Abstract
Chromatographic purification of the n-hexane and dichloromethane extracts of Nuxia oppositifolia aerial parts, growing in Saudi Arabia, resulted in the isolation and characterization of three new labdane-type diterpene acids, 2β-acetoxy-labda-7-en-15-oic acid (1), 2β-acetoxy-7-oxolabda-8-en-15-oic acid (2), 2β-acetoxy-6-oxolabda-7-en-15-oic acid (3), and one new seco-triterpene, 3,4-seco olean-12-en-3,30 dioic acid (4), together with 10 known lupane, oleanane and ursane-type triterpenes, as well as the common phytosterols, β-sitosterol and stigmasterol (5–16). Their structures have been assigned on the basis of different spectroscopic techniques including 1D and 2D NMR. Moreover, 13 of the isolated compounds were tested on the human cancer cell lines HeLa (cervical), A549 (lung) and MDA (breast), and most of the compounds showed potent cytotoxic activities in vitro.
1. Introduction
The genus Nuxia (family Buddlejaceae) comprises ca. 40 species of shrubs and trees distributed over the southern region of the Arabian peninsula, tropical Africa (Madagascar, Comoro and the Mascarene Islands) and South Africa [1]. Nuxia is represented in Saudi Arabia by only two species, viz. N. oppositifolia Benth and N. congesta Fresen, with limited distribution in the southwestern parts of the Hijaz area [2,3]. Several Nuxia species are plants of economic and medicinal interest with a rich diversity of ethnobotanical uses. Some species are used for the treatment of urine albumin, venereal disease, and as purgatives [4]. Leaves of N. sphaerocephala Baker are used in the traditional medicine of Madagascar to treat malarial splenomegaly and infantile hydrocephalus [5].
The dichloromethane leaf extract of N. verticillata, endemic to the Mascarene Archipelago, had potential anti-proliferative activity and selectively inhibited cancer cell growth in comparison with normal cells [4]. Some of the Nuxia plants have been studied phytochemically, revealing the presence of phenylpropanoid glycosides such as verbascoside [6], in addition to clerodane and labdane diterpenoids, and pentacyclic triterpenes [5].
Recently, various studies have shown pentacyclic triterpenes as valuable candidates to treat cancer by several modes of action [7]. Reviewing literature revealed that few phytochemical and biological studies have been conducted on N. oppositifolia, where only two compounds, namely verbascoside and the acylated diglycoside 2′′-acetyl-3′′-benzoyl-nuxioside, have been reported [6] These facts encouraged us to reinvestigate the chemical constituents and biological activities of N. oppositifolia, aiming to find new entities able to cure contemporary diseases such as cancer. In this paper, we describe the isolation and structural elucidation of four new compounds and 11 known compounds as well as assessment of in vitro cytotoxic activities of some of the isolated compounds against three cancer cell lines.
2. Results and Discussion
2.1. Phytochemical Study
As part of our search for novel medicinal compounds from plants, we investigated the aerial parts of N. oppositifolia. Chromatographic separation and purification resulted in the isolation and full identification of three new labdane diterpenoic acids (1–3), a new seco-triterpene, 3,4-seco olean-12-en-3,30 dioic acid (4), along with nine known triterpenes, and the common phytosterols β-sitosterol and stigmasterol (5–16) (Figure 1).
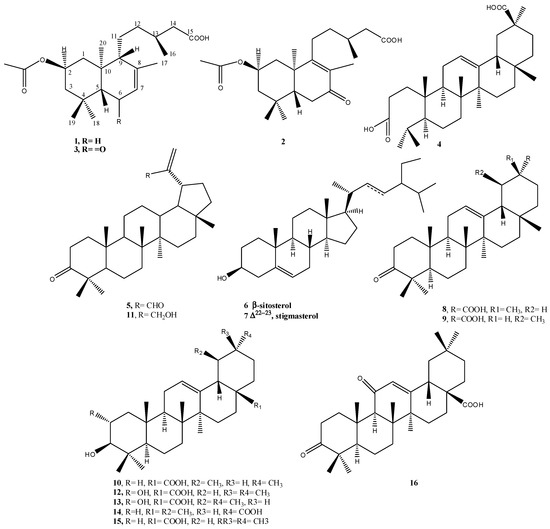
Figure 1.
Structures of isolated compounds from aerial parts of N. oppositifolia.
The molecular formula of compound 1, obtained as a viscous liquid, was determined to be C22H36O4 by HR-EI-MS analysis (m/z 364.2614 [M+]) with five degrees of unsaturation. IR showed intense absorption bands at 3470 (OH), 1745 (CO2R), 1715 (CO2H) and 1665 (C=C), suggesting the presence of OH, two carbonyl groups assigned to an ester and free carboxylic acid groups and an olefinic double bond, respectively. The carbonyl groups showed signals at δC 170.8 and 179.0 ppm in 13C-NMR. 1H-NMR revealed the presence of six methyl groups at δH 2.00, 1.62, 0.93, 0.92, 0.87 and 0.79 ppm, assigned to acetate, olefinic (Me-17), secondary methyl (Me-16), and three tertiary methyl groups, Me-19, Me-18 and Me-20, respectively. The presence of a trisubstituted double bond was evident from an olefinic methyl group at δH 1.62 ppm allylically coupled to the olefinic proton at δH 5.34 (H-5). Additionally, the 1H-NMR spectra showed a downfield triplet centered at 4.95 ppm and correlated it directly to a carbon signal at δC 68.9 in an HSQC experiment, and assigned it to H-2. The 13C-NMR and distortionless enhancement by polarization transfer (DEPT) experiment afforded a total of 22 carbon atoms ascribed to six methyl, six methylene, five methine groups and five quaternary carbon atoms, two of them appearing at δC 170.8 and 179.0 ppm, assigned to an acetate carbonyl at C-2 and a free carboxylic acid carbonyl at C15. Based on the above spectral evidence, the structure of (1) was proposed to be a labdane-type diterpene with an acetoxyl group, a trisubstituted double bond and a free carboxylic acid group [5]. The 1H-1H COSY experiment showed cross-peak correlations between the downfield proton at δH 4.94 (H-2) and both protons at positions H-1 (α and β), H-2 (α and β) at δH 1.00, 2.13, 1.17 and 1.71, respectively, positioning the acetoxyl group at C-2. An additional COSY correlation was detected between the secondary methyl at δH 0.93 (Me-16) and the multiplet proton at δH 1.80 (H-13). Moreover, it showed a correlation between the broad olefinic proton at δH 5.34 (H-7) and both signals at δH 1.90 (H-6) and 1.62 (Me-17). The exact positions for the acetoxyl at C-2 and the free carboxylic acid groups at C-14 were proved through interpretation of the HMBC experiment where significant 2,3J correlations were observed from the methine proton at 4.94 ppm (H-2) and the acetoxyl carbonyl carbon at 170.8 ppm; from Me-16 protons at 0.93 ppm to C-12 and C-14; and from the methylene protons at 1.80, 2.07 (H-13, H-14) and the free carbonyl of the carboxylic acid group at C-15 (δC 179.0). Finally, the location of the double bond at C-7 was confirmed, also from the HMBC experiment, showing two and three bond correlations from the olefinic proton at δH 5.34 (C-7) to both C-5 and C-9 at 49.5 and 54.3 ppm, respectively (Figure 2). The relative stereochemistry was determined by the NOESY experiment where significant cross-peaks were observed between Me-20, Me-18 and H-2, demonstrating that all are on the lower face of the molecule (α-form), while Me-19, H-5, H-9 and Me-16 are on the opposite side of the molecule (β- form). Compound 1, identified as 2β-acetoxy-labda-7-en-15-oic acid, is reported here for the first time from a natural source. To the best of our knowledge, the deacetylated derivative of compound 1, 2α-hydroxylabda-7-en-15 oic acid, has been isolated before from the aerial parts of Brickellia lanciniata [8].

Figure 2.
Key HMBC correlations of compound 1.
Compound 2, obtained as a viscous liquid, showed in HR-EI-MS [M+] 378.2406 (calcd. for 378.2400, C22H34O5). The spectral data of compound 2 showed, like compound 1, the skeleton of a labdane-type diterpene [5]. IR and UV suggested the presence of an α,β-unsaturated carbonyl group (IR 1662 cm−1, UV 246 nm). The 1H- and 13C-NMR results (Table 1 and Table 2) also showed spectral data similar to 1 with six methyl groups, one assigned to acetate and the other five assigned to the labdane diterpene skeleton, distinguished into one secondary methyl group at δH 0.95 (d, J = 6.2 Hz) and four tertiary methyl groups, one of which is an olefinic methyl appearing as a singlet at δH 1.64. It showed also, like compound 1, an acetoxyl group located at position 2, a downfield proton centered at 5.00 ppm (H-2). The absence of an olefinic proton in the 1H-NMR spectrum and the presence of two downfield quaternary carbons at δC 130.0 (C-8) and 167.1 (C-9) proved the existence of a tetrasubstituted double bond conjugated with a carbonyl carbon at δC 199.0 (C-7) (α,β-unsaturated double bond). The significant cross-peak correlations observed in 1H-1H COSY between both H-1 (δH 1.27 and 1.18) and H-2 (δH 4.98) were also clear in compound 2 and confirmed the position of the acetoxyl group at C-2. The main difference between 1 and 2 was observed in the 13C-NMR and DEPT experiment where the number of methine carbons in compound 2 was less by two (three in 2 instead of five in 1), while the number of quaternary carbon atoms increased to six in 2 vs. five in 1. The HMBC experiment verified the exact structure of compound 2 as 2β-acetoxy-7-oxolabda-8-en-15-oic acid, through two- and three-bond correlations observed from olefinic methyl (Me-17) at δH 1.64 to C-9 (δC 167.0), C-7 (δC 199.9) and C-8 (δC 130.0); from Me-20 protons (δH 1.06) to C-5 (δC 49.5) and C-9 (δC 130.0); from both α and β methylene protons at C-11 (δH 2.03, 2.19) to C-8 and C-9; from Me-16 (δH 0.95) to C-12 (δC 35.2) and C-14 (δC 40.9); and finally from H-13 at δH 1.95 to C-14 (δC 41.5) and C-15 (δC 177.9). Like compound 1, the relative stereochemistry was determined by interpretation of the NOESY experiment. Compound 2 is reported here for the first time from a natural source.

Table 1.
1H-NMR spectral data (500 MHz, CDCl3) of compounds 1–4 a.
Table 2.
13C-NMR spectral data (125 MHz, CDCl3) of compounds 1–4 a.
Compound 3, isolated as a viscous liquid, showed the same [M+] 378.2406 in HR-EI-MS as 2. The presence of a carbonyl of unsaturated ketone was suggested by intense IR absorption bands at 1737, 1662 cm−1 and UVλmax at 246 nm. NMR data showed, like compounds 1 and 2, the basic skeleton of labdane-type diterpenes [5], also with an acetate moiety, a free carboxylic acid and an unsaturated ketone (conjugated with an olefinic double bond) observed at δC 170.7, 177.9 and 199.1 ppm in 13C-NMR.
The 13C-NMR and DEPT experiments showed a close resemblance to 1 where a total of 22 carbon atoms were counted and ascribed to six methyl, five methylene, five methine groups, and six quaternary carbon atoms. The above data revealed that 3 is an analogue to compound 1, except that the number of methylene carbons was decreased by one and subsequently the number of quaternary carbons was increased by 1. The rest of the NMR data were closely similar to 1. The 1H-NMR showed, also like 1, the presence of a singlet olefinic proton at δH 5.70 assigned to H-7, a downfield oxygenated proton at 4.90 ppm assigned to H-2, a singlet olefinic methyl at 1.82 ppm assigned to Me-17 with allylic coupling in the COSY experiment, with the olefinic proton at C-7. A significant singlet signal appeared at δH 1.97, integrated with one proton and assigned to H-5. Comparing the chemical shift and the splitting pattern of H-5 in 3 with that of 1 pointed to the existence of a nearby electron-withdrawing group (CO at C-6) in compound 3. The HMBC experiment was able to verify the structure of 3 as 2β-acetoxy-labda-7-ene-6-oxo-15 oic acid by clear two- and three-bond cross-peaks observed from Me-17 at δH 1.82 to C-7 (128.5 ppm) and C-8 (158.9 ppm); from H-7 at δH 5.70 to C-5 (62.5 ppm) and C-9 (56.4 ppm); from H-1 at δH 2.03 to the acetate carbonyl (170.7 ppm); from H-3 at δH 1.61 to C-2 (67.9 ppm); from Me-16 to C-12 (38.9 ppm) and C-14 (41.5); from H-13 at δH 1.93 to C-15 (177.9 ppm); and finally from Me-20 at δH 0.82 to C-5, C-9 and C-10. Additionally, the 2D NOESY experiment was useful for the determination of the relative stereochemistry of 3 where significant cross-peaks were observed between Me-18, M-20 and H-2, demonstrating that all are on the lower face of the molecule, while Me-19, H-5 and Me-16 are on the opposite side of the molecule (β-form). Therefore, the final structure of 3 was established as 2β-acetoxy-6-oxolabda-7-en-15-oic acid, isolated here for the first time from natural source as an isomer of 2.
Compound 4, isolated as a white powder, had the molecular formula C30H48O4 as deduced from HR-EI-MS m/z 472.3553 [M+] (calcd. for 472.3553). The basic skeleton of 4 was proved to be a triterpene based on the assignment of the data obtained from MS and NMR. HR-EI-MS showed a molecular ion peak at m/z at 472 calculated for C30H48O4 with seven degrees of unsaturation, three of which were accounted for by one olefinic and two carbonyls of free carboxylic acids; the rest of the unsaturation number were allocated to four cyclic rings, indicating that 4 is a seco pentacyclic triterpene with two carboxylic acid moieties. Characteristic fragments were observed in EI-MS at m/z 248, 238 and 203, suggesting the presence of an oleanane-type triterpene with a double bond at C-12 (∆12). The presence of a significant fragment at m/z 248 is an explicit pattern of fragmentation characteristic of oleanane-type triterpenes conforming to rings D and E [9,10] while the fragments at m/z 233 and 203 were due to the loss of the methyl and carboxylic acid groups from the fragment at m/z 248. The 1H-NMR results showed that a broad singlet olefinic proton appeared at δH 5.19 (brs, H-12), five tertiary methyl groups (Me-25, 26, 27, 28 and 29) and two secondary methyl groups (Me-23 and 24); the latter were coupled to protons at δH 1.71 (m, H-4) and 0.96 (brd, J = 11.7, H-5) in the COSY experiment (Figure 3), indicating the presence of a separate isopropyl moiety at C-5. A total of 30 carbons were observed in the 13C-NMR spectrum, differentiated into seven methyl, 10 methylene, five methane groups and eight quaternary carbon atoms with the aid of the HSQC and DEPT experiments. Three downfield quaternary carbon atoms appeared at δC 174.8, 177.9 and 144.2 ppm, assigned to C-3, C-30 and C-13. The remaining quaternary carbon atoms resonated at δC 31.6, 39.0, 39.5, 41.6 and 43.1, assigned to carbon atoms 17, 8, 10, 14 and 20, respectively. The above data proved the presence of a ∆12 seco oleanan–type triterpene with two free carboxylic acid groups at C-3 and C-30. The HMBC experiment revealed two- and three-bond correlations observed from H-1 methylene protons at δH 1.54 to C-3, C-5; from H-12 (δH 5.19) to C-9, C-14 and C-18; from H-2 at 2.04 ppm to C-1 and C-3; from H-11 at δH 1.80 m to C-12 and C-13; from Me-29 protons at δH 1.04 to C-20 and C-30; from H-19 at δH 1.59 (brd, J = 13.4) to C-21 and C-29; from H-5 to C-23 and C-24; and finally from H-4 to C-5, C-6 and C-10. The latter correlations proved the attachment of the isopropyl moiety at C-5 (Figure 3). The upfield shift of the methyl protons at 1.06 (Me-29) verified the location of the second free carboxylic acid group to be at C-30 [11]. The presence of the free carboxylic acid group at C-3 and the isopropyl moiety at C-5 verified that compound 4 is a new 3,4-seco oleanane triterpene and its final structure is 3,4-seco-olean-12 ene-3,30 dioic acid. It is worth mentioning that an isomer of compound 4 has been isolated from Junellia tridens [9], where the locations of the dicarboxylic acid groups were at positions 3 and 28 rather than 3 and 30 as in 4.

Figure 3.
Key HMBC and COSY correlations of compound 4.
The known compounds were identified as 3-oxolup-20(29)-en-30-al (3-oxolupenal) (5) [12,13], β-sitosterol and stigmasterol (6, 7), 3-oxoolean-12-en-29α-oic acid (katononic acid) (8) [14,15], urs-12-en-3-one-30-oic acid (ifflaionic acid) (9) [16,17], ursolic acid (10) [5] 30-hydroxylup-20(29)-en-3-one (3-oxolupenol) (11) [12,18], 2α,3β-dihydroxyolean-12-ene-28-oic acid (maslinic acid) (12), 2α-hydroxy-ursolic acid (asiatic acid) (13) [19], 3-hydroxyurs-12-en-30-oic acid (plectranthoic acid A) (14) [20], oleanolic acid (15) [21], and 3,11-dioxoolean-12-en-28-oic acid (16) [22]. The identities of the known compounds were determined by analysis of their spectroscopic data and comparison with those reported in the literature.
Notably, all isolated compounds, except 6, 10, 11 and 15, are reported here from the genus Nuxia for the first time.
2.2. Cytotoxicity Assay
Most of the isolated compounds were evaluated for their in vitro cytotoxic activities against human cervical (HeLa), lung (A549) and breast (MDA) cancer cell lines. Among these, compound 8 possessed the most significant activity (IC50 = 29.35 μg/mL) against HeLa cells, while compound 4 showed the highest cytotoxic potential against A549 cells (IC50 = 53.78 μg/mL) (Table 3).
Table 3.
IC50 (μg/mL) values of isolated compounds from N. oppositifolia.
3. Materials and Methods
3.1. General Procedures
A high-resolution mass spectrophotometer, Jeol JMS-700, was used for accurate mass determination with electron impact mode of ionization at 70 ev. Direct probe was used with temperature ramp setting, initial temperature 50 °C rise, rate of 32 °C per minute and final temperature set up to 350 °C. The IR spectra were recorded on JASCO 320-A spectrometers, optical activity was measured on Jasco P-2000 Polarimeter, Japan while the 1H- and 13C-NMR spectra were recorded at the NMR Unit at the College of Pharmacy, Prince Sattam Bin Abdulaziz University, on an Ultra Shield Plus 500 MHz (Bruker, Billerica, MA, USA) spectrometer operating at 500 MHz for proton and 125 MHz for carbon, respectively. The chemical shift values are reported in δ (ppm) relative to the internal standard TMS; the coupling constants (J) are reported in Hertz (Hz). The 2D NMR experiments (COSY, HSQC, HMBC, and NOESY) were obtained using a standard Bruker program.
Isolations of compounds was performed using Silica gel 60 (0.063–0.200 mm, Merck, Darmstadt Germany) and Sephadex LH-20 (Fluka, Buchs, Switzerland) for open column chromatographic separations, while Lichroprep RP-18 (25–40 µm, Merck) reversed phase material was used for vacuum liquid chromatography (VLC). Centrifugal preparative thin layer chromatography (CPTLC) was performed on a Chromatotron device (Harrison Research, Palo Alto, CA, USA). Plates coated with 1 and 2 mm of silica gel 60, 0.04–0.06 mm were used. All solvents used were of analytical grade. TLC was performed on pre-coated silica gel F254 plates (E. Merck, Darmstadt, Germany); detection performed by spraying with p-anisaldehyde/H2SO4 reagent at 254 nm. All chemicals were purchased from Sigma Chemical Company (St. Louis, MO, USA). The absorbance was read on a microplate reader (ELX 800, Bio-Tek Instruments, Winooski, VT, USA) at 549 nm.
3.2. Plant Material
Aerial part (leaves, stems and flowers) of N. oppositifolia was collected from Wadi Lajab in Jazan province of Saudi Arabia in March 2012 and identified at the Pharmacognosy Department, College of Pharmacy, King Saud University. A voucher specimen (Voucher # 15501) was deposited at the Pharmacognosy Department, College of Pharmacy, King Saud University.
3.3. Extraction and Isolation
The dried and powdered aerial parts of N. oppositifolia (900 g) were extracted by maceration with 80% ethanol (4 × 2 L) at room temperature. The combined obtained ethanolic extract was filtered and concentrated under reduced pressure at 40 °C using a rotary evaporator. The dried ethanolic extract (105 g) was redissolved in 40% ethanol and successively partitioned for several times with n-hexane (3 × 500 mL), chloroform (3 × 500 mL) and n-butanol (3 × 500 mL) to provide the corresponding extracts.
The n-hexane fraction (17.6 g) was subjected to column chromatography on pre-packed silica gel column (40 mm i.d. × 350 mm) and eluted with n-hexane-ethyl acetate gradient. Collected fractions were examined with TLC and similar ones were pooled together into four fractions (A–D). Fraction A, eluted with 5% EtOAc/n-hexane, was further purified with a Chromatotron device (0.5% EtOAc/CHCl3, 2 mm) to yield pure compounds 5 (170 mg), 6 and 7. Part of fraction B, eluted with 10% EtOAc/n-hexane, afforded upon crystallization, compounds 8, 9 and 15 in pure form, whereas another part of the same fraction gave compound 1 upon further purification using RP-18 CC with 5% H2O/MeOH. Fraction C eluted with 20% EtOAc/n-hexane, afforded compound 10 after solvent evaporation, while subsequent centrifugal purification with chromatotron using 25% EtOAc/n-hexane provided compound 11 in a clear form. Fraction D eluted with 30% EtOAc/n-hexane afforded two main subtractions Da, Db. Subfraction Da was subjected to RP-18 CC using 5% H2O/MeOH to afford compounds 13 and 14, while elution with 10% H2O/MeOH gave compound 12. Subfraction Db was applied to Centrifugal radial TLC using 0.5% MeOH/CHCl3 to afford compound 4. The chloroform fraction (8 g) was loaded on a silica gel CC and eluted with increasing amounts of MeOH/CHCl3 yielding four major fractions (E–H). The fraction eluted with 1% MeOH/CHCl3 (E) was subjected to sephadex LH-20 column to afford sub-fractions a–c. Subsequent purification of the sub-fraction a by RP-18 CC (MeOH/H2O, 90:10) yielded compound 15 (5 mg) directly in pure form, while compounds 2 and 3 were obtained in pure form after further chromatotron purification with 7% ethanol in n-hexane.
3.4. Spectral Data of New Compounds
2β-Acetoxy-labda-7-ene-15(E)-oic acid (1), 80 mg; viscous; 1H- and 13C-NMR: see Table 1 and Table 2; [α]D = −7.0, (CHCl3), IR (KBr) νmax cm−1: 3470, 2960, 1745, 1715, 1665, 1463, 1380, 1245, 1027; HREIMS: m/z = 364.2614 calc. for C22H36O4.
2β-Acetoxy-7-oxolabda-8-ene-15(E)-oic acid (2), 70 mg; viscous; 1H- and 13C-NMR: see Table 1 and Table 2; [α]D = 0, (CHCl3), IR (KBr)νmax cm−1: 3450, 2960, 29232873, 1733 (O-C=O), 1662. 1465, 1377, 1161, 1050; HREIMS: m/z = 378.2406 calc. for C22H34O5.
3.5. Spectral Data of Known Compounds
Further information about the known compounds (5–16) can be found in Supplementary Materials.
3.6. Cytotoxicity Assay
3.6.1. Cell Culture
Human cancer cell lines: HeLa (cervical), A549 (lungs) and MDA MB321 (breast) were maintained in T75 flasks in DMEM (F12/GlutaMax) medium (Invitrogen, Carlsbad, CA, USA), supplemented with 10% heat-inactivated bovine serum (Gibco, Bigcabin, OK, USA) and 1× penicillin-streptomycin (Gibco) at 37 °C in a humified chamber with 5% CO2 supply.
3.6.2. Cytotixicty Assay
Cells were seeded (105 cells/well) in 96-well flat-bottom plates (Becton-Dickinson Labware) a day before treatment and grown overnight. Compounds were dissolved in DMSO (Sigma, Saint Louis, MO, USA) and finally prepared as 1.0 mg/mL and 5.0 mg/mL stocks, respectively in culture media. The final concentration of DMSO never exceeded 0.1% in the treatment doses. Three different doses of compounds (50, 25 and 12.5 μg/mL) were further prepared by diluting the stocks in culture media, and cells were treated (in triplicate/dose). Doxorubicin was included as standard reference drug. The treated cultures were further incubated for 48 h, and cell viability test was performed using TACS MTT Cell Proliferation and Viability Assay Kit (TACS) as per manufacturer’s instructions. The optical density (OD) was recorded at 570 nm in a microplate reader (BioTek, ELx800). The cell survival fraction was calculated as (A − B)/A, where A and B are the OD of untreated and treated cells, respectively. The 50% cell survival (IC50) values were estimated using the best fit regression curve method in Excel software.
3.6.3. Microscopy
A direct visual investigation was made under an inverted microscope (40× and 100×, Richter Optica, Carlsbad, CA, USA) to observe any morphological changes in the cells cultured with different treatment doses at 24 and 48 h.
4. Conclusions
Sixteen compounds, including three new labdane-type diterpenic acids (1–3) and one new 3,4-seco-triterpenoic acid (4), were isolated from the n-hexane and chloroform-soluble fractions of the ethanol extract of the aerial parts of Nuxia oppositifolia. Of these, most of the tested compounds showed promising cytotoxic activities against human cancer cells in vitro. The results of our study therefore justify the use of N. oppositifolia in traditional medicine.
Supplementary Materials
Supplementary materials are available online.
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding the work through the Research Group Project no. RG-1437-021.
Author Contributions
S.M.A.-M. and A.A.E.-G. deigned and supervised the study, identified the structures of the isolated compounds and wrote/revised the manuscript. M.K.P. and M.S.A-D carried out the bioactivity assays and revised the manuscript. M.S.A.-S., M.S.A.-D. and O.A.B. interpret and assigned the NMR data and help in preparing/revising the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Johnson, L. Review of the systematics of scrophulariaceae s.L. And their current disposition. Aust. Syst. Bot. 2006, 19, 289–307. [Google Scholar]
- Al-Abbasi, T.M.; Al-Farhan, A.H.; Al-Khulaidi, W.; Hall, M.; Lewellyn, O.A.; Miller, A.G.; Patzelt, A. Important plant areas in the Arabian peninsula. Edinb. J. Bot. 2010, 67, 25–35. [Google Scholar] [CrossRef]
- Migahid, A.M. Flora of Saudi Arabia, 3rd ed.; University Libraries, King Saud University: Riyadh, Saudi Arabia, 1989; Volume II. [Google Scholar]
- Jonville, M.C.; Kodja, H.; Strasberg, D.; Pichette, A.; Ollivier, E.; Frederich, M.; Angenot, L.; Legault, J. Antiplasmodial, anti-inflammatory and cytotoxic activities of various plant extracts from the Mascarene archipelago. J. Ethnopharmacol. 2011, 136, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Mambu, L.; Grellier, P.; Florent, L.; Joyeau, R.; Ramanitrahasimbola, D.; Rasoanaivo, P.; Frappier, F. Clerodane and labdane diterpenoids from Nuxia phaerocephala. Phytochemistry 2006, 67, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.R. Chemotaxonomy of the genus Nuxia (buddlejaceae). Stud. Plant Sci. 1999, 6, 379–382. [Google Scholar]
- Laszczyk, M. Pentacyclic triterpenes of the lupane, oleanane and ursane group as tools in cancer therapy. Planta Med. 2009, 15, 1549–1560. [Google Scholar] [CrossRef] [PubMed]
- Jakupovic, J.; Tsichritzis, F.; Tamayo-Castillo, G.; Castro, V.; Bohlmann, F.; Boldt, E. Diterpenes from Fleischmannia hymenophylla and Brickellia laciniata. Phytochemistry 1989, 28, 2741–2744. [Google Scholar] [CrossRef]
- Caldwell, C.G.; Franzblau, S.G.; Suarez, E.; Timmermann, B.N. Oleanane triterpenes from Junellia tridens. J. Nat. Prod. 2000, 63, 1611–1614. [Google Scholar] [CrossRef] [PubMed]
- Shiojima, K.; Arai, Y.; Masuda, K.; Takase, Y.; Ageta, T.; Ageta, H. Mass spectra of pentacyclic triterpenoids. Chem. Pharm. Bull. 1992, 40, 1683–1690. [Google Scholar] [CrossRef]
- Ngounou, F.N.; Lontsi, D.; Sondengam, B.L. Myrianthinic acid: A new triterpenoid from Myrianthus arboreus. Phytochemistry 1988, 27, 301–303. [Google Scholar] [CrossRef]
- De Souza e Silva, S.R.; de Fátima Silva, G.D.; de Almeida Barbosa, L.C.; Duarte, L.P.; Filho, S.A.V. Lupane pentacyclic triterpenes isolated from stems and branches of Maytenus imbricata (celastraceae). Helv. Chim. Acta 2005, 88, 1102–1109. [Google Scholar] [CrossRef]
- Wijeratne, D.B.T.; Kumar, V.; Uvais, M.; Sultanbawa, S.; Balasubramaniam, S. Triterpenes from Gymnosporia emarginata. Phytochemistry 1982, 21, 2422–2423. [Google Scholar] [CrossRef]
- Tanaka, T.; Koyano, T.; Kowithayakorn, T.; Fujimoto, H.; Okuyama, E.; Hayashi, M.; Komiyama, K.; Ishibashi, M. New multiflorane-type triterpenoid acids from Sandoricum indicum. J. Nat. Prod. 2001, 64, 1243–1245. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, J.R.; Silva, G.D.F.; Pedersoli, J.L.; Alves, R.J. Friedelane and oleanane triterpenoids from bark wood of Austroplenckia populnea. Phytochemistry 1990, 29, 3259–3261. [Google Scholar] [CrossRef]
- Chiu-Ming, C.; Ming-Tyan, C. 6-methoxybenzoxazolinone and triterpenoids from roots of Scoparia dulcis. Phytochemistry 1976, 15, 1997–1999. [Google Scholar] [CrossRef]
- Bosson, J.; Galbraith, M.; Ritchie, E.; Taylor, W. The chemical constituents of Australian Flindersia species. Xviii. The structure of ifflaionic acid. Aust. J. Chem. 1963, 16, 491–498. [Google Scholar] [CrossRef]
- Bohlmann, F.; Jakupovic, J. Neue sesquiterpene, triterpene, flavanone und andere aromatische verbindungen aus Flourensia heterolepis. Phytochemistry 1979, 18, 1189–1194. [Google Scholar] [CrossRef]
- Lan, X.; Wu, H.; Wang, W. Chemical constituents from Sinacalia davidii. Zhongguo Zhong Yao Za Zhi 2010, 35, 1001–1003. [Google Scholar] [PubMed]
- Razdan, T.K.; Kachroo, V.; Harkar, S.; Koul, G.L. Plectranthoic acid a & b, two new triterpenoids from Plectranthus rugosus. Tetrahedron 1982, 38, 991–992. [Google Scholar]
- Mahato, S.B.; Kundu, A.B. 13C-NMR Spectra of pentacyclic triterpenoids—A compilation and some salient features. Phytochemistry. 1994, 37, 1517–1575. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, L.; Liu, J.Y.; Cai, P.L.; Yang, S.L. Chemical constituents of Patrinia scabiosaefolia. Zhong Cao Yao 2011, 42, 1477–1480. [Google Scholar]
- Sample Availability: Not available.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).