Versatile Chemical Derivatizations to Design Glycol Chitosan-Based Drug Carriers



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
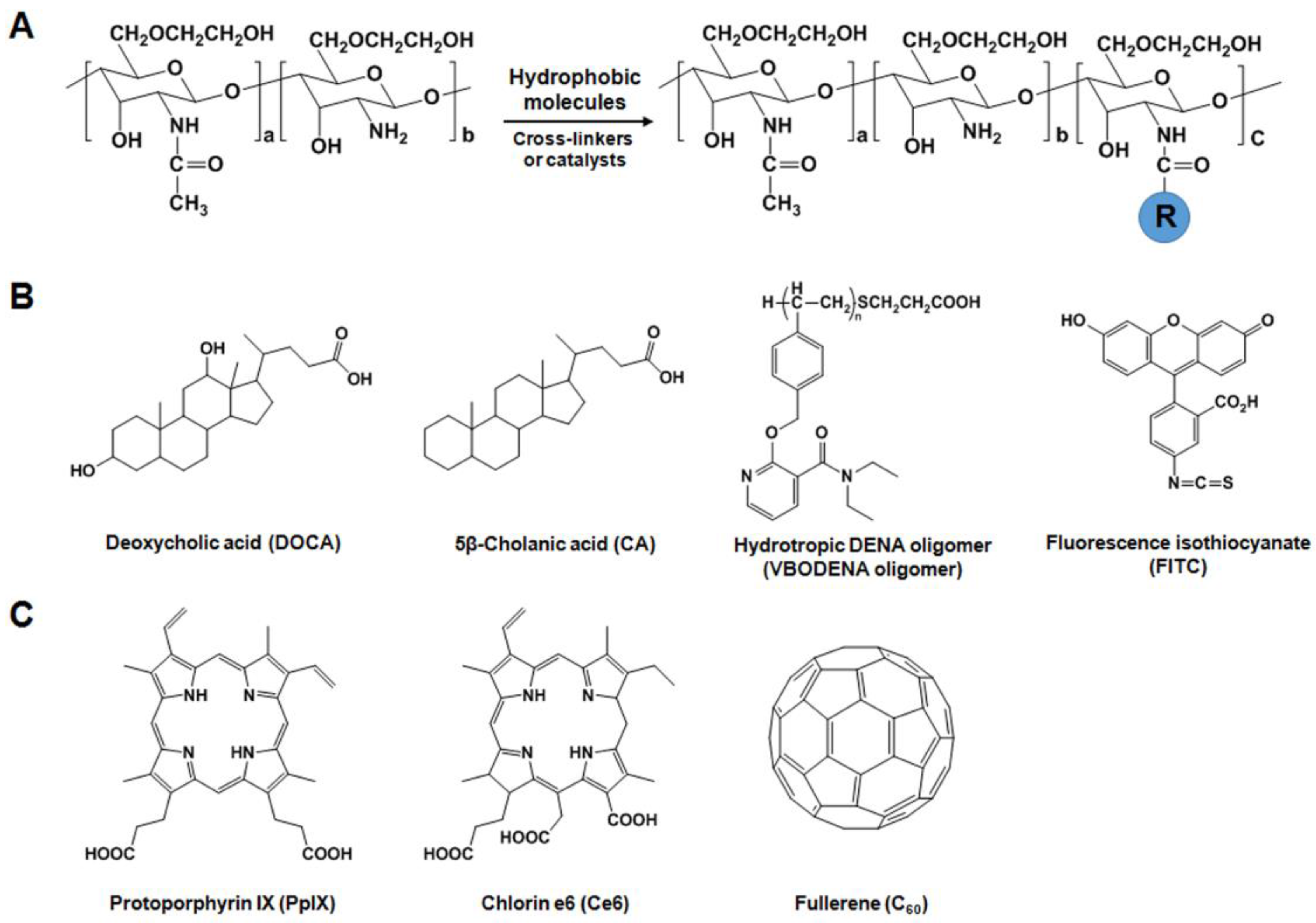
2. Versatile Chemical Derivatizations of Glycol Chitosan
2.1. Preparation of GC
2.2. Design of GC Derivatives as Drug Carriers
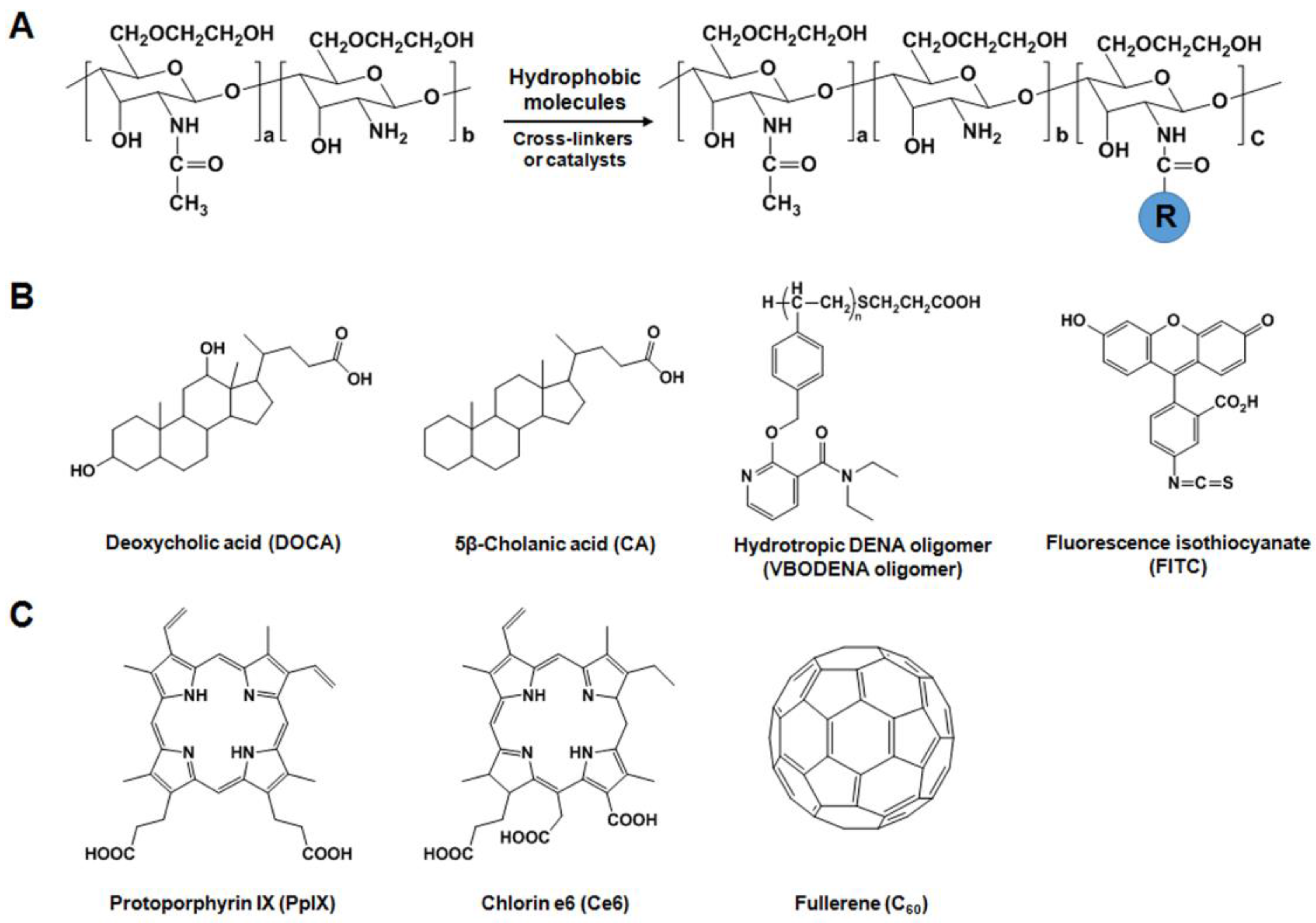
2.2.1. Hydrophobically-Modified GC (HGC) Derivatives for Anticancer Drug Delivery
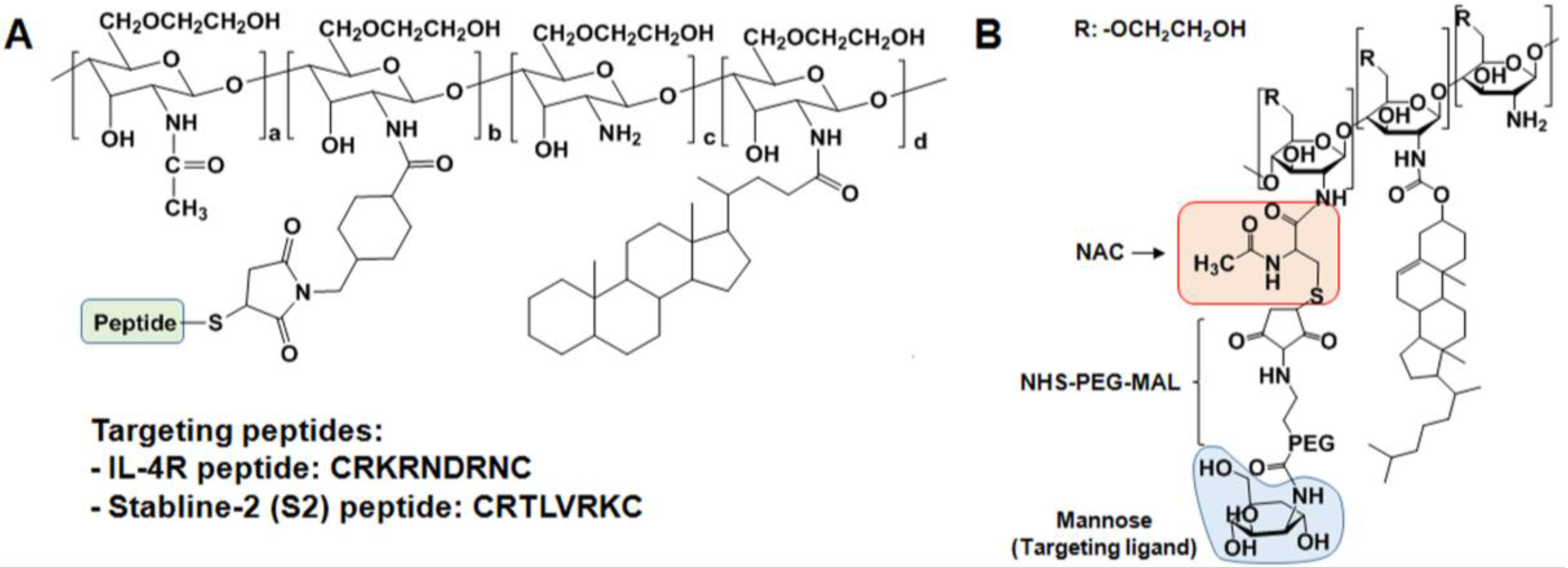
2.2.2. GC Derivatives for Nucleic Acid Delivery
2.2.3. GC Derivatives for Photodynamic Therapy (PDT)
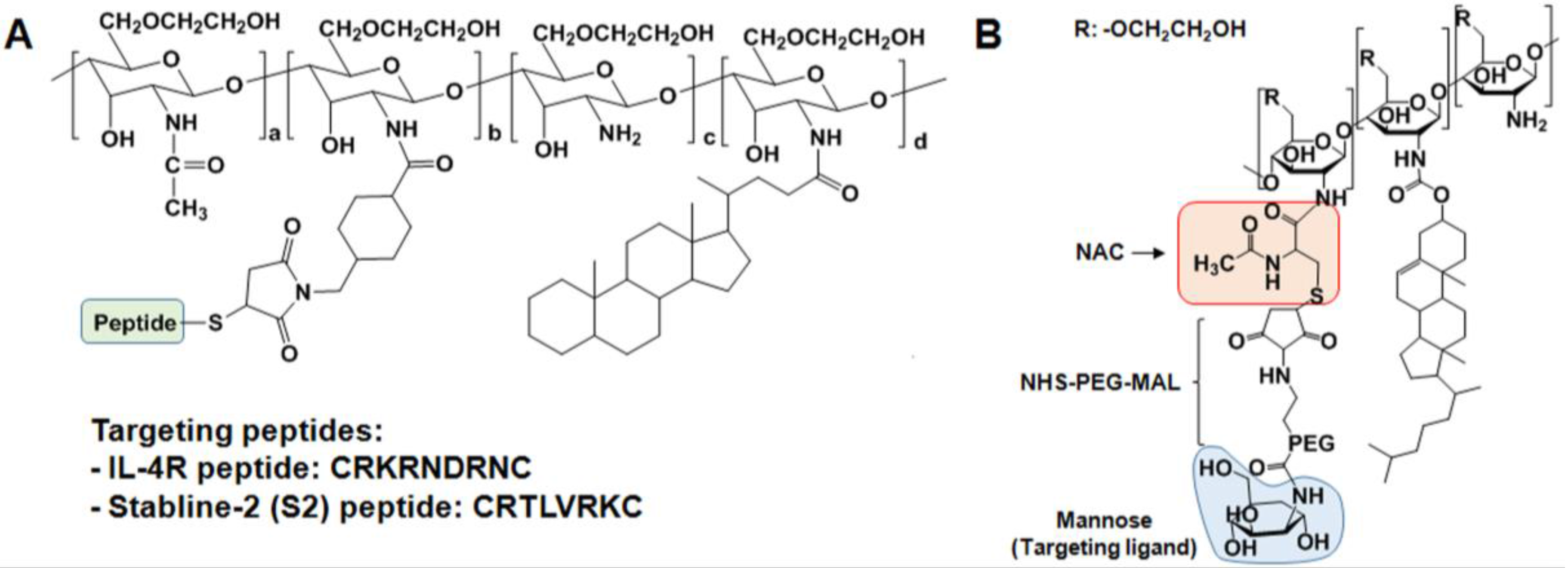
2.3. Specific Receptor Targetable GC Derivatives
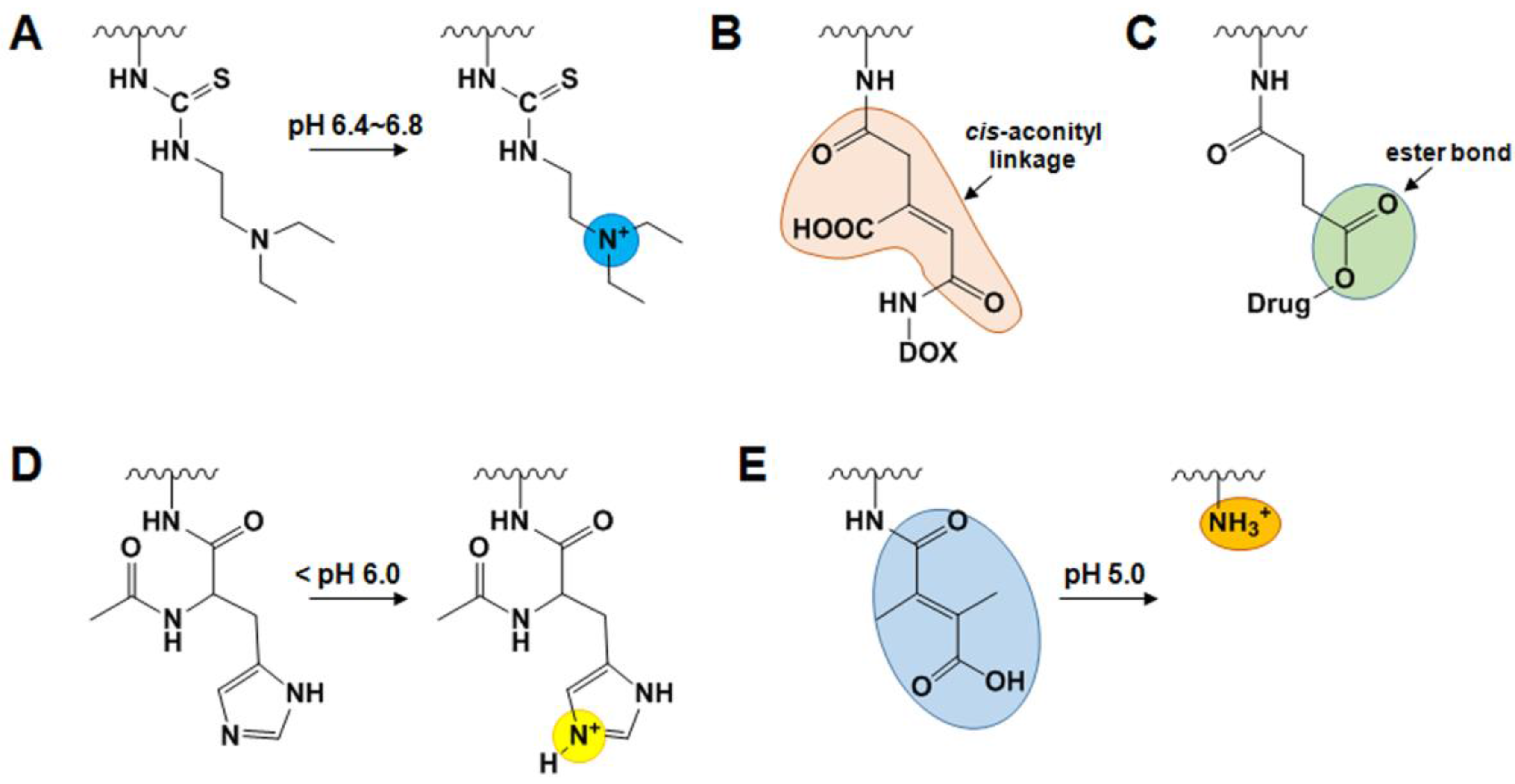
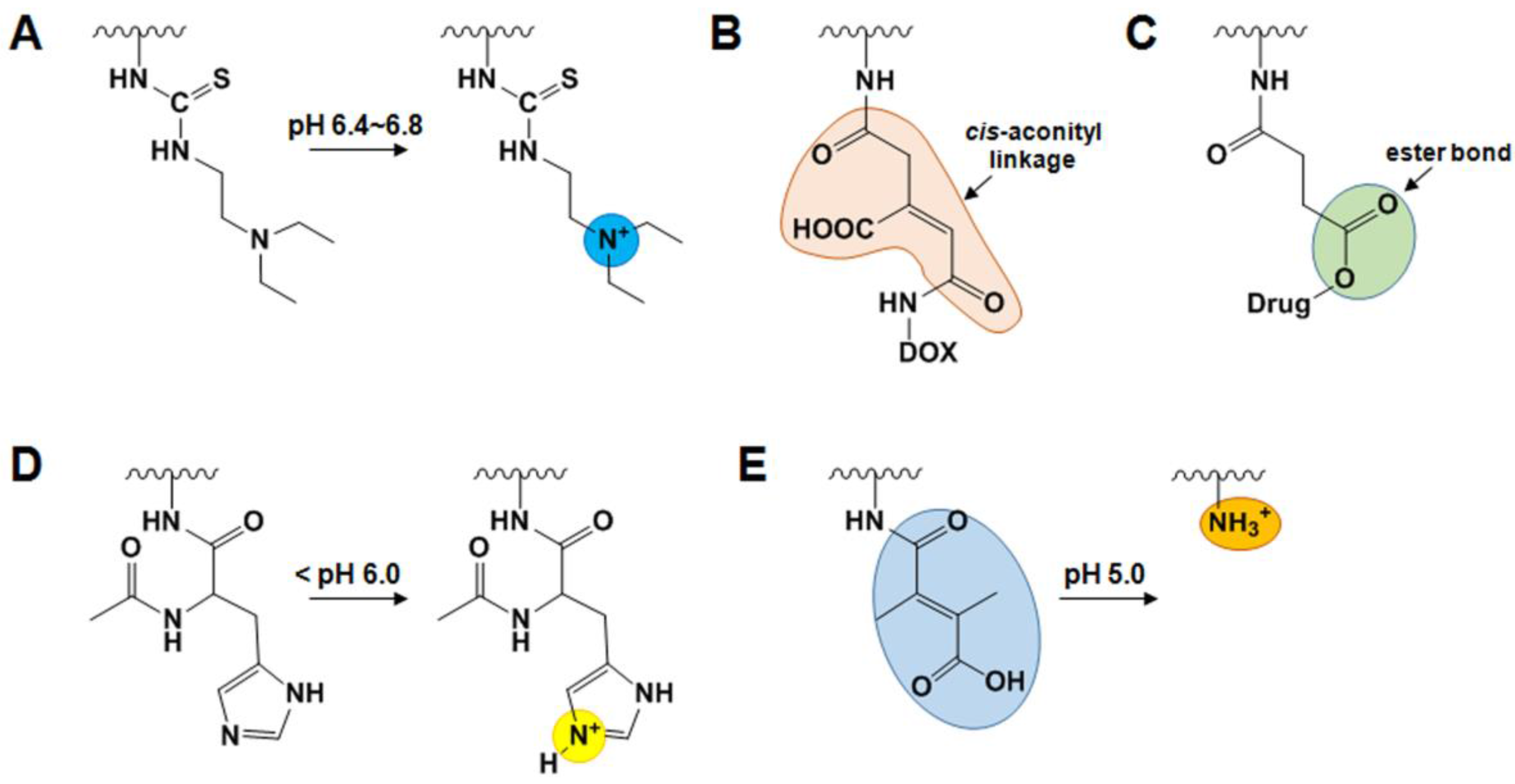
2.4. Endogenous Stimuli-Responsive GC Derivatives
2.4.1. pH-Sensitive GC Derivatives
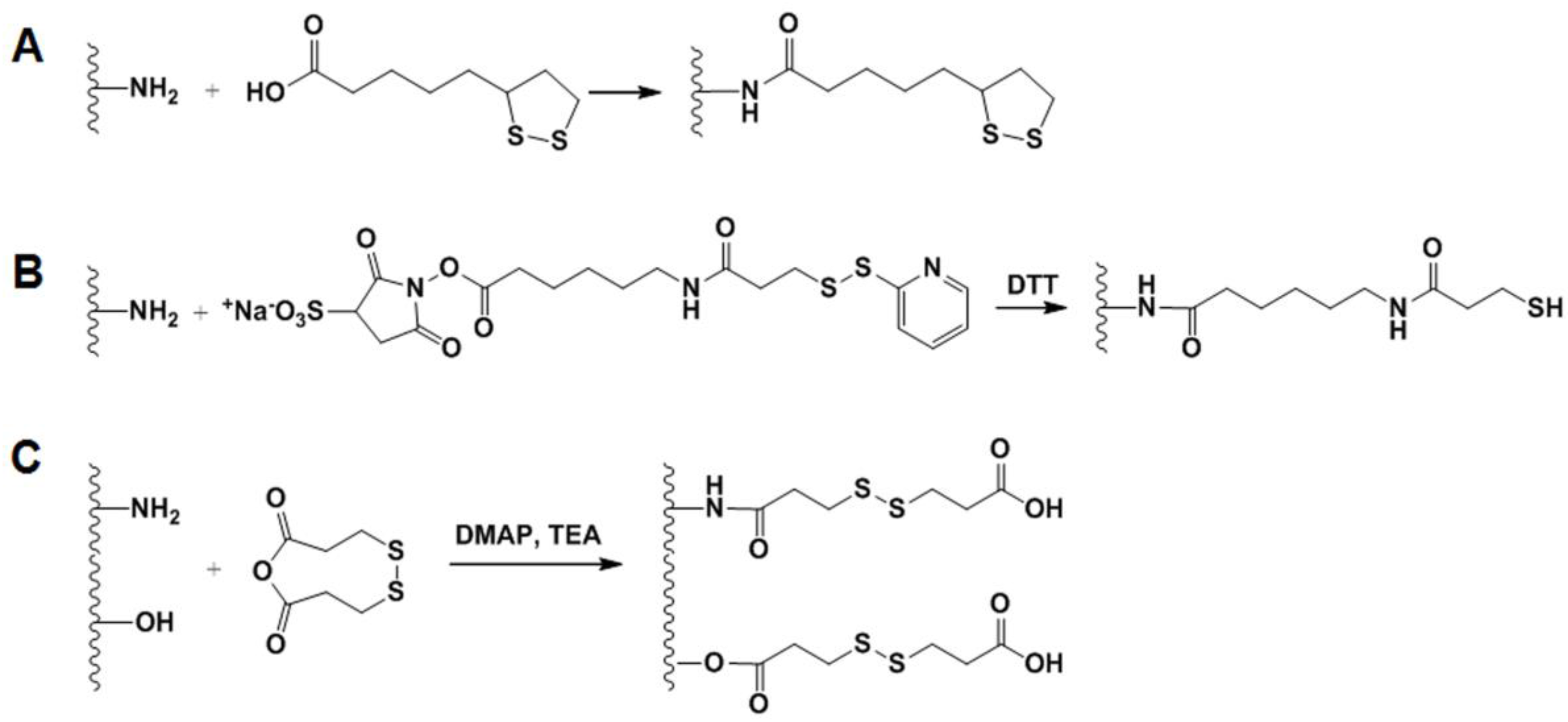
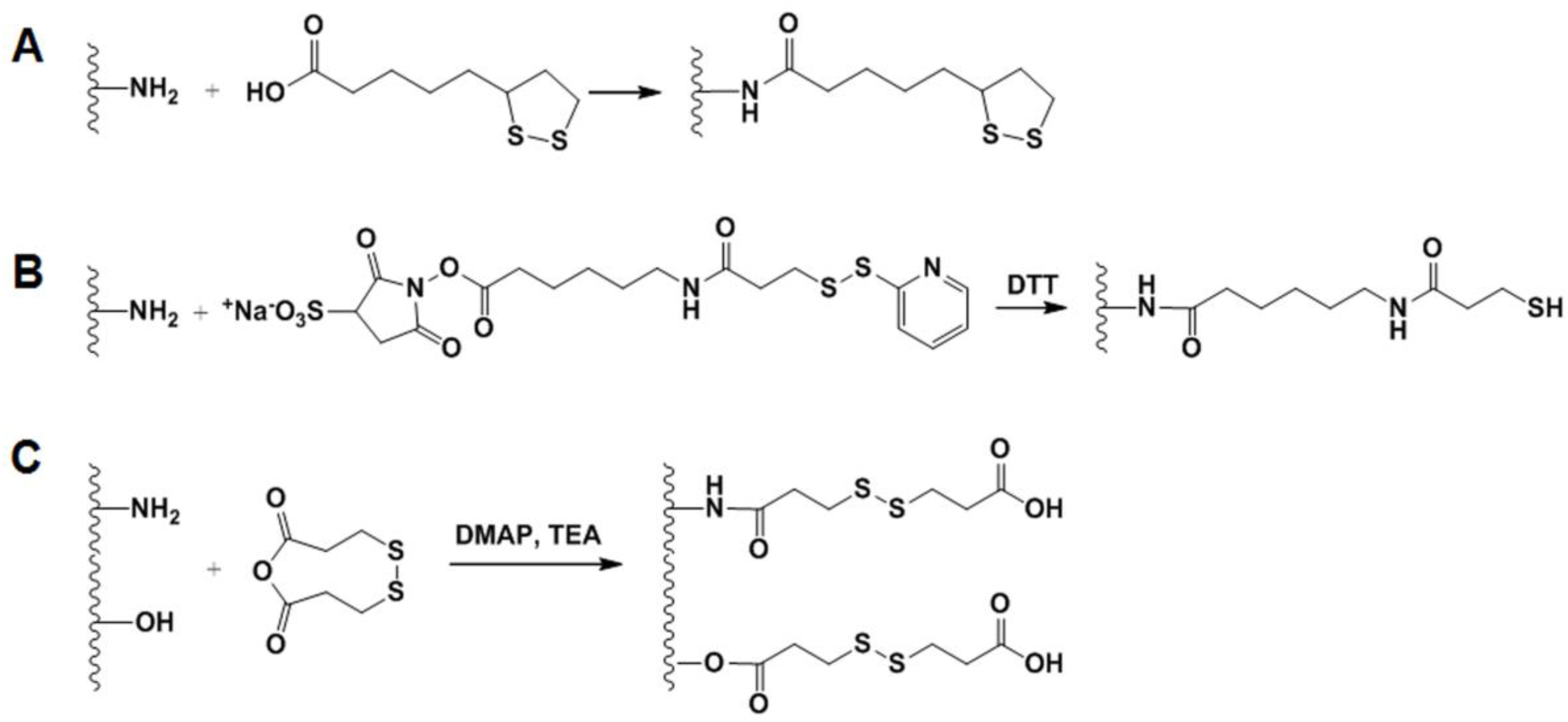
2.4.2. GSH-Sensitive GC Derivatives
2.5. External Stimuli-Sensitive GC Derivatives
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Xiao, Z.; Valencia, P.M.; Radovic-Moreno, A.F.; Farokhzad, O.C. Targeted polymeric therapeutic nanoparticles: Design, development and clinical translation. Chem. Soc. Rev. 2012, 41, 2971–3010. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.K.; Park, O.K.; Lee, A.; Yang, D.H.; Park, K. Glycol chitosan-based fluorescent theranostic nanoagents for cancer therapy. Mar. Drugs 2014, 12, 6038–6057. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2003, 2, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jiao, Y.; Wang, Y.; Zhou, C.; Zhang, Z. Polysaccharides-based nanoparticles as drug delivery systems. Adv. Drug Deliv. Rev. 2008, 60, 1650–1662. [Google Scholar] [CrossRef] [PubMed]
- Riva, R.; Ragelle, H.; des Rieux, A.; Duhem, N.; Jérôme, C.; Préat, V. Chitosan and chitosan derivatives in drug delivery and tissue engineering. Adv. Polym. Sci. 2011, 244, 19–44. [Google Scholar]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Illum, L. Chitosan and its use as a pharmaceutical excipient. Pharm. Res. 1998, 15, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Varshosaz, J. The promise of chitosan microspheres in drug delivery systems. Expert Opin. Drug Deliv. 2007, 4, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Madihally, S.V.; Matthew, H.W. Porous chitosan scaffolds for tissue engineering. Biomaterials 1999, 20, 1133–1142. [Google Scholar] [CrossRef]
- Ong, S.Y.; Wu, J.; Moochhala, S.M.; Tan, M.H.; Lu, J. Development of a chitosan-based wound dressing with improved hemostatic and antimicrobial properties. Biomaterials 2008, 29, 4323–4332. [Google Scholar] [CrossRef] [PubMed]
- Mourya, V.K.; Inamdar, N.N. Chitosan-modifications and applcations: Opportunities galore. React. Funct. Polym. 2008, 68, 1013–1051. [Google Scholar] [CrossRef]
- Trapani, A.; Sitterberg, J.; Bakowsky, U.; Kissel, T. The potential of glycol chitosan nanoparticles as carrier for low water soluble drugs. Int. J. Pharm. 2009, 375, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Saravanakumar, G.; Kim, K.; Kwon, I.C. Targeted delivery of low molecular drugs using chitosan and its derivatives. Adv. Drug Deliv. Rev. 2010, 62, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.K.; Shapka, S.N.; Amsden, B.G. Structure, depolymerization, and cytotompatibility evaluation of glycol chitosan. J. Biomed. Mater. Res. A 2007, 83, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.K.; Dutta, J.; Tripathi, V.S. Chitin and chitosan: Chemistry, properties and appications. J. Sci. Ind. Res. 2004, 63, 20–31. [Google Scholar]
- Son, Y.J.; Jang, J.S.; Cho, Y.W.; Chung, H.; Park, R.W.; Kwon, I.C.; Kim, I.S.; Park, J.Y.; Seo, S.B.; Park, C.R.; et al. Biodistribution and anti-tumor efficacy of doxorubicin loaded glycol-chitosan nanoaggregates by epr effect. J. Control. Release 2003, 91, 135–145. [Google Scholar] [CrossRef]
- Hyung Park, J.; Kwon, S.; Lee, M.; Chung, H.; Kim, J.H.; Kim, Y.S.; Park, R.W.; Kim, I.S.; Bong Seo, S.; Kwon, I.C.; et al. Self-assembled nanoparticles based on glycol chitosan bearing hydrophobic moieties as carriers for doxorubicin: In vivo biodistribution and anti-tumor activity. Biomaterials 2006, 27, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, Y.S.; Kim, S.; Park, J.H.; Kim, K.; Choi, K.; Chung, H.; Jeong, S.Y.; Park, R.W.; Kim, I.S.; et al. Hydrophobically modified glycol chitosan nanoparticles as carriers for paclitaxel. J. Control. Release 2006, 111, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kwon, S.; Nam, J.O.; Park, R.W.; Chung, H.; Seo, S.B.; Kim, I.S.; Kwon, I.C.; Jeong, S.Y. Self-assembled nanoparticles based on glycol chitosan bearing 5beta-cholanic acid for rgd peptide delivery. J. Control. Release 2004, 95, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, Y.S.; Park, K.; Kang, E.; Lee, S.; Nam, H.Y.; Kim, K.; Park, J.H.; Chi, D.Y.; Park, R.W.; et al. Self-assembled glycol chitosan nanoparticles for the sustained and prolonged delivery of antiangiogenic small peptide drugs in cancer therapy. Biomaterials 2008, 29, 1920–1930. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.S.; Lee, J.E.; Chung, H.; Kwon, I.C.; Jeong, S.Y. Self-assembled nanoparticles containing hydrophobically modified glycol chitosan for gene delivery. J. Control. Release 2005, 103, 235–243. [Google Scholar] [PubMed]
- Lee, K.Y.; Kwon, I.C.; Kim, Y.H.; Jo, W.H.; Jeong, S.Y. Preparation of chitosan self-aggregates as a gene delivery system. J. Control. Release 1998, 51, 213–220. [Google Scholar] [CrossRef]
- Saravanakumar, G.; Min, K.H.; Min, D.S.; Kim, A.Y.; Lee, C.M.; Cho, Y.W.; Lee, S.C.; Kim, K.; Jeong, S.Y.; Park, K.; et al. Hydrotropic oligomer-conjugated glycol chitosan as a carrier of paclitaxel: Synthesis, characterization, and in vivo biodistribution. J. Control. Release 2009, 140, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.; Min, K.H.; Lee, S.C.; Park, J.H.; Park, K.; Jeong, S.Y.; Choi, K.; Kwon, I.C.; Kim, K. Enhanced drug-loading and therapeutic efficacy of hydrotropic oligomer-conjugated glycol chitosan nanoparticles for tumor-targeted paclitaxel delivery. J. Control. Release 2013, 172, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, Y.S.; Park, K.; Lee, S.; Nam, H.Y.; Min, K.H.; Jo, H.G.; Park, J.H.; Choi, K.; Jeong, S.Y.; et al. Antitumor efficacy of cisplatin-loaded glycol chitosan nanoparticles in tumor-bearing mice. J. Control. Release 2008, 127, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Min, K.H.; Park, K.; Kim, Y.S.; Bae, S.M.; Lee, S.; Jo, H.G.; Park, R.W.; Kim, I.S.; Jeong, S.Y.; Kim, K.; et al. Hydrophobically modified glycol chitosan nanoparticles-encapsulated camptothecin enhance the drug stability and tumor targeting in cancer therapy. J. Control. Release 2008, 127, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.Y.; Kim, I.S.; Kwon, I.C.; Kim, Y.H. Tumor targetability and antitumor effect of docetaxel-loaded hydrophobically modified glycol chitosan nanoparticles. J. Control. Release 2008, 128, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.W.; Guo, H.Y.; Chen, Z.; Yu, Z.W.; Wang, Z.; Wu, F.G. In situ visualization of lipid raft domains by fluorescent glycol chitosan derivatives. Langmuir ACS J. Surf. Colloids 2016, 32, 6739–6745. [Google Scholar] [CrossRef] [PubMed]
- Huh, M.S.; Lee, S.Y.; Park, S.; Lee, S.; Chung, H.; Lee, S.; Choi, Y.; Oh, Y.K.; Park, J.H.; Jeong, S.Y.; et al. Tumor-homing glycol chitosan/polyethylenimine nanoparticles for the systemic delivery of sirna in tumor-bearing mice. J. Control. Release 2010, 144, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Park, H.I.; Choi, J.S. Novel glycol chitosan-based polymeric gene carrier synthesized by a michael addition reaction with low molecular weight polyethylenimine. Carbohydr. Polym. 2016, 137, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.; Lee, Y.H.; Lee, S.; Han, J.; Ko, K.S.; Choi, J.S. Characterization of glycol chitosan grafted with low molecular weight polyethylenimine as a gene carrier for human adipose-derived mesenchymal stem cells. Carbohydr. Polym. 2016, 153, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Park, K.; Oh, Y.K.; Kwon, S.H.; Her, S.; Kim, I.S.; Choi, K.; Lee, S.J.; Kim, H.; Lee, S.G.; et al. Tumor specificity and therapeutic efficacy of photosensitizer-encapsulated glycol chitosan-based nanoparticles in tumor-bearing mice. Biomaterials 2009, 30, 2929–2939. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Koo, H.; Jeong, H.; Huh, M.S.; Choi, Y.; Jeong, S.Y.; Byun, Y.; Choi, K.; Kim, K.; Kwon, I.C. Comparative study of photosensitizer loaded and conjugated glycol chitosan nanoparticles for cancer therapy. J. Control. Release 2011, 152, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Chiang, L.Y.; Hamblin, M.R. Photodynamic therapy with fullerenes in vivo: Reality or a dream? Nanomedicine 2011, 6, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.W.; Wilson, S.R.; Schuster, D.I. Biological applications of fullerenes. Bioorg. Med. Chem. 1996, 4, 767–779. [Google Scholar] [CrossRef]
- Kwag, D.S.; Oh, N.M.; Oh, Y.T.; Oh, K.T.; Youn, Y.S.; Lee, E.S. Photodynamic therapy using glycol chitosan grafted fullerenes. Int. J. Pharm. 2012, 431, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.V.; Prescher, J.A.; Sletten, E.M.; Baskin, J.M.; Miller, I.A.; Agard, N.J.; Lo, A.; Bertozzi, C.R. Copper-free click chemistry in living animals. Proc. Natl. Acad. Sci. USA 2010, 107, 1821–1826. [Google Scholar] [CrossRef] [PubMed]
- Prescher, J.A.; Bertozzi, C.R. Chemistry in living systems. Nat. Chem. Biol. 2005, 1, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Agard, N.J.; Prescher, J.A.; Bertozzi, C.R. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 2004, 126, 15046–15047. [Google Scholar] [CrossRef] [PubMed]
- Ngo, J.T.; Champion, J.A.; Mahdavi, A.; Tanrikulu, I.C.; Beatty, K.E.; Connor, R.E.; Yoo, T.H.; Dieterich, D.C.; Schuman, E.M.; Tirrell, D.A. Cell-selective metabolic labeling of proteins. Nat. Chem. Biol. 2009, 5, 715–717. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Al Zaki, A.; Hui, J.Z.; Muzykantov, V.R.; Tsourkas, A. Multifunctional nanoparticles: Cost versus benefit of adding targeting and imaging capabilities. Science 2012, 338, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Gartner, Z.J.; Bertozzi, C.R. Programmed assembly of 3-dimensional microtissues with defined cellular connectivity. Proc. Natl. Acad. Sci. USA 2009, 106, 4606–4610. [Google Scholar] [CrossRef] [PubMed]
- Bertozzi, C.R.; Kiessling, L.L. Chemical glycobiology. Science 2001, 291, 2357–2364. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Koo, H.; Na, J.H.; Han, S.J.; Min, H.S.; Lee, S.J.; Kim, S.H.; Yun, S.H.; Jeong, S.Y.; Kwon, I.C.; et al. Chemical tumor-targeting of nanoparticles based on metabolic glycoengineering and click chemistry. ACS Nano 2014, 8, 2048–2063. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the epr effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Kang, J.H.; Toita, R.; Katayama, Y. Bio and nanotechnological strategies for tumor-targeted gene therapy. Biotechnol. Adv. 2010, 28, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Dufes, C.; Muller, J.M.; Couet, W.; Olivier, J.C.; Uchegbu, I.F.; Schatzlein, A.G. Anticancer drug delivery with transferrin targeted polymeric chitosan vesicles. Pharm. Res. 2004, 21, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Yhee, J.Y.; Son, S.; Kim, S.H.; Park, K.; Choi, K.; Kwon, I.C. Self-assembled glycol chitosan nanoparticles for disease-specific theranostics. J. Control. Release 2014, 193, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Zitzmann, S.; Mier, W.; Schad, A.; Kinscherf, R.; Askoxylakis, V.; Kramer, S.; Altmann, A.; Eisenhut, M.; Haberkorn, U. A new prostate carcinoma binding peptide (dup-1) for tumor imaging and therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 139–146. [Google Scholar]
- Park, K.; Hong, H.Y.; Moon, H.J.; Lee, B.H.; Kim, I.S.; Kwon, I.C.; Rhee, K. A new atherosclerotic lesion probe based on hydrophobically modified chitosan nanoparticles functionalized by the atherosclerotic plaque targeted peptides. J. Control. Release 2008, 128, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Bae, S.M.; Na, M.H.; Shin, H.; Yang, Y.J.; Min, K.H.; Choi, K.Y.; Kim, K.; Park, R.W.; Kwon, I.C.; et al. Facilitated intracellular delivery of peptide-guided nanoparticles in tumor tissues. J. Control. Release 2012, 157, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.Y.; Kim, J.H.; Oh, G.T.; Lee, B.H.; Kwon, I.C.; Kim, I.S. Molecular targeting of atherosclerotic plaques by a stabilin-2-specific peptide ligand. J. Control. Release 2011, 155, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Galis, Z.S.; Sukhova, G.K.; Lark, M.W.; Libby, P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J. Clin. Investig. 1994, 94, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tung, C.H.; Mahmood, U.; Ntziachristos, V.; Gyurko, R.; Fishman, M.C.; Huang, P.L.; Weissleder, R. In vivo imaging of proteolytic activity in atherosclerosis. Circulation 2002, 105, 2766–2771. [Google Scholar] [CrossRef] [PubMed]
- Kolodgie, F.D.; Petrov, A.; Virmani, R.; Narula, N.; Verjans, J.W.; Weber, D.K.; Hartung, D.; Steinmetz, N.; Vanderheyden, J.L.; Vannan, M.A.; et al. Targeting of apoptotic macrophages and experimental atheroma with radiolabeled annexin v: A technique with potential for noninvasive imaging of vulnerable plaque. Circulation 2003, 108, 3134–3139. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, J.; Hilderbrand, S.A.; Waterman, P.; Heinecke, J.W.; Weissleder, R.; Libby, P. A fluorescent probe for the detection of myeloperoxidase activity in atherosclerosis-associated macrophages. Chem. Biol. 2007, 14, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Park, K.; Ryu, J.; Lee, J.J.; Lee, M.W.; Cho, H.S.; Nam, H.S.; Park, O.K.; Song, J.W.; Kim, T.S.; et al. Intravascular optical imaging of high-risk plaques in vivo by targeting macrophage mannose receptors. Sci. Rep. 2016, 6, 22608. [Google Scholar] [CrossRef] [PubMed]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Mo, R.; Gu, Z. Tumor microenvironment and intracellular signal-activated nanomaterials for anticancer drug delivery. Mater. Today 2016, 9, 274–283. [Google Scholar] [CrossRef]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.T.; Boucrot, E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2011, 12, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Mayor, S.; Pagano, R.E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Oh, N.M.; Oh, K.T.; Baik, H.J.; Lee, B.R.; Lee, A.H.; Youn, Y.S.; Lee, E.S. A self-organized 3-diethylaminopropyl-bearing glycol chitosan nanogel for tumor acidic ph targeting: In vitro evaluation. Colloids Surf. B Biointerfaces 2010, 78, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Baik, H.J.; Oh, Y.T.; Oh, K.T.; Youn, Y.S.; Lee, E.S. A smart polysaccharide/drug conjugate for photodynamic therapy. Angew. Chem. Int. Ed. 2011, 50, 1644–1647. [Google Scholar] [CrossRef] [PubMed]
- Mathiyalagan, R.; Subramaniyam, S.; Kim, Y.J.; Kim, Y.C.; Yang, D.C. Ginsenoside compound k-bearing glycol chitosan conjugates: Synthesis, physicochemical characterization, and in vitro biological studies. Carbohydr. Polym. 2014, 112, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Han, T.H.; Lee, K.Y.; Han, S.S.; Hwang, J.J.; Moon, D.H.; Kim, S.Y.; Cho, Y.W. N-acetyl histidine-conjugated glycol chitosan self-assembled nanoparticles for intracytoplasmic delivery of drugs: Endocytosis, exocytosis and drug release. J. Control. Release 2006, 115, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Park, K.; Park, S.; Kim, G.C.; Kim, H.J.; Lee, S.; Kil, H.; Oh, S.J.; Chi, D.; Kim, K.; et al. Tumor targeting efficiency of bare nanoparticles does not mean the efficacy of loaded anticancer drugs: Importance of radionuclide imaging for optimization of highly selective tumor targeting polymeric nanoparticles with or without drug. J. Control. Release 2010, 147, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, D.J.; Kwag, D.S.; Lee, U.Y.; Youn, Y.S.; Lee, E.S. Acid ph-activated glycol chitosan/fullerene nanogels for efficient tumor therapy. Carbohydr. Polym. 2014, 101, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Pompella, A.; Visvikis, A.; Paolicchi, A.; De Tata, V.; Casini, A.F. The changing faces of glutathione, a cellular protagonist. Biochem. Pharmacol. 2003, 66, 1499–1503. [Google Scholar] [CrossRef]
- Saito, G.; Swanson, J.A.; Lee, K.D. Drug delivery strategy utilizing conjugation via reversible disulfide linkages: Role and site of cellular reducing activities. Adv. Drug Deliv. Rev. 2003, 55, 199–215. [Google Scholar] [CrossRef]
- Hu, Y.W.; Du, Y.Z.; Liu, N.; Liu, X.; Meng, T.T.; Cheng, B.L.; He, J.B.; You, J.; Yuan, H.; Hu, F.Q. Selective redox-responsive drug release in tumor cells mediated by chitosan based glycolipid-like nanocarrier. J. Control. Release 2015, 206, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Kang, Z.; Xue, L.; Shang, Y.; Su, Z.; Sun, H.; Ping, Q.; Mo, R.; Zhang, C. A collaborative assembly strategy for tumor-targeted sirna delivery. J. Am. Chem. Soc. 2015, 137, 6000–6010. [Google Scholar] [CrossRef] [PubMed]
- Oh, I.H.; Min, H.S.; Li, L.; Tran, T.H.; Lee, Y.K.; Kwon, I.C.; Choi, K.; Kim, K.; Huh, K.M. Cancer cell-specific photoactivity of pheophorbide a-glycol chitosan nanoparticles for photodynamic therapy in tumor-bearing mice. Biomaterials 2013, 34, 6454–6463. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Yu, J.; Feng, X.; Li, W.; Wang, Y.; Jin, H.; Huang, H.; Liu, Y.; Fan, D. Reduction-responsive core-crosslinked micelles based on a glyco-chitosan-lipoic acid conjugate for triggered release of doxorubicin. RSC Adv. 2016, 6, 31391–31400. [Google Scholar] [CrossRef]
- Lee, S.Y.; Huh, M.S.; Lee, S.; Lee, S.J.; Chung, H.; Park, J.H.; Oh, Y.K.; Choi, K.; Kim, K.; Kwon, I.C. Stability and cellular uptake of polymerized sirna (poly-sirna)/polyethylenimine (pei) complexes for efficient gene silencing. J. Control. Release 2010, 141, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Huh, M.S.; Lee, S.Y.; Min, S.; Lee, S.; Koo, H.; Chu, J.U.; Lee, K.E.; Jeon, H.; Choi, Y.; et al. Tumor-homing poly-sirna/glycol chitosan self-cross-linked nanoparticles for systemic sirna delivery in cancer treatment. Angew. Chem. Int. Ed. 2012, 51, 7203–7207. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Lee, A.; Hwang, S.R.; Park, J.S.; Jang, J.; Huh, M.S.; Jo, D.G.; Yoon, S.Y.; Byun, Y.; Kim, S.H.; et al. Tnf-alpha gene silencing using polymerized sirna/thiolated glycol chitosan nanoparticles for rheumatoid arthritis. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Yook, S.; Yhee, J.Y.; Yoon, H.Y.; Kim, M.G.; Ku, S.H.; Kim, S.H.; Park, J.H.; Jeong, J.H.; Kwon, I.C.; et al. Co-delivery of vegf and bcl-2 dual-targeted sirna polymer using a single nanoparticle for synergistic anti-cancer effects in vivo. J. Control. Release 2015, 220, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Yhee, J.Y.; Song, S.; Lee, S.J.; Park, S.G.; Kim, K.S.; Kim, M.G.; Son, S.; Koo, H.; Kwon, I.C.; Jeong, J.H.; et al. Cancer-targeted mdr-1 sirna delivery using self-cross-linked glycol chitosan nanoparticles to overcome drug resistance. J. Control. Release 2015, 198, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.G.; Jo, S.D.; Yhee, J.Y.; Lee, B.S.; Lee, S.J.; Park, S.G.; Kang, S.W.; Kim, S.H.; Jeong, J.H. Synergistic anti-tumor effects of bevacizumab and tumor targeted polymerized vegf sirna nanoparticles. Biochem. Biophys. Res. Commun. 2017, 489, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Ku, S.H.; Kim, M.J.; Lee, S.J.; Kim, H.C.; Kim, K.; Kim, S.H.; Kwon, I.C. Rolling circle transcription-based polymeric sirna nanoparticles for tumor-targeted delivery. J. Control. Release 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Taranejoo, S.; Chandrasekaran, R.; Cheng, W.; Hourigan, K. Bioreducible pei-functionalized glycol chitosan: A novel gene vector with reduced cytotoxicity and improved transfection efficiency. Carbohydr. Polym. 2016, 153, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Huang, W.; Wang, D.; Huang, X.; Zhu, X.; Yan, D. Chitosan-based nanocarriers with ph and light dual response for anticancer drug delivery. Biomacromolecules 2013, 14, 2601–2610. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.O.; Li, Z.; Shim, H.E.; Cho, I.S.; Nurunabi, M.; Park, H.; Lee, K.Y.; Moon, S.H.; Kim, K.S.; Kang, S.W.; et al. Bioinspired tuning of glycol chitosan for 3d cell culture. NPG Asia Mater. 2016, 8, e309. [Google Scholar] [CrossRef]
- Feng, J.; Chen, Y.; Li, F.; Cui, L.; Shi, N.; Kong, W.; Zhang, Y. Synthesis, characterization and in vitro evaluation of a novel glycol chitosan-edta conjugate to inhibit aminopeptidase-mediated deradation of thymopoietin oligopeptides. Molecules 2017, 22, 1253. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.E.; Kim, H.-J.; Rhee, J.-K.; Park, K. Versatile Chemical Derivatizations to Design Glycol Chitosan-Based Drug Carriers. Molecules 2017, 22, 1662. https://doi.org/10.3390/molecules22101662
Kim SE, Kim H-J, Rhee J-K, Park K. Versatile Chemical Derivatizations to Design Glycol Chitosan-Based Drug Carriers. Molecules. 2017; 22(10):1662. https://doi.org/10.3390/molecules22101662
Chicago/Turabian StyleKim, Sung Eun, Hak-Jun Kim, Jin-Kyu Rhee, and Kyeongsoon Park. 2017. "Versatile Chemical Derivatizations to Design Glycol Chitosan-Based Drug Carriers" Molecules 22, no. 10: 1662. https://doi.org/10.3390/molecules22101662