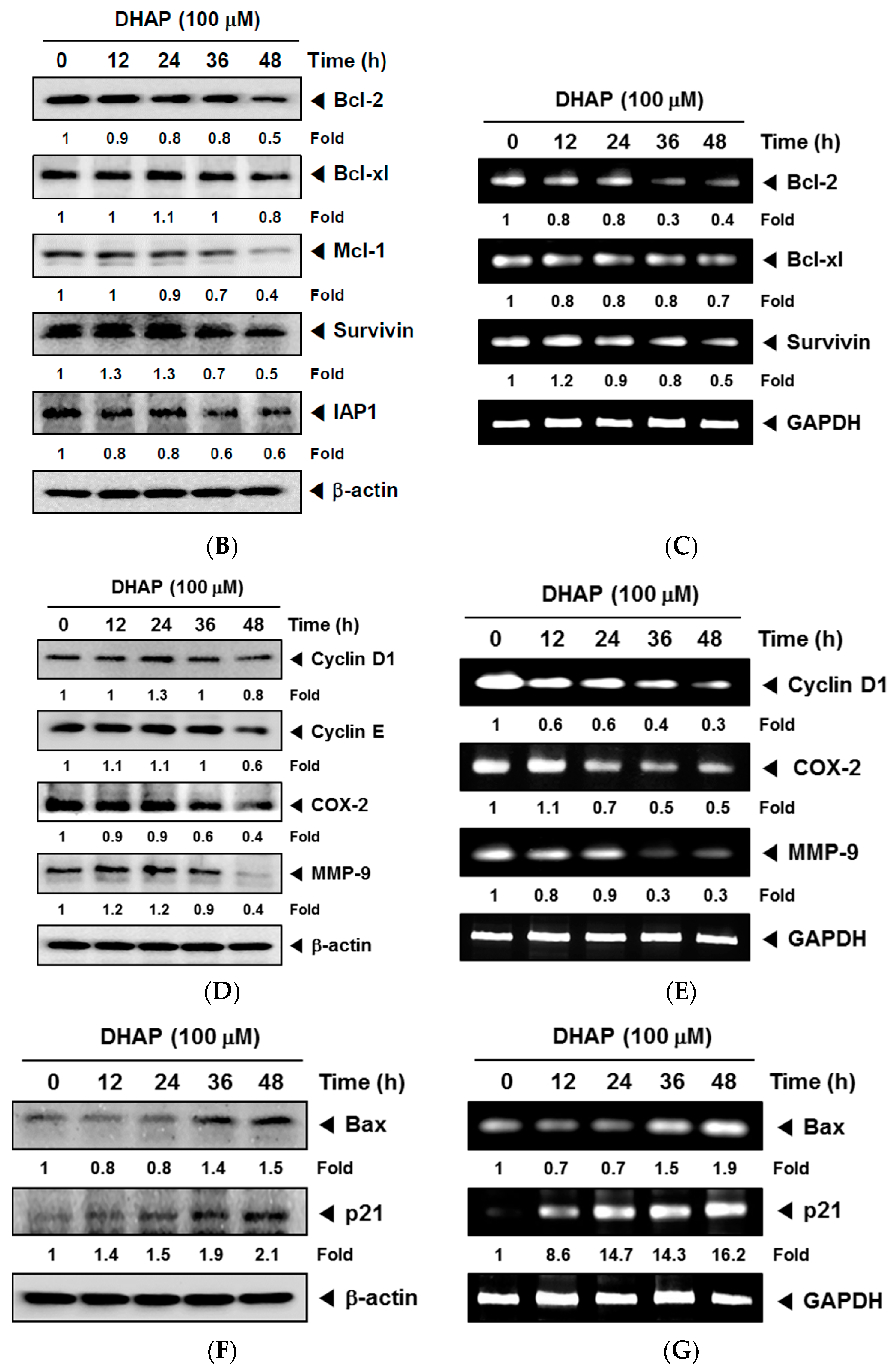
2.1. DHAP Modulates the Expression of Certain Proteins Connected to Apoptosis, Metastasis, and Proliferation
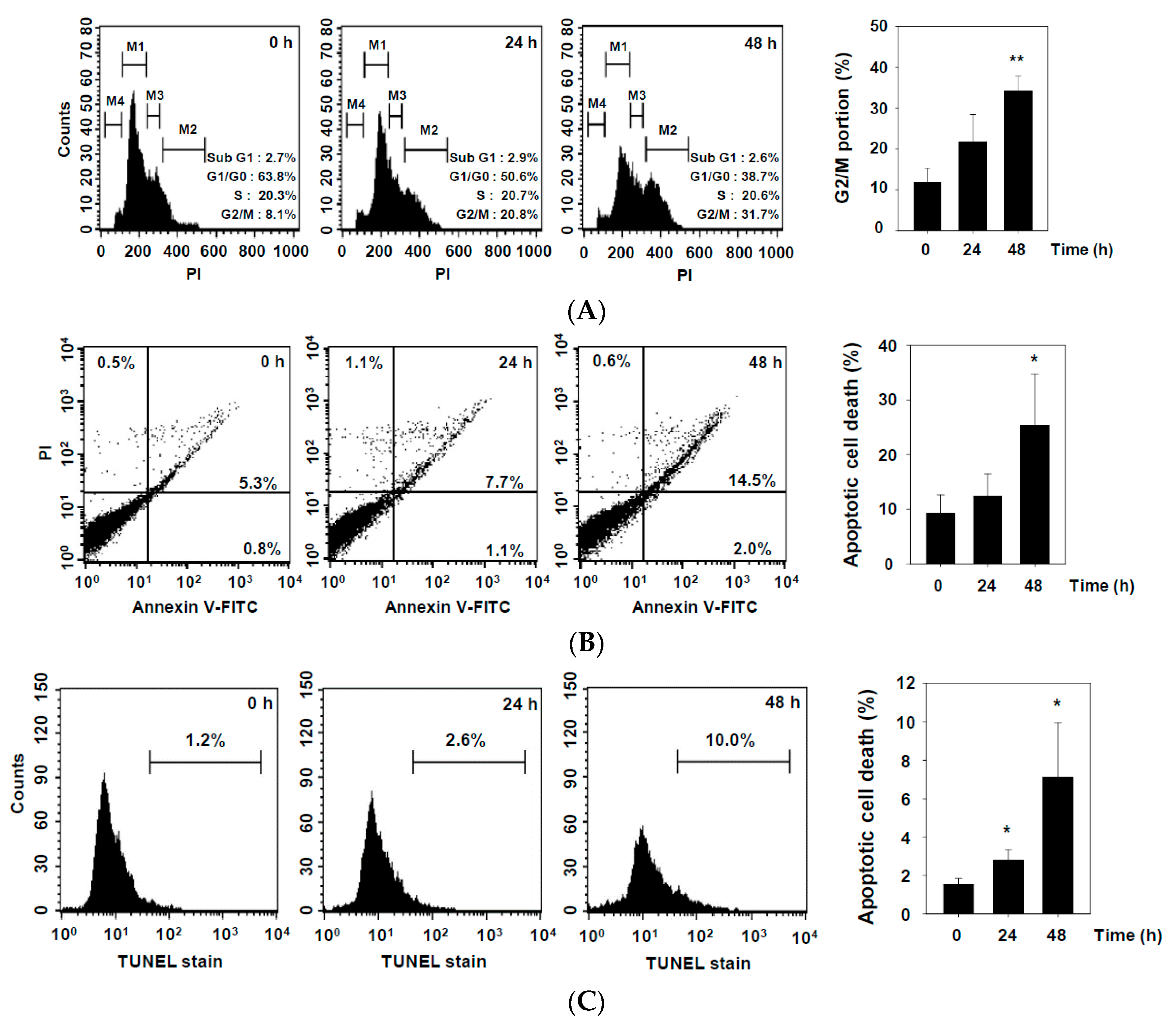
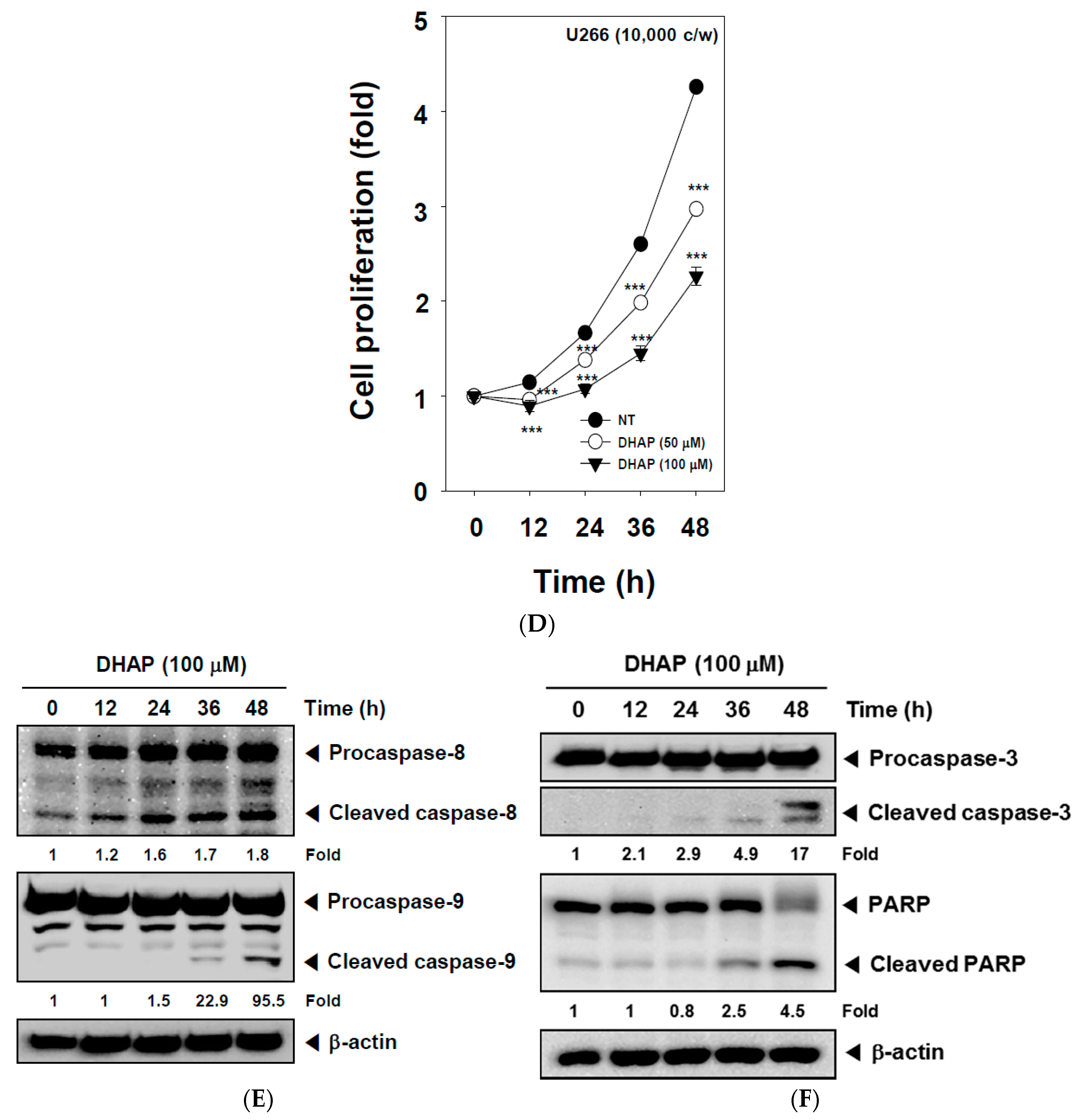
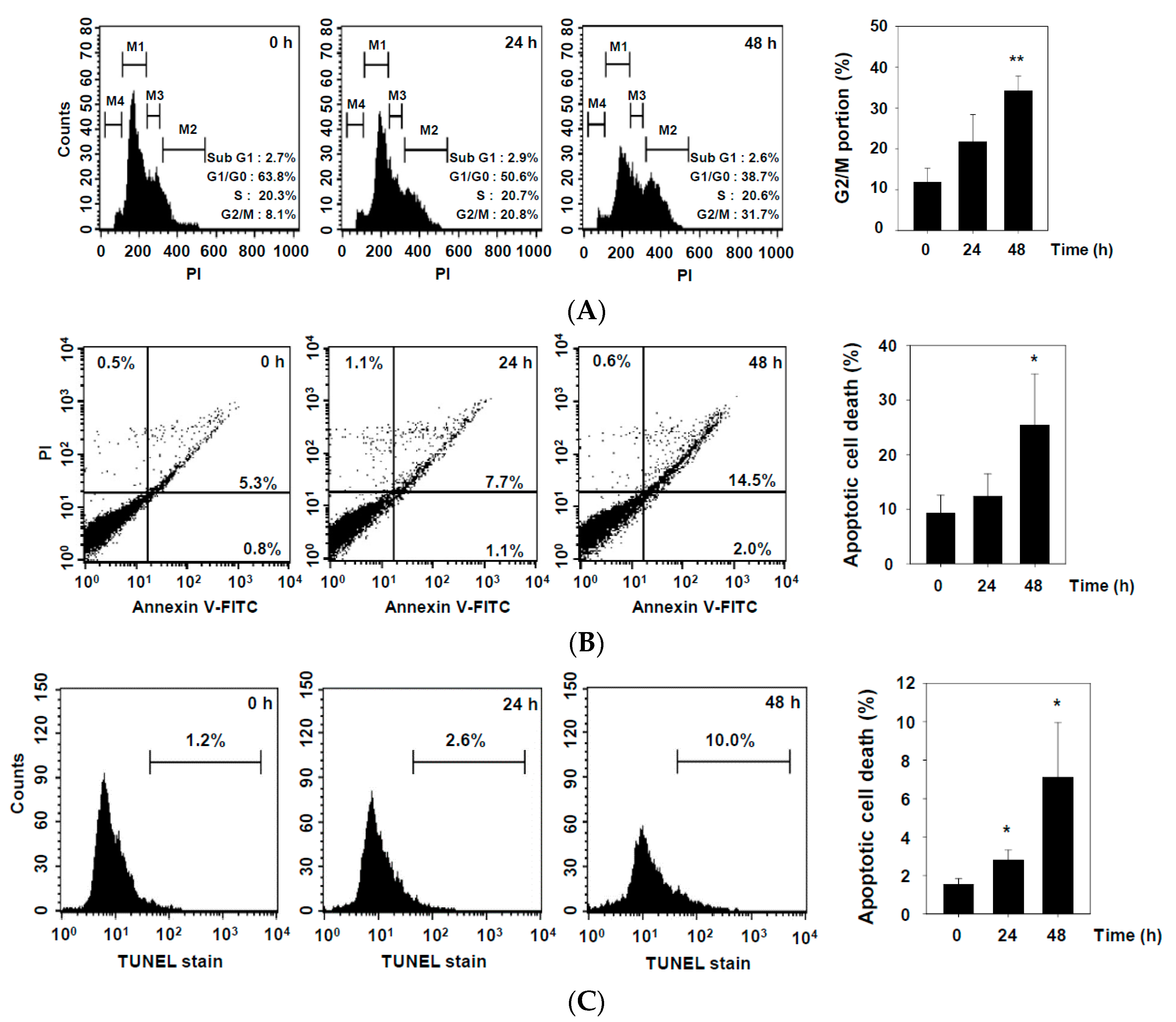
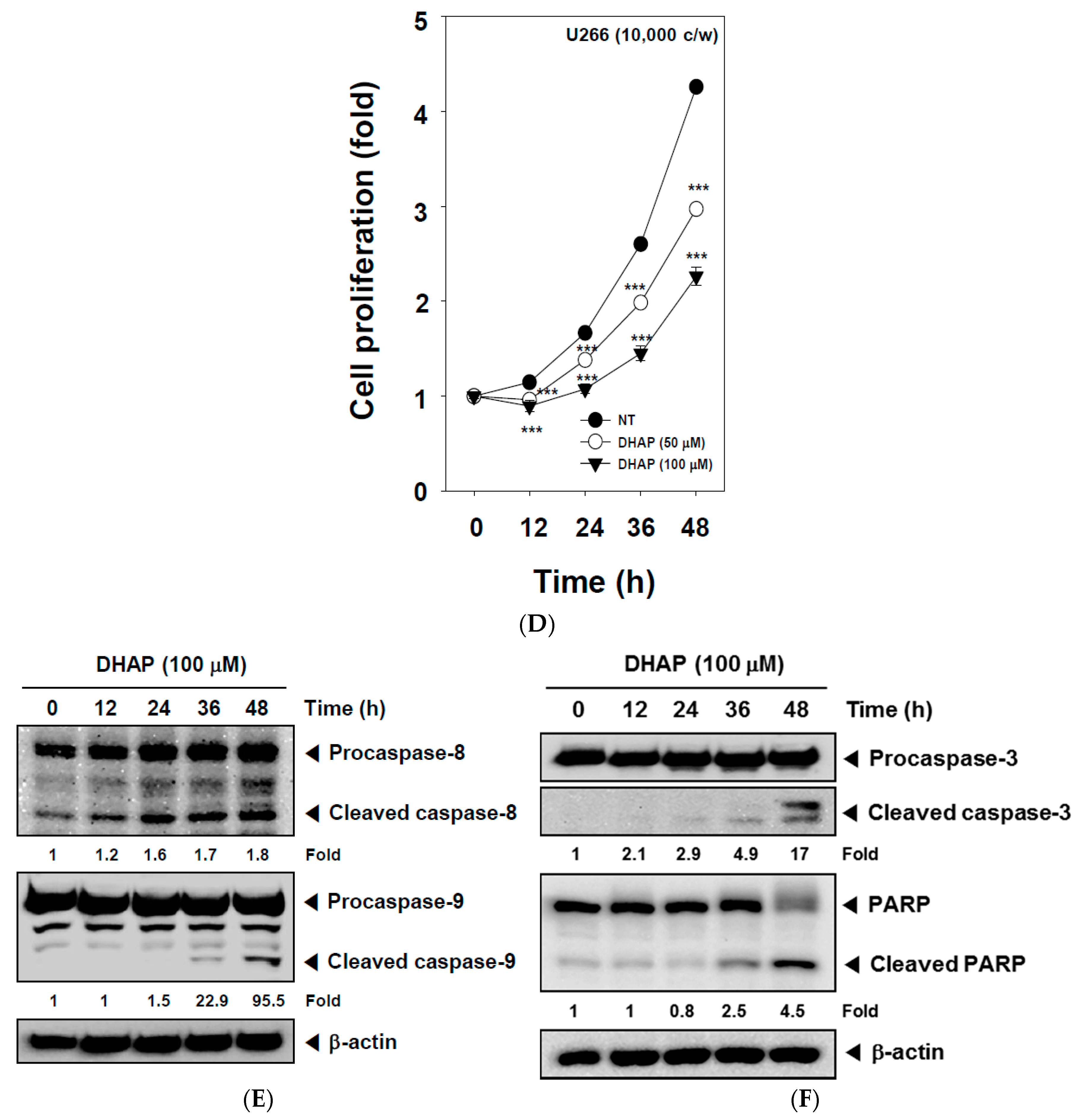
2.2. DHAP Inhibits Cell Proliferation and Induces Apoptosis in U266 Cells
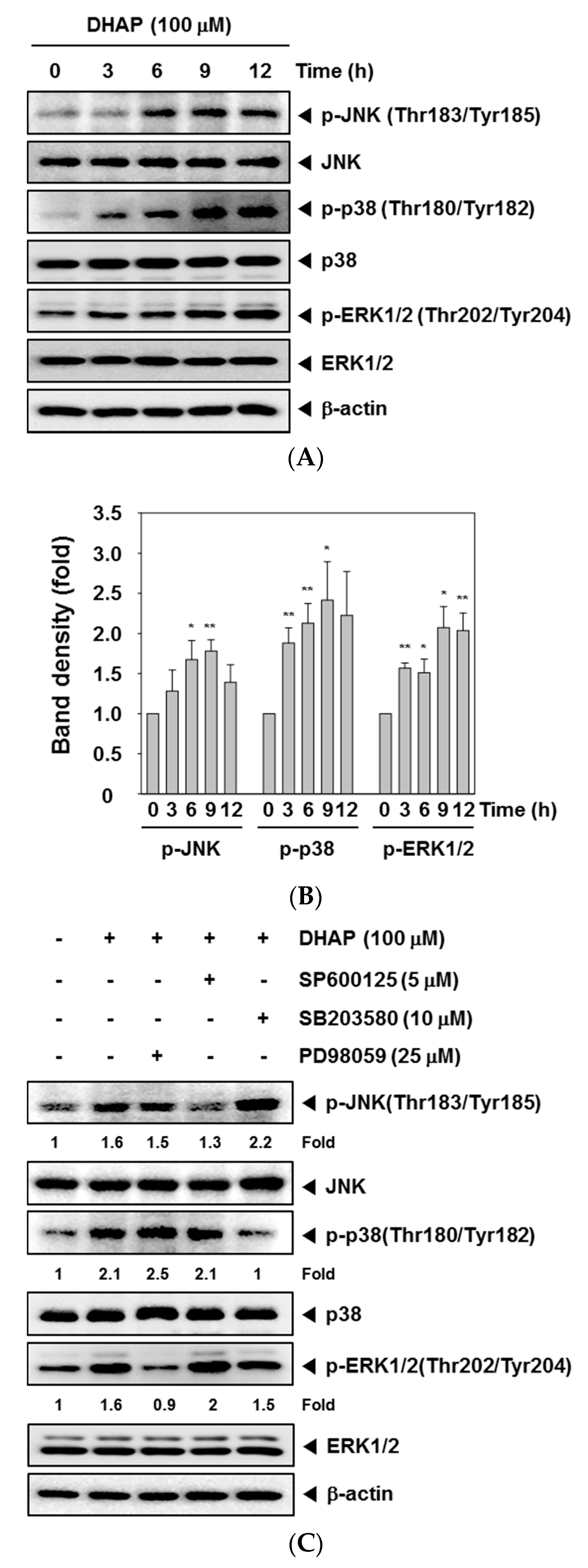
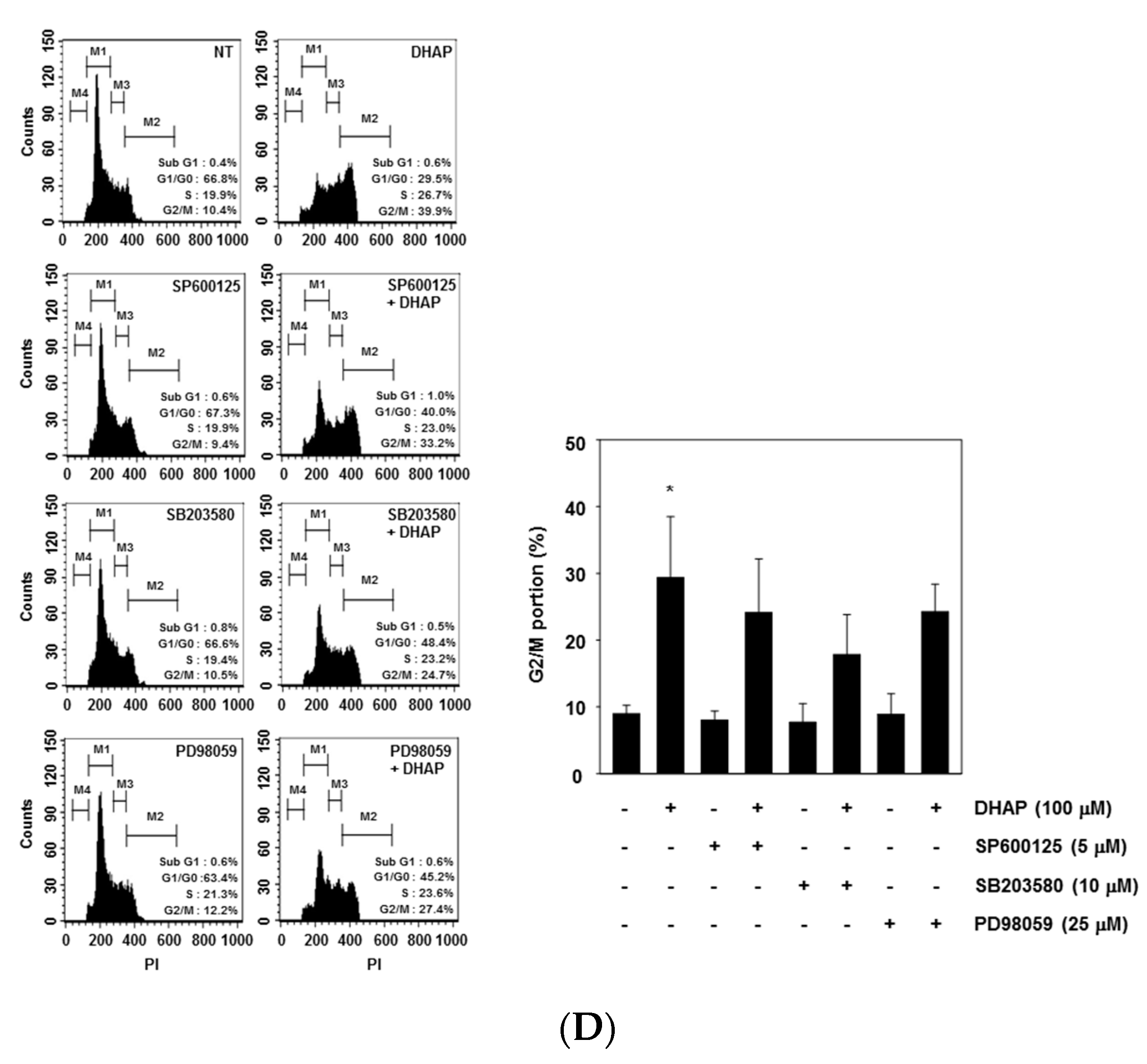
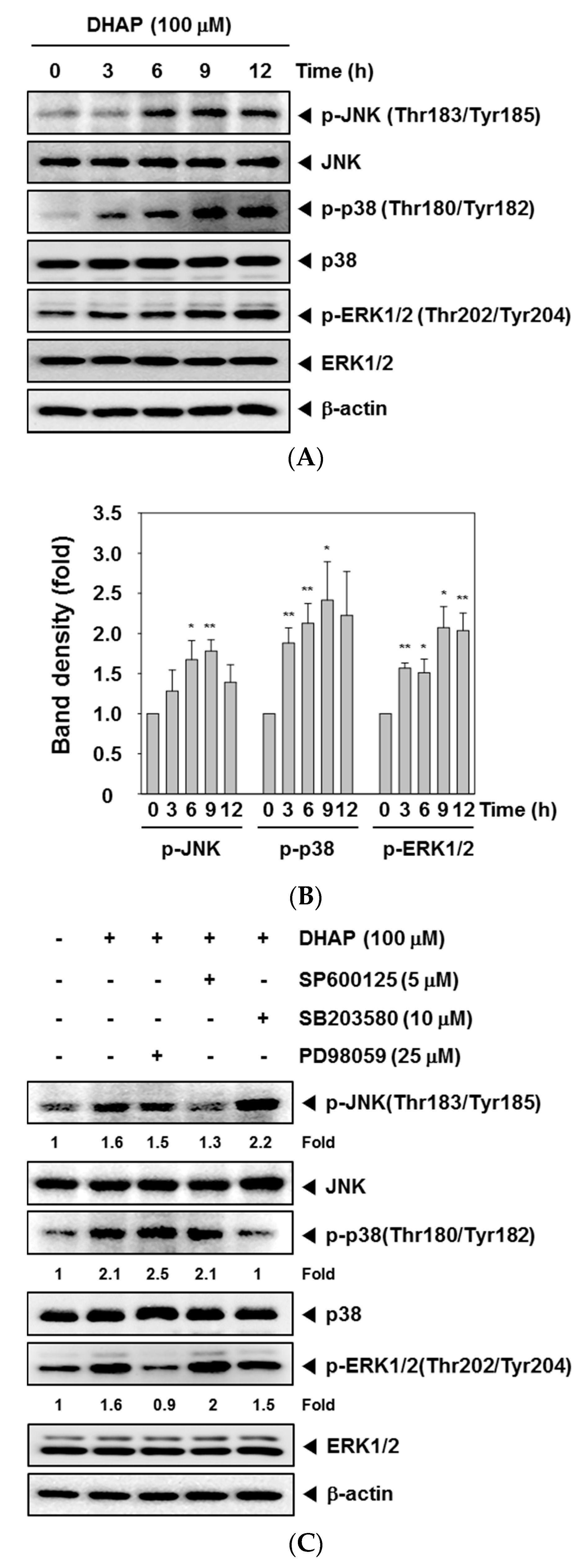
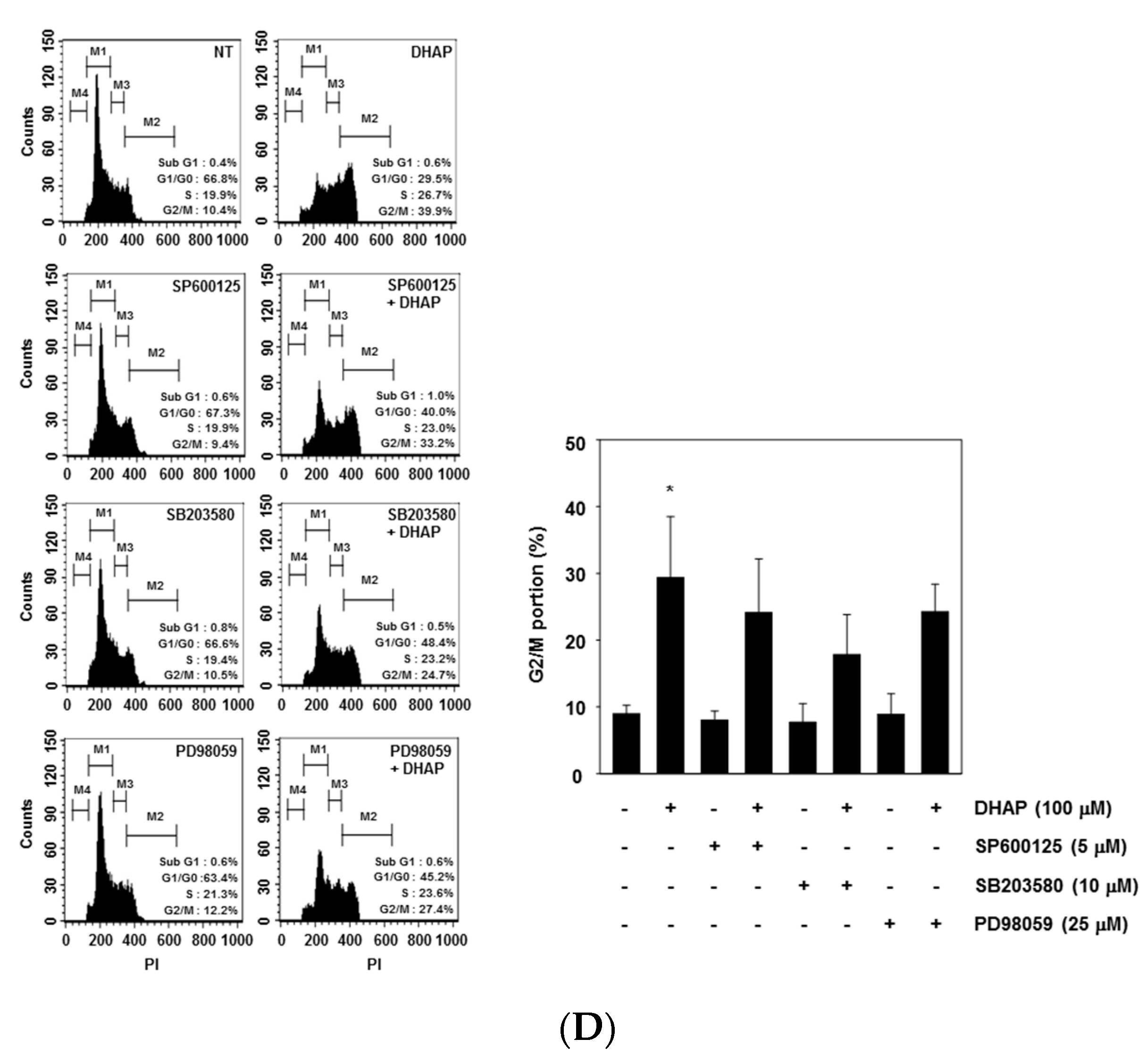
2.3. DHAP Activates MAPK Signaling Pathways
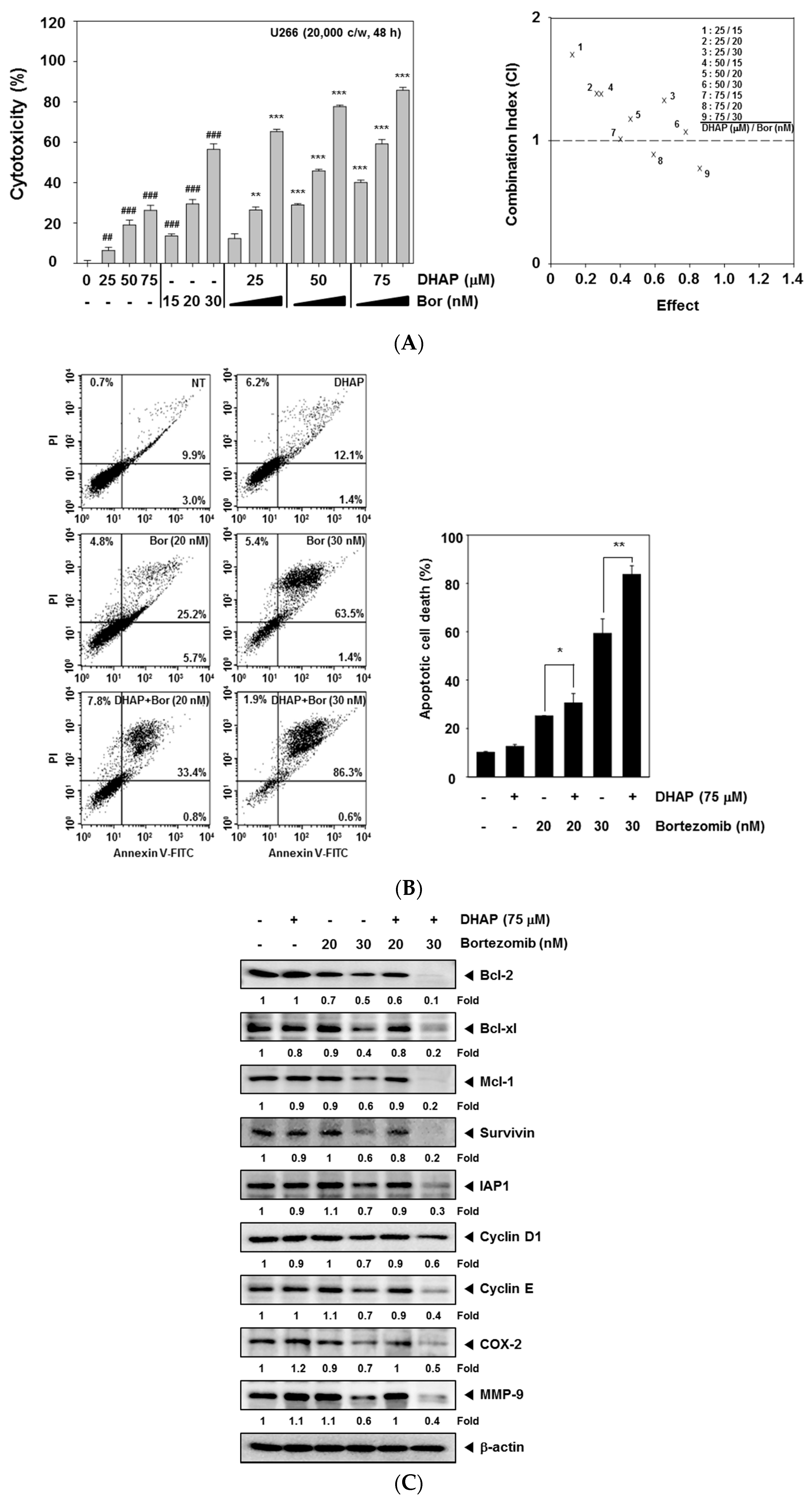
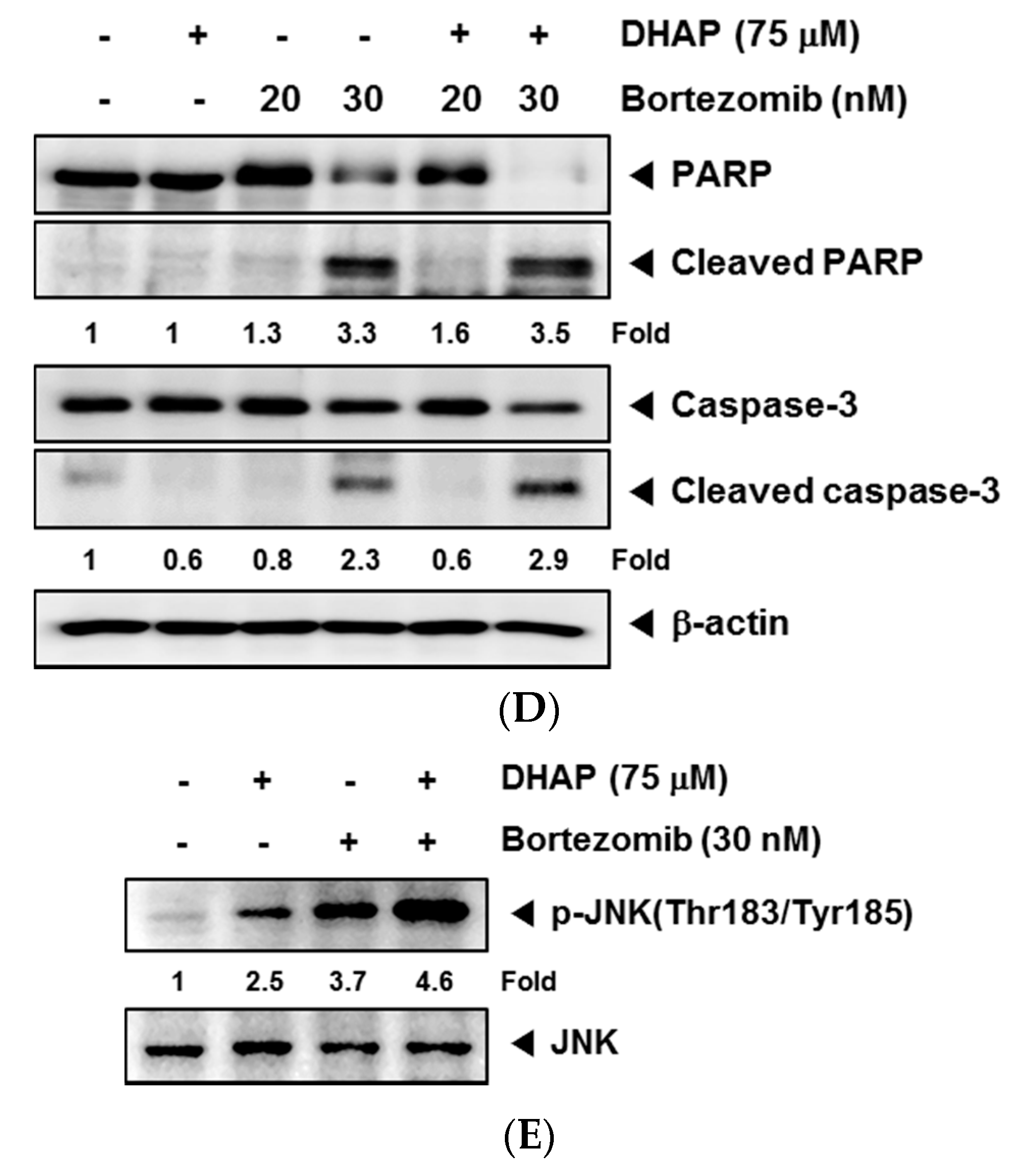
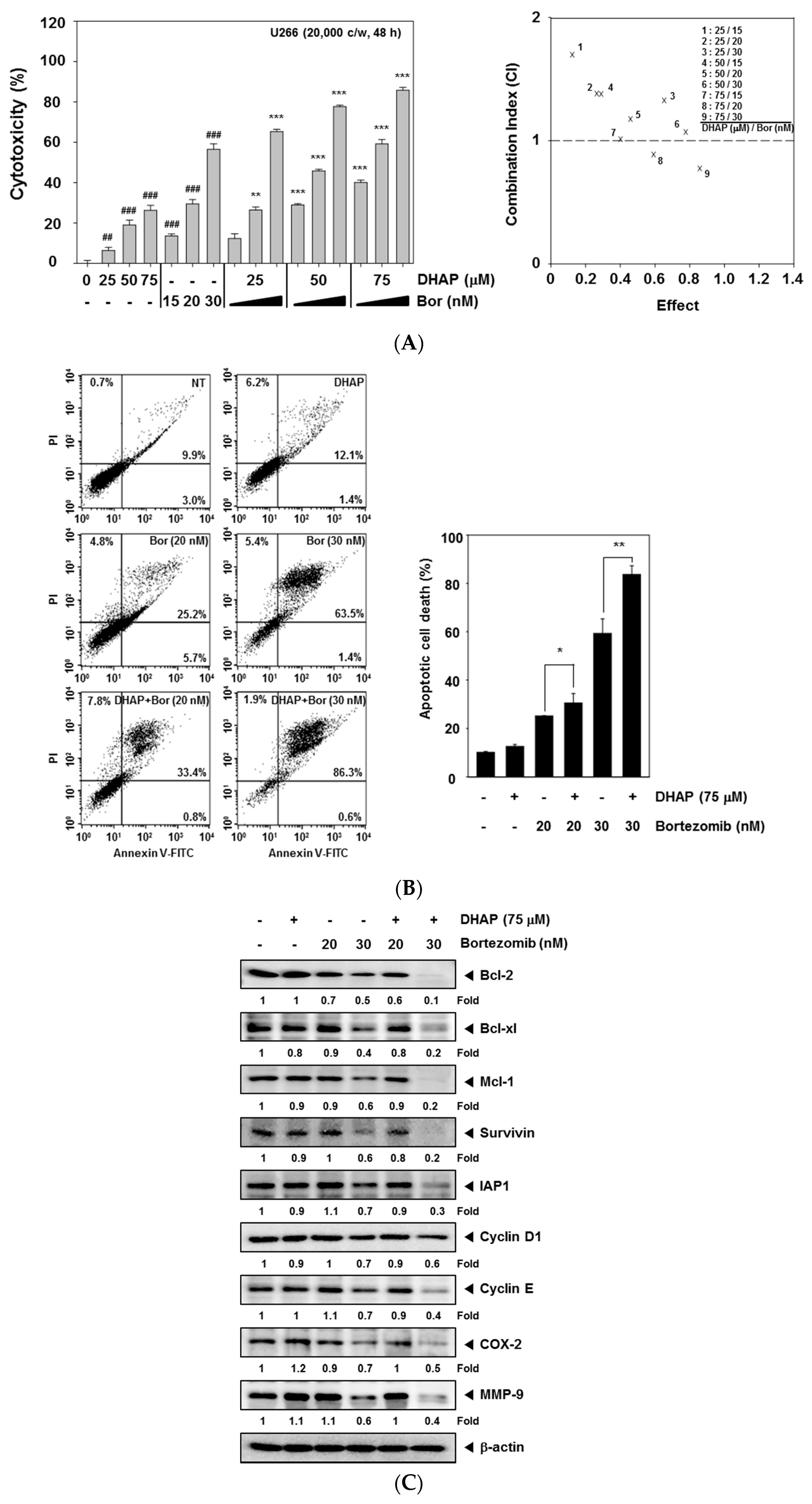
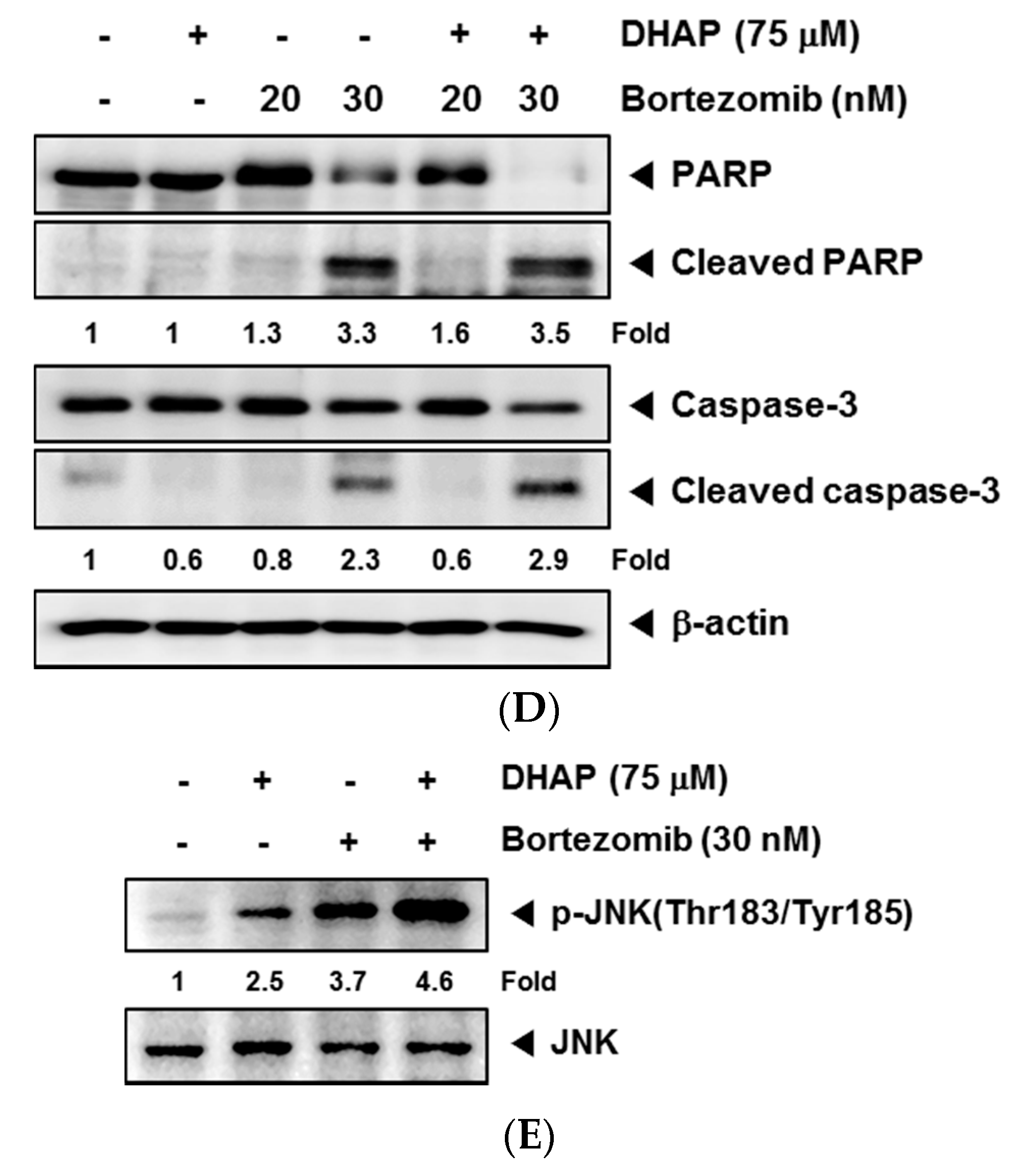
2.4. DHAP Causes Potentiation of the Apoptotic Effect of Bortezomib in U266 Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines and Cell Culture
4.3. Western Blot Analysis
4.4. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.5. MTT Assay
4.6. Cell Cycle Analysis
4.7. Annexin V Assay
4.8. TUNEL Assay
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yokozawa, T.; Kim, H.Y.; Yamabe, N. Amelioration of diabetic nephropathy by dried Rehmanniae Radix (Di Huang) extract. Am. J. Chin. Med. 2004, 32, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.J.; Kim, H.R.; Kim, D.S.; Woo, E.R.; Cho, Y.G.; Chae, S.W. Saeng-Ji-Hwang has a protective effect on adriamycin-induced cytotoxicity in cardiac muscle cells. Life Sci. 2005, 76, 2027–2042. [Google Scholar] [CrossRef] [PubMed]
- Lau, T.W.; Lam, F.F.; Lau, K.M.; Chan, Y.W.; Lee, K.M.; Sahota, D.S.; Ho, Y.Y.; Fung, K.P.; Leung, P.C.; Lau, C.B. Pharmacological investigation on the wound healing effects of Radix Rehmanniae in an animal model of diabetic foot ulcer. J. Ethnopharmacol. 2009, 123, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Jung, H.W.; Lee, J.Y.; Kim, J.S.; Kang, S.S.; Kim, Y.S.; Park, Y.K. 2,5-dihydroxyacetophenone isolated from Rehmanniae Radix Preparata inhibits inflammatory responses in lipopolysaccharide-stimulated RAW264.7 macrophages. J. Med. Food 2012, 15, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A. Cancer: Inflaming metastasis. Nature 2009, 457, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Shanmugam, M.K.; Ramachandran, L.; Kumar, A.P.; Tergaonkar, V. Multifaceted link between cancer and inflammation. Biosci. Rep. 2012, 32, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chai, E.Z.; Siveen, K.S.; Shanmugam, M.K.; Arfuso, F.; Sethi, G. Analysis of the intricate relationship between chronic inflammation and cancer. Biochem. J. 2015, 468, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Makin, G.; Dive, C. Apoptosis and cancer chemotherapy. Trends Cell Biol. 2001, 11, S22–S26. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Targeting apoptosis pathways in cancer therapy. Curr. Cancer Drug Targets 2004, 4, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Kannaiyan, R.; Sethi, G. Targeting cell signaling and apoptotic pathways by dietary agents: Role in the prevention and treatment of cancer. Nutr. Cancer 2011, 63, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.H.; Sethi, G.; Kuo, P.L. Novel medicines and strategies in cancer treatment and prevention. Biomed. Res. Int. 2014, 2014, 474078. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A.; Sethi, G. Bioactive natural products in cancer prevention and therapy: Progress and promise. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2016; Volume 40–41, pp. 1–3. [Google Scholar]
- Shanmugam, M.K.; Lee, J.H.; Chai, E.Z.; Kanchi, M.M.; Kar, S.; Arfuso, F.; Dharmarajan, A.; Kumar, A.P.; Ramar, P.S.; Looi, C.Y.; et al. Cancer prevention and therapy through the modulation of transcription factors by bioactive natural compounds. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2016; Volume 40–41, pp. 35–47. [Google Scholar]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.F.; Weng, C.J.; Sethi, G.; Hu, D.N. Natural bioactives and phytochemicals serve in cancer treatment and prevention. Evid. Based Complement. Alternat. Med. 2013, 2013, 698190. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.S.; Yang, S.F.; Sethi, G.; Hu, D.N. Natural bioactives in cancer treatment and prevention. Biomed. Res. Int. 2015, 2015, 182835. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-kappaB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Boutros, T.; Chevet, E.; Metrakos, P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: Roles in cell growth, death, and cancer. Pharmacol. Rev. 2008, 60, 261–310. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Rajendran, P.; Sethi, G. Thymoquinone inhibits proliferation, induces apoptosis and chemosensitizes human multiple myeloma cells through suppression of signal transducer and activator of transcription 3 activation pathway. Br. J. Pharmacol. 2010, 161, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.S.; Mustafa, N.; Li, F.; Kannaiyan, R.; Ahn, K.S.; Kumar, A.P.; Chng, W.J.; Sethi, G. Thymoquinone overcomes chemoresistance and enhances the anticancer effects of bortezomib through abrogation of NF-kappaB regulated gene products in multiple myeloma xenograft mouse model. Oncotarget 2014, 5, 634–648. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.G.; Murillo, G.; Naithani, R.; Peng, X. Cancer chemoprevention by natural products: How far have we come? Pharm. Res. 2010, 27, 950–961. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Dhanalakshmi, S.; Agarwal, R. Phytochemicals as cell cycle modulators a less toxic approach in halting human cancers. Cell Cycle 2002, 1, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Kannaiyan, R.; Manu, K.A.; Chen, L.; Li, F.; Rajendran, P.; Subramaniam, A.; Lam, P.; Kumar, A.P.; Sethi, G. Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-Jun N-terminal kinase and suppression of PI3 K/Akt signaling pathways. Apoptosis 2011, 16, 1028–1041. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Lee, J.H.; Kim, C.; Ko, J.H.; Ryu, S.H.; Lee, S.G.; Yang, W.M.; Um, J.Y.; Chinnathambi, A.; Alharbi, S.A.; et al. Ginkgolic Acid C 17:1, Derived from Ginkgo biloba Leaves, Suppresses Constitutive and Inducible STAT3 Activation through Induction of PTEN and SHP-1 Tyrosine Phosphatase. Molecules 2017, 22, 276. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Bortner, D.M.; Rosenberg, M.P. Induction of mammary gland hyperplasia and carcinomas in transgenic mice expressing human cyclin E. Mol. Cell. Biol. 1997, 17, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, W.; Gao, Y.F.; Su, X.Q.; Zhai, Z.H. Senescence-like changes induced by expression of p21(waf1/Cip1) in NIH3T3 cell line. Cell Res. 2002, 12, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Coqueret, O. New roles for p21 and p27 cell-cycle inhibitors: A function for each cell compartment? Trends Cell Biol. 2003, 13, 65–70. [Google Scholar] [CrossRef]
- Cho, S.D.; Li, G.; Hu, H.; Jiang, C.; Kang, K.S.; Lee, Y.S.; Kim, S.H.; Lu, J. Involvement of c-Jun N-terminal kinase in G2/M arrest and caspase-mediated apoptosis induced by sulforaphane in DU145 prostate cancer cells. Nutr. Cancer 2005, 52, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Shen, G.; Yuan, X.; Kim, J.H.; Gopalkrishnan, A.; Keum, Y.S.; Nair, S.; Kong, A.N. ERK and JNK signaling pathways are involved in the regulation of activator protein 1 and cell death elicited by three isothiocyanates in human prostate cancer PC-3 cells. Carcinogenesis 2006, 27, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, D.; DeSimone, J.; Hankewych, M.; Kousnetzova, T.; Chen, Y.H. Decitabine induces cell cycle arrest at the G1 phase via p21(WAF1) and the G2/M phase via the p38 MAP kinase pathway. Leuk. Res. 2003, 27, 999–1007. [Google Scholar] [CrossRef]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.M.; Hallahan, A.R. p38 MAP kinase: A convergence point in cancer therapy. Trends Mol. Med. 2004, 10, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Sah, J.F.; Balasubramanian, S.; Eckert, R.L.; Rorke, E.A. Epigallocatechin-3-gallate inhibits epidermal growth factor receptor signaling pathway evidence for direct inhibition of ERK1/2 and AKT kinases. J. Biol. Chem. 2004, 279, 12755–12762. [Google Scholar] [CrossRef] [PubMed]
- Bacus, S.S.; Gudkov, A.V.; Lowe, M.; Lyass, L.; Yung, Y.; Komarov, A.P.; Keyomarsi, K.; Yarden, Y.; Seger, R. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene 2001, 20, 147–155. [Google Scholar] [CrossRef] [PubMed]
- She, Q.B.; Bode, A.M.; Ma, W.Y.; Chen, N.Y.; Dong, Z. Resveratrol-induced activation of p53 and apoptosis is mediated by extracellular-signal-regulated protein kinases and p38 kinase. Cancer Res. 2001, 61, 1604–1610. [Google Scholar] [PubMed]
- Nguyen, T.T.; Tran, E.; Nguyen, T.H.; Do, P.T.; Huynh, T.H.; Huynh, H. The role of activated MEK-ERK pathway in quercetin-induced growth inhibition and apoptosis in A549 lung cancer cells. Carcinogenesis 2004, 25, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Kannaiyan, R.; Hay, H.S.; Rajendran, P.; Li, F.; Shanmugam, M.K.; Vali, S.; Abbasi, T.; Kapoor, S.; Sharma, A.; Kumar, A.P.; et al. Celastrol inhibits proliferation and induces chemosensitization through down-regulation of NF-kappaB and STAT3 regulated gene products in multiple myeloma cells. Br. J. Pharmacol. 2011, 164, 1506–1521. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Cho, S.K.; Kapoor, S.; Kumar, A.; Vali, S.; Abbasi, T.; Kim, S.H.; Sethi, G.; Ahn, K.S. β-caryophyllene oxide inhibits constitutive and inducible STAT3 signaling pathway through induction of the SHP-1 protein tyrosine phosphatase. Mol. Carcinog. 2014, 53, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, C.; Kim, S.H.; Sethi, G.; Ahn, K.S. Farnesol inhibits tumor growth and enhances the anticancer effects of bortezomib in multiple myeloma xenograft mouse model through the modulation of STAT3 signaling pathway. Cancer Lett. 2015, 360, 280–293. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ko, J.-H.; Lee, J.H.; Jung, S.H.; Lee, S.-G.; Chinnathambi, A.; Alharbi, S.A.; Yang, W.M.; Um, J.-Y.; Sethi, G.; Ahn, K.S. 2,5-Dihydroxyacetophenone Induces Apoptosis of Multiple Myeloma Cells by Regulating the MAPK Activation Pathway. Molecules 2017, 22, 1157. https://doi.org/10.3390/molecules22071157
Ko J-H, Lee JH, Jung SH, Lee S-G, Chinnathambi A, Alharbi SA, Yang WM, Um J-Y, Sethi G, Ahn KS. 2,5-Dihydroxyacetophenone Induces Apoptosis of Multiple Myeloma Cells by Regulating the MAPK Activation Pathway. Molecules. 2017; 22(7):1157. https://doi.org/10.3390/molecules22071157
Chicago/Turabian StyleKo, Jeong-Hyeon, Jae Hwi Lee, Sang Hoon Jung, Seok-Geun Lee, Arunachalam Chinnathambi, Sulaiman Ali Alharbi, Woong Mo Yang, Jae-Young Um, Gautam Sethi, and Kwang Seok Ahn. 2017. "2,5-Dihydroxyacetophenone Induces Apoptosis of Multiple Myeloma Cells by Regulating the MAPK Activation Pathway" Molecules 22, no. 7: 1157. https://doi.org/10.3390/molecules22071157