Novel FXa Inhibitor Identification through Integration of Ligand- and Structure-Based Approaches

 ,
,
Abstract
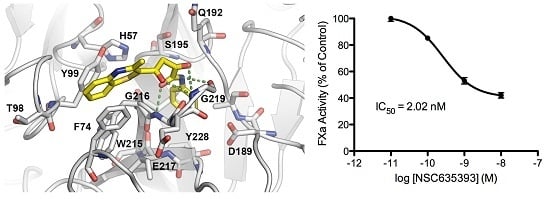
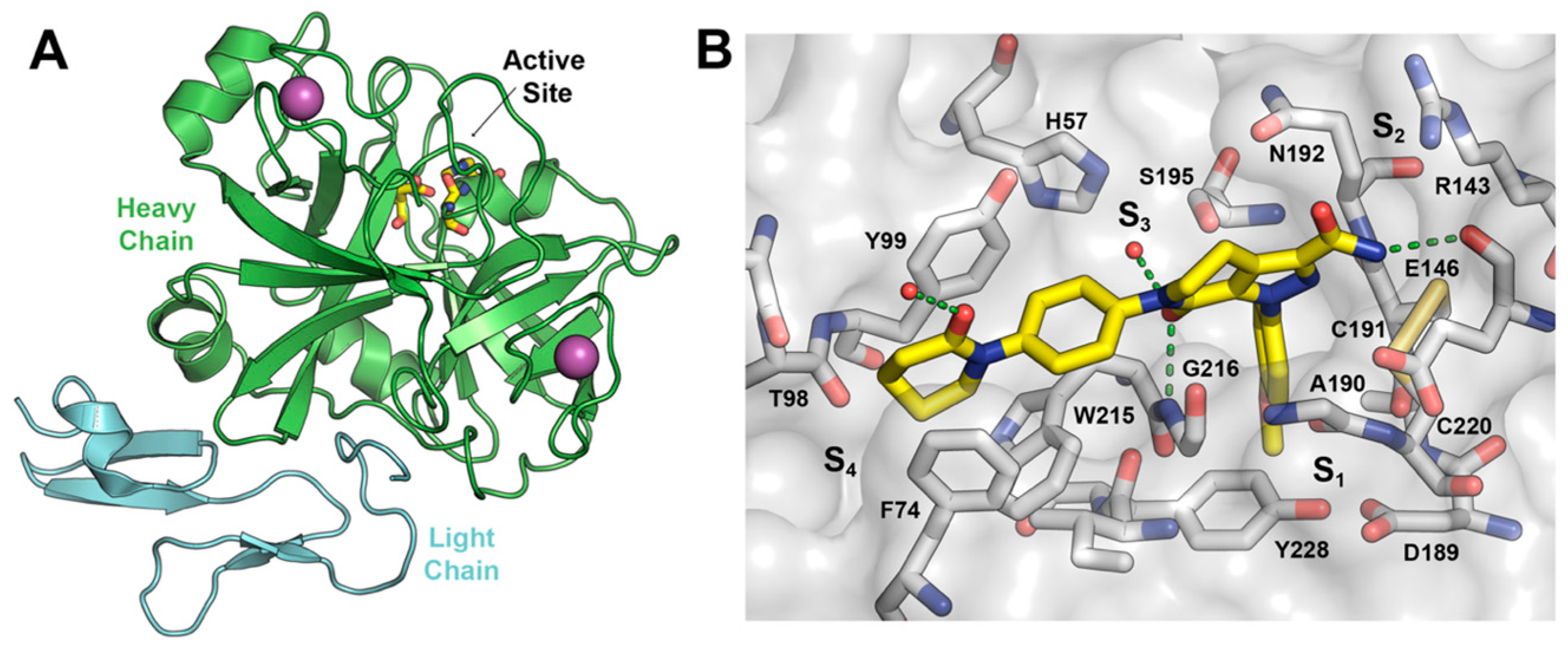
:
1. Introduction
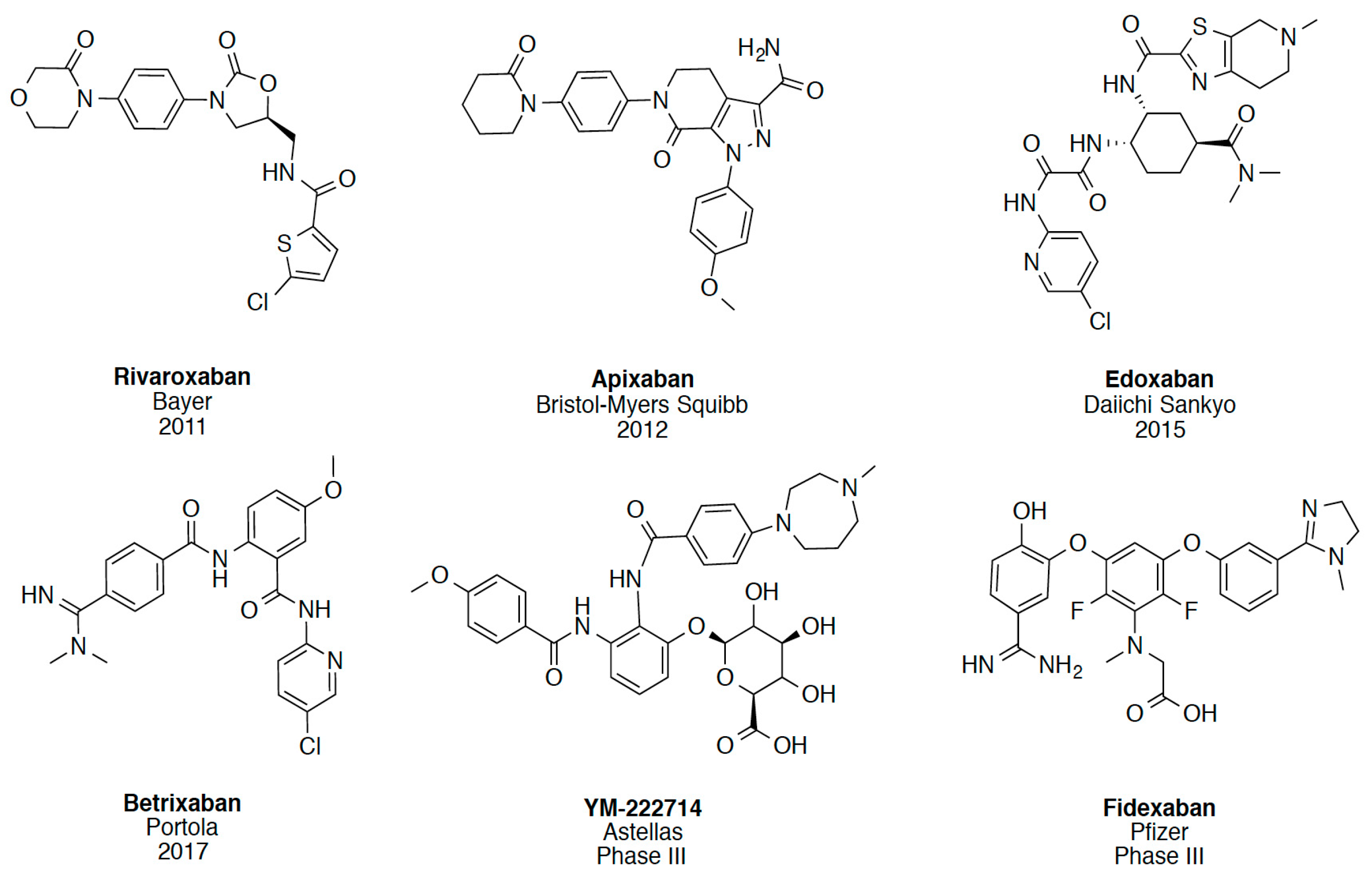
2. Results and Discussion
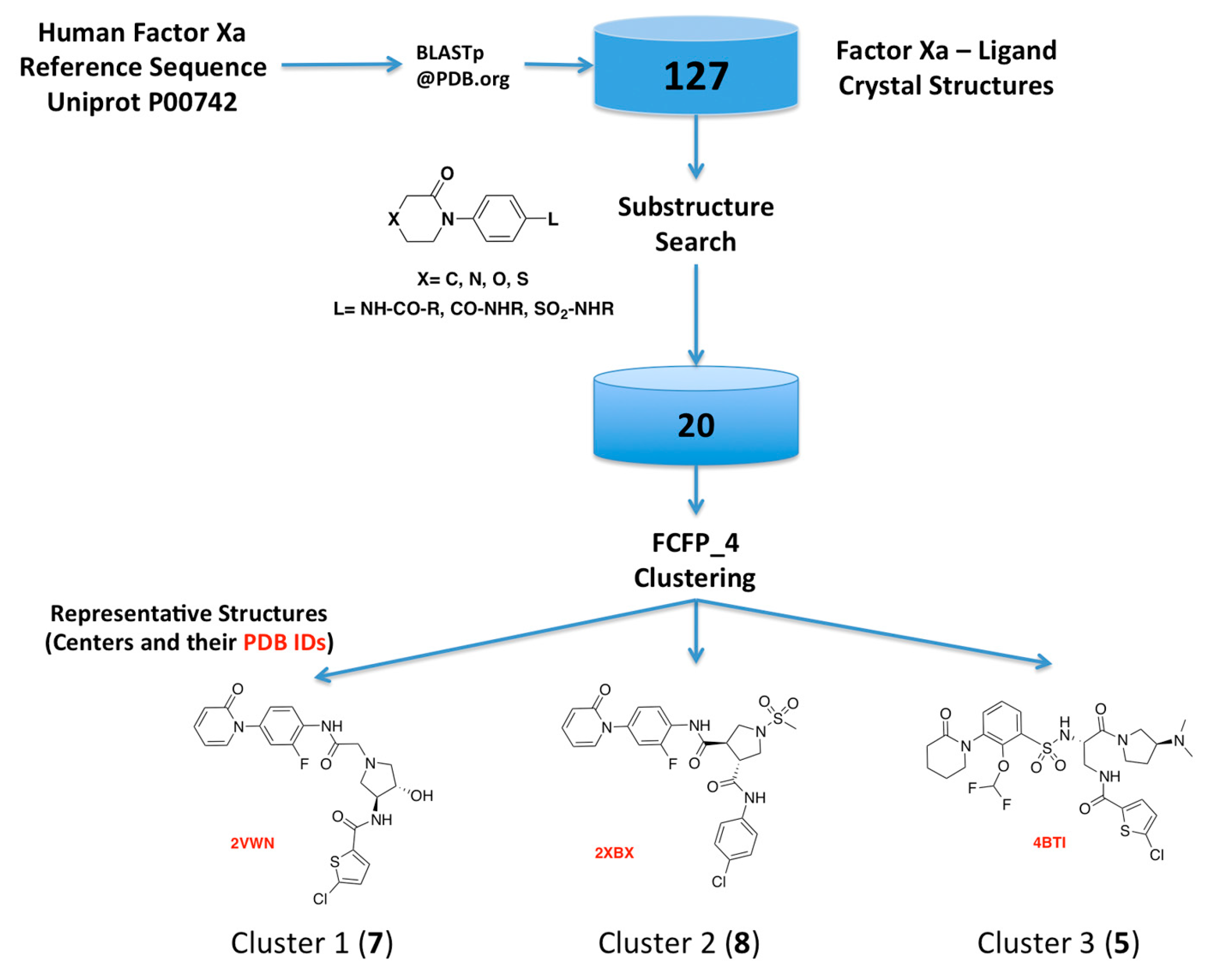
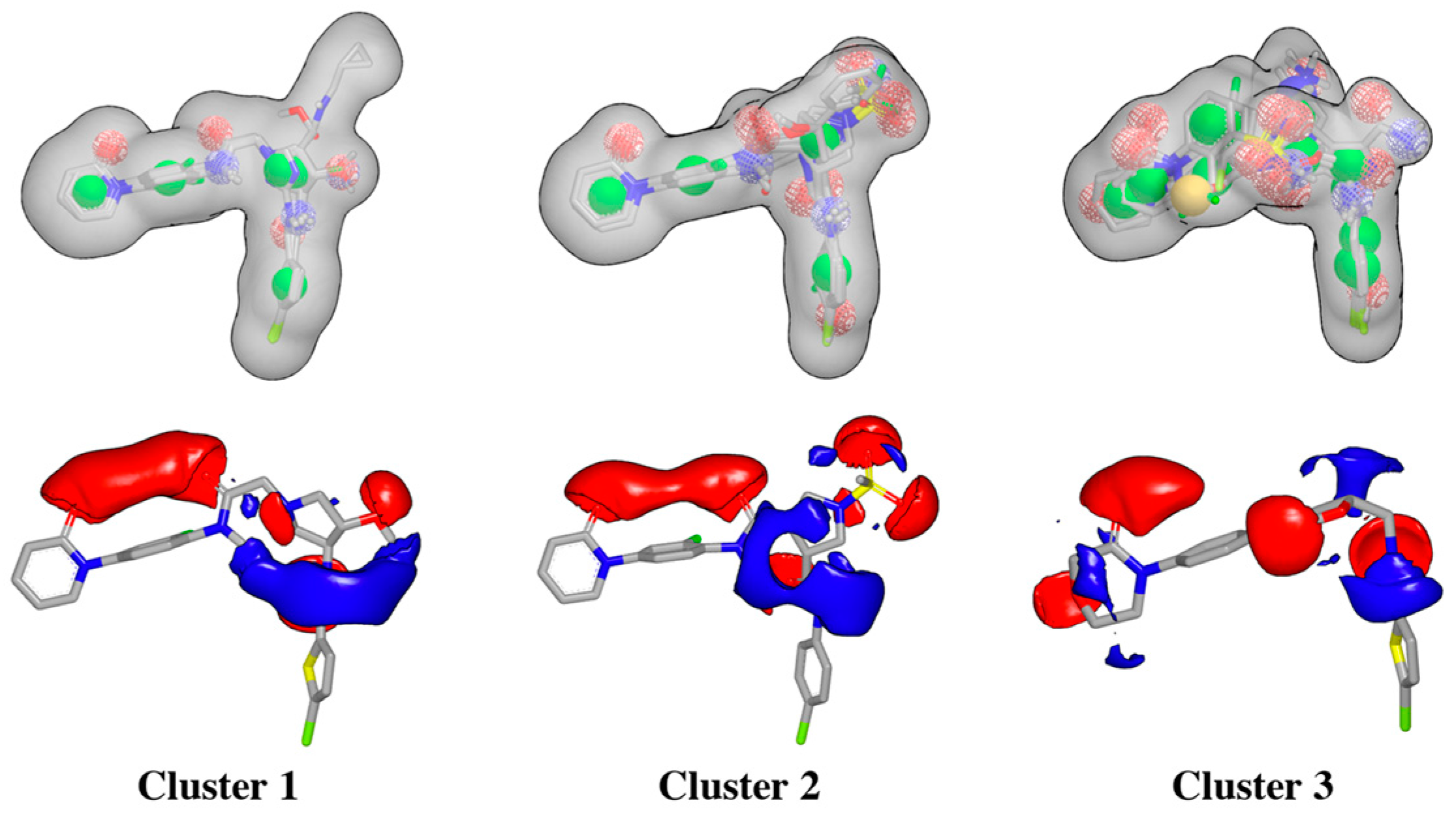
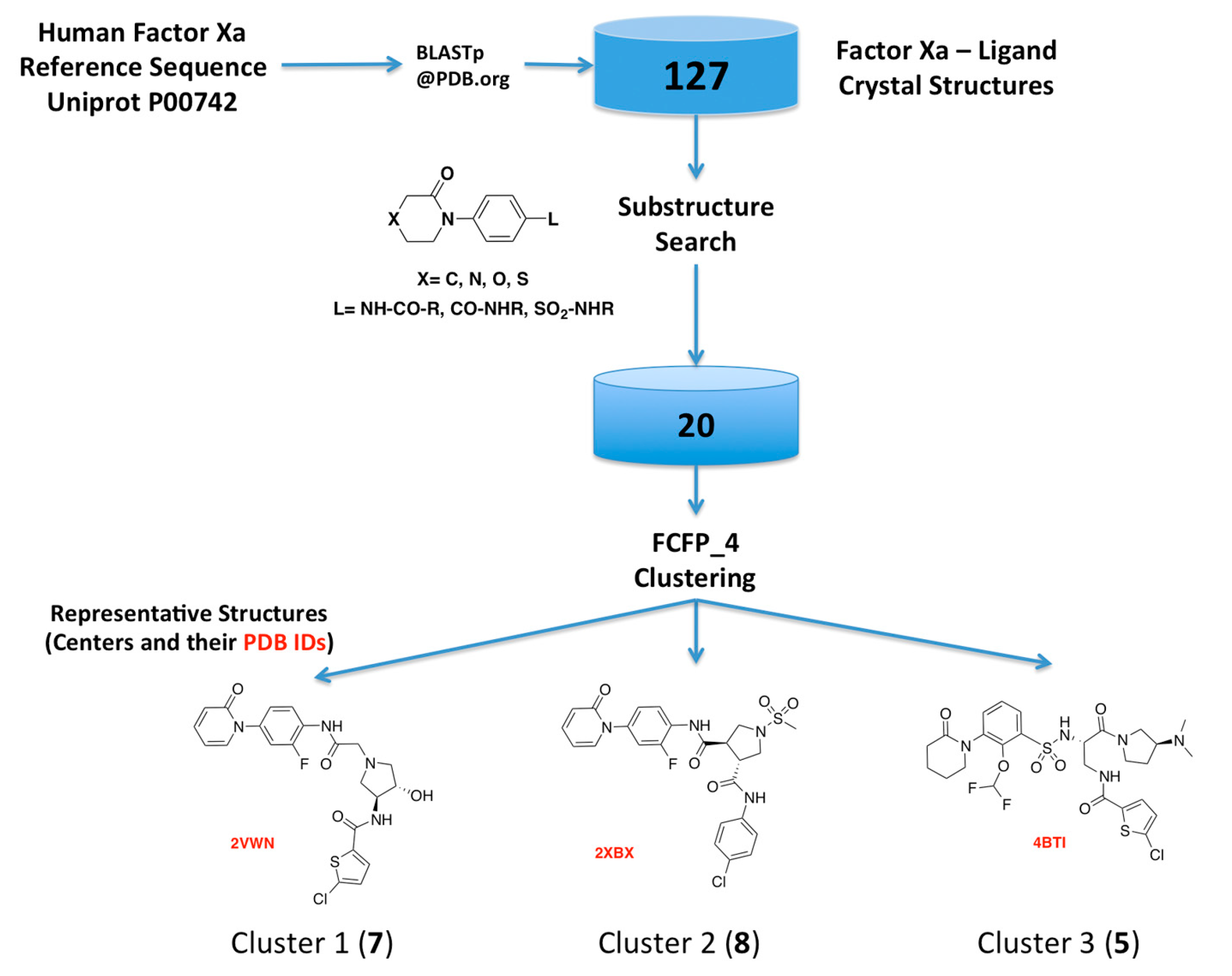
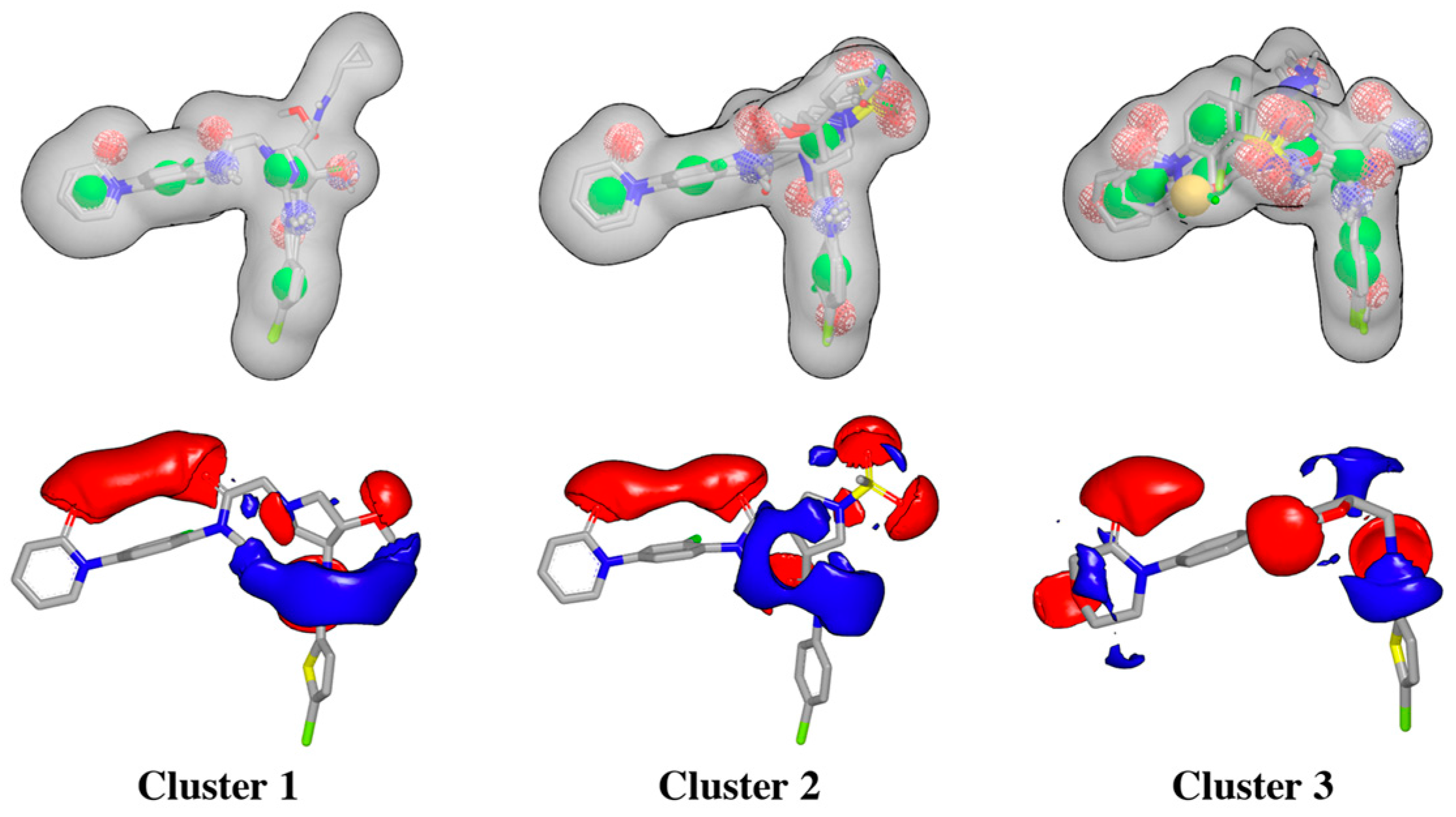
2.1. Protein-Ligand Complexes Selection and Ligand Clustering
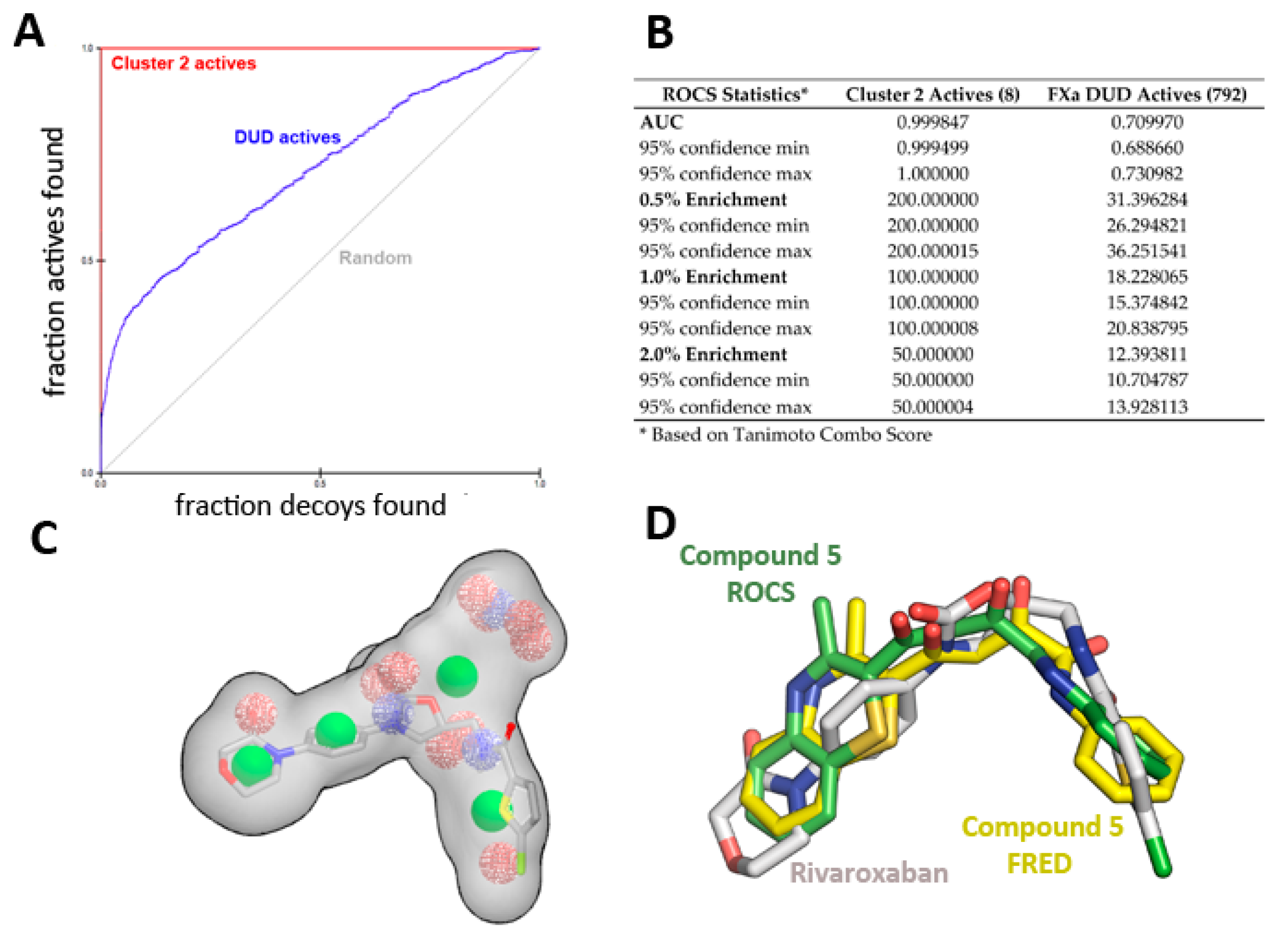
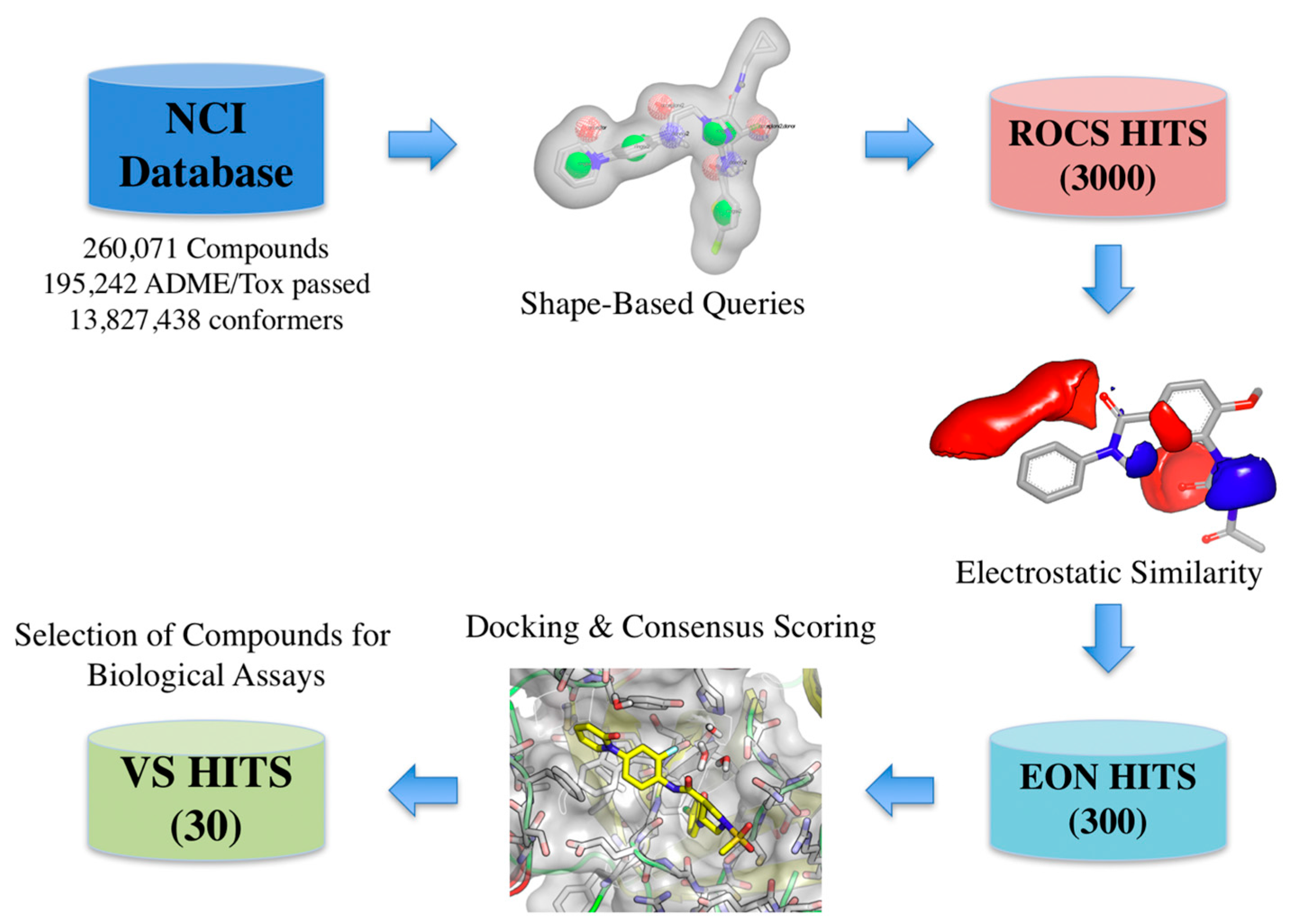
2.2. Virtual Screening and Compound Selection
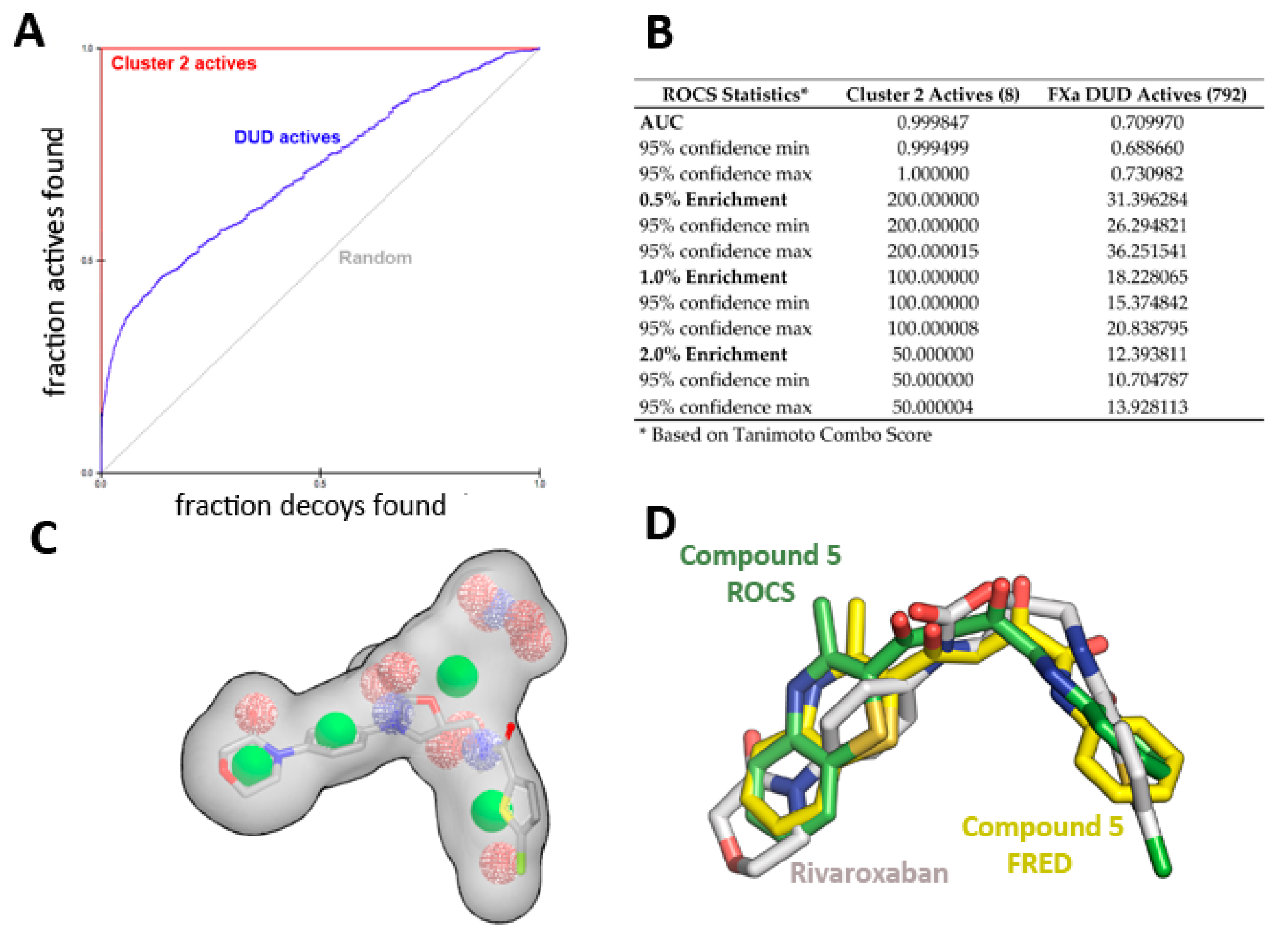
Shape and Electrostatic-Based Queries Development and Validation
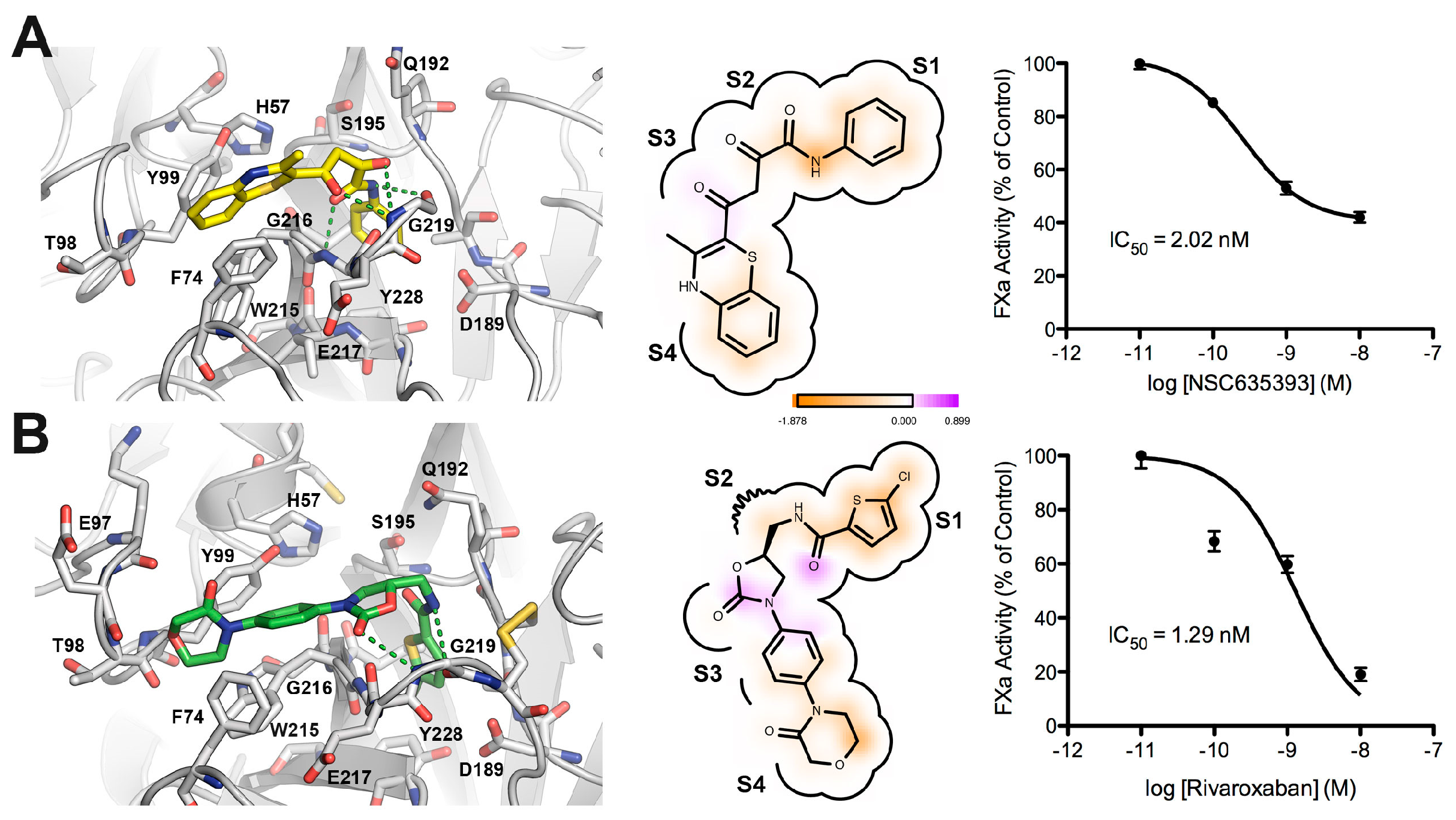
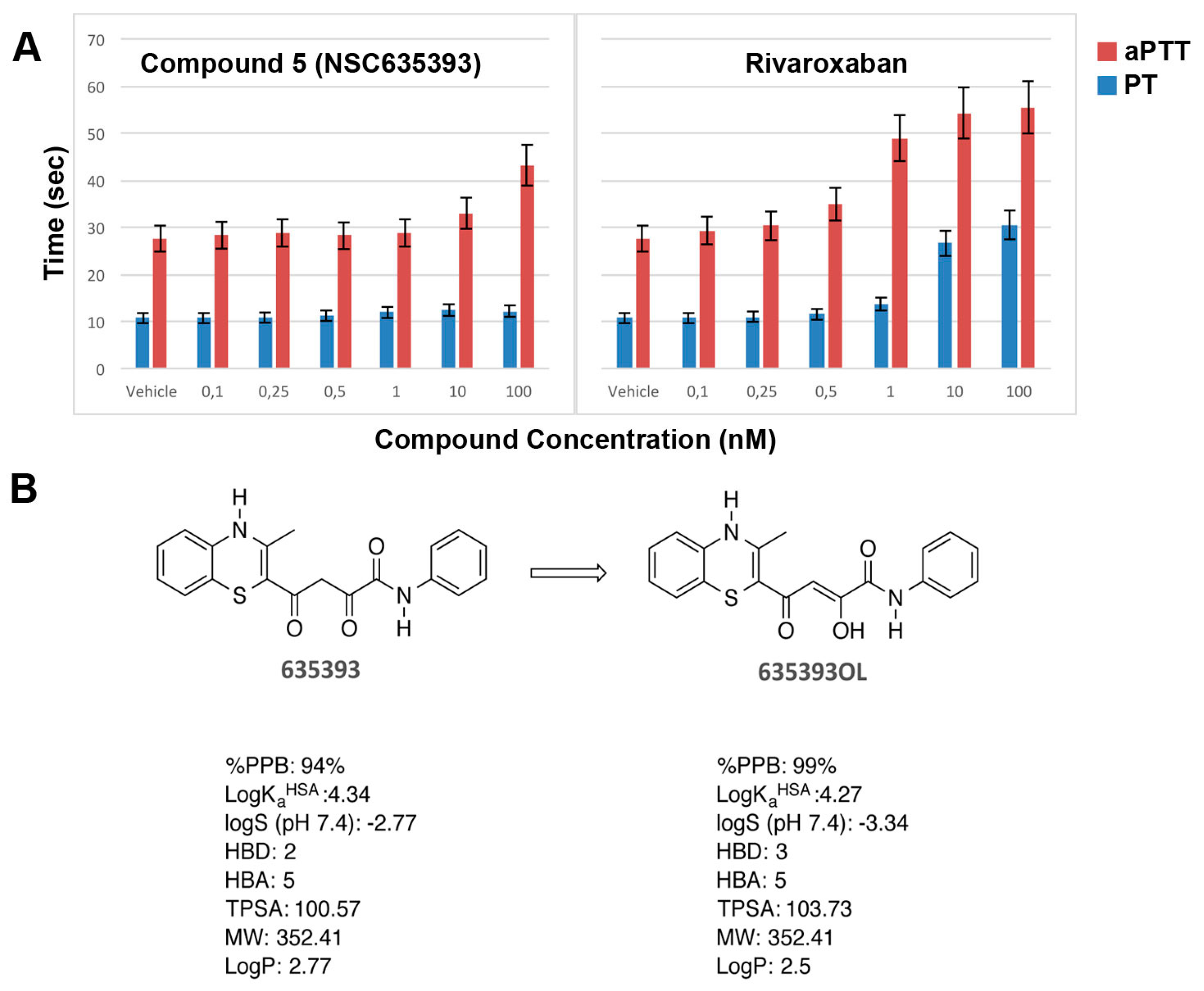
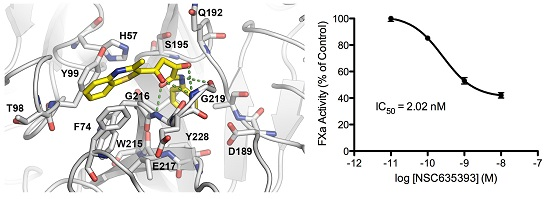
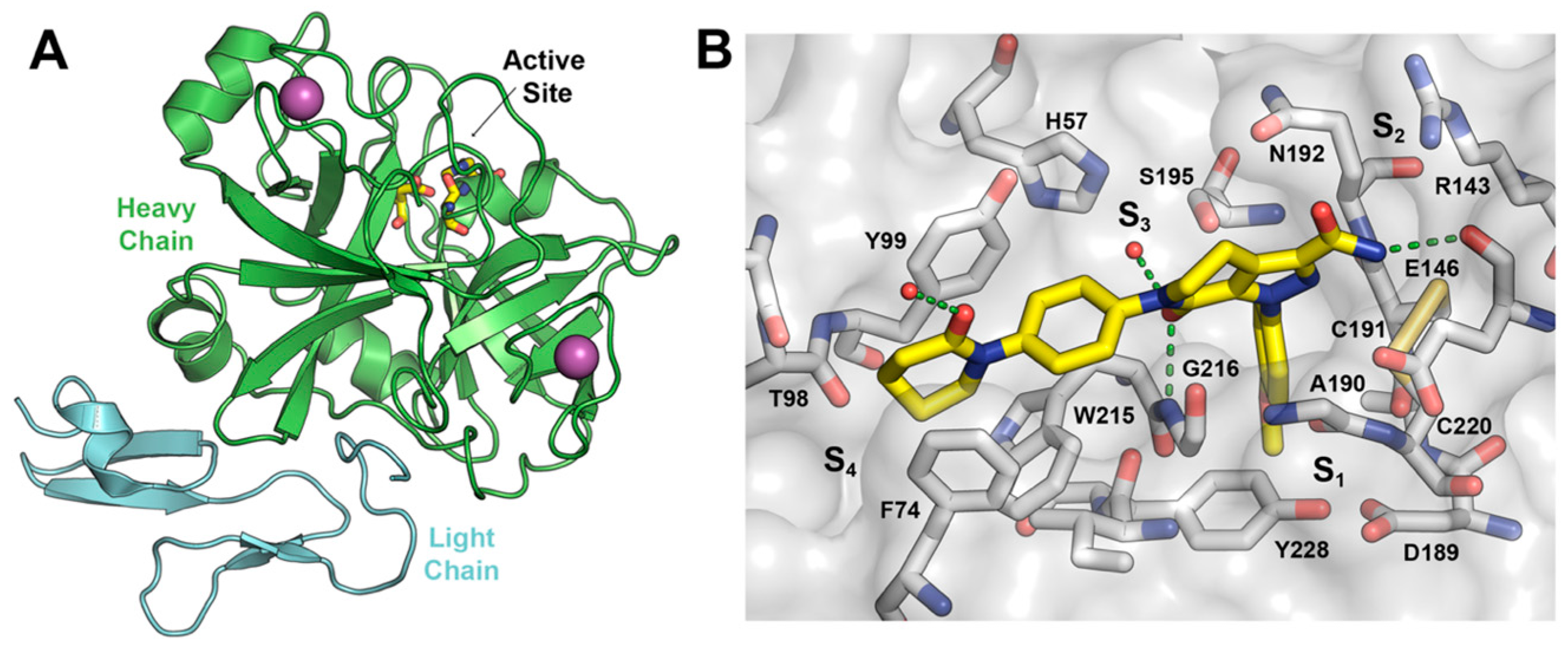
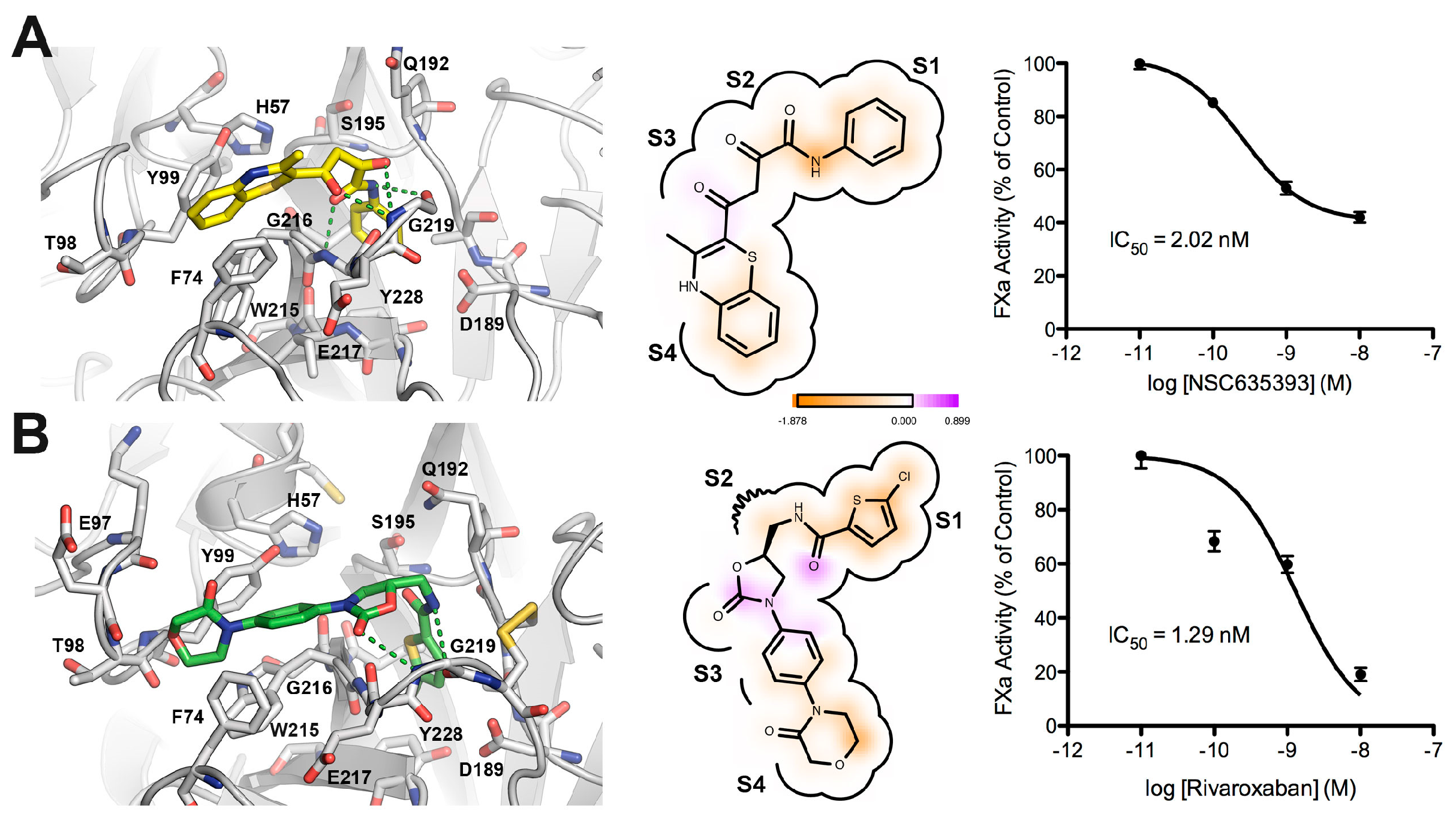
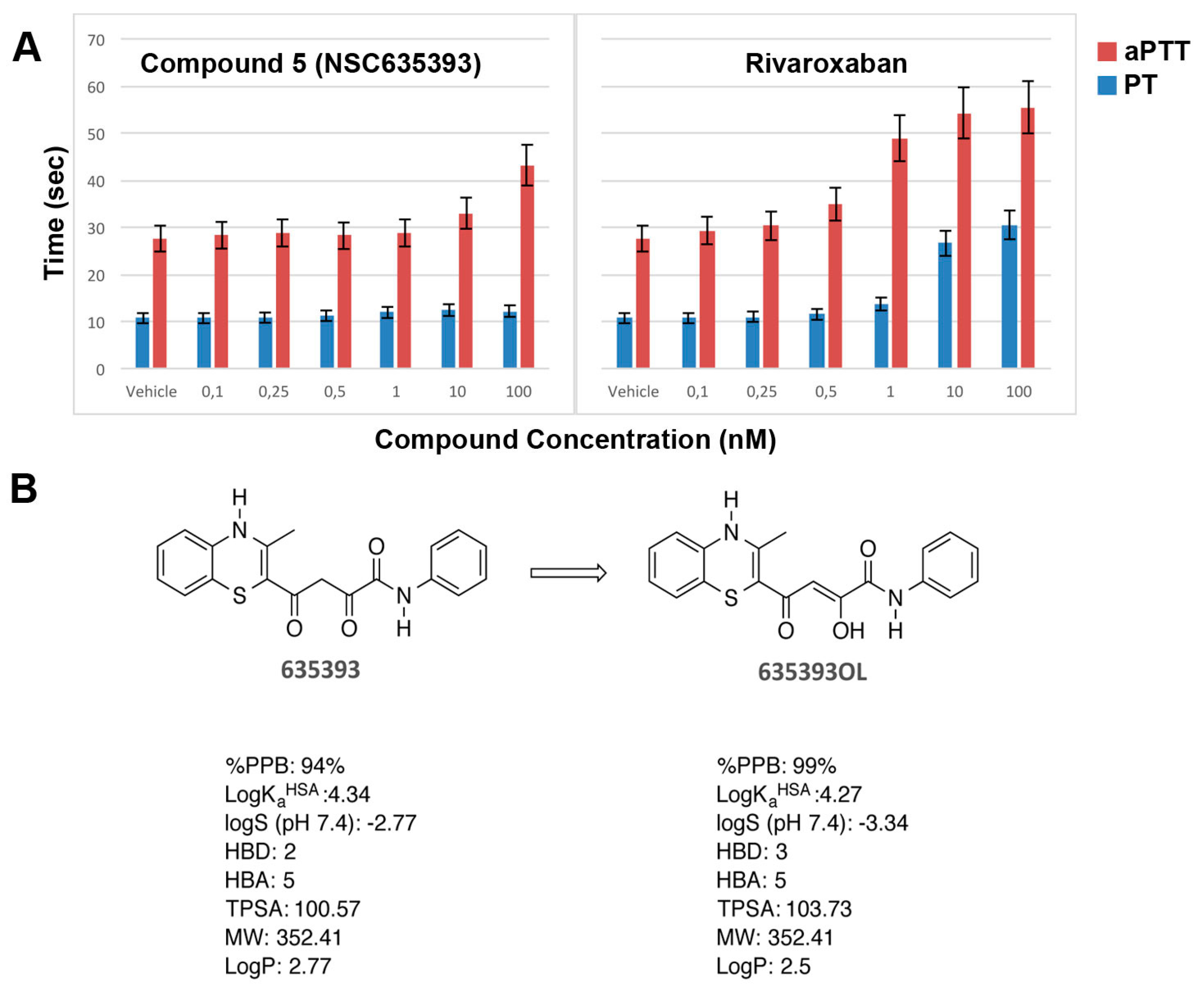
2.3. Biological Evaluation of FXa Inhibition
3. Materials and Methods
3.1. FXa-Ligand Structure Retrieval and Standardization
3.2. Shape-Based Query Generation, Validation, and Screening with Electrostatic Similarity Filtering
3.3. Docking of Primary Shape/Electrostatic-Based Hits
3.4. Inhibition of FXa In Vitro
3.5. In Vitro Coagulation Assays
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mani, H.; Lindhoff-Last, E. New oral anticoagulants in patients with nonvalvular atrial fibrillation: A review of pharmacokinetics, safety, efficacy, quality of life, and cost effectiveness. Drug Des. Dev. Ther. 2014, 8, 789–798. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Global Status Report on Noncommunicable Diseases; WHO Publishing: Geneva, Switzerland, 2014. [Google Scholar]
- Lee, Y.K.; Player, M.R. Developments in factor Xa inhibitors for the treatment of thromboembolic disorders. Med. Res. Rev. 2011, 31, 202–283. [Google Scholar] [CrossRef] [PubMed]
- Alquwaizani, M.; Buckley, L.; Adams, C.; Fanikos, J. Anticoagulants: A Review of the Pharmacology, Dosing, and Complications. Curr. Emerg. Hosp. Med. Rep. 2013, 1, 83–97. [Google Scholar] [CrossRef] [PubMed]
- De Candia, M.; Lopopolo, G.; Altomare, C. Novel factor Xa inhibitors: A patent review. Expert Opin. Ther. Pat. 2009, 19, 1535–1580. [Google Scholar] [CrossRef] [PubMed]
- Ansell, J.E. Reversing the Effect of Oral Anticoagulant Drugs: Established and Newer Options. Am. J. Cardiovasc. Drugs 2016, 16, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ghadimi, K.; Dombrowski, K.E.; Levy, J.H.; Welsby, I.J. Andexanet alfa for the reversal of Factor Xa inhibitor related anticoagulation. Expert Rev. Hematol. 2016, 9, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Mavrakanas, T.; Bounameaux, H. The potential role of new oral anticoagulants in the prevention and treatment of thromboembolism. Pharmacol. Ther. 2011, 130, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Quan, M.L.; Glunz, P.W.; Smallheer, J.M. Advances in Anticoagulants A2—Chackalamannil, Samuel. In Comprehensive Medicinal Chemistry III; Rotella, D., Ward, S.E., Eds.; Elsevier: Oxford, UK, 2017; pp. 600–627. [Google Scholar]
- Acanfora, D.; Acanfora, C.; Scicchitano, P.; Longobardi, M.; Furgi, G.; Casucci, G.; Lanzillo, B.; Dentamaro, I.; Zito, A.; Incalzi, R.A.; et al. Safety and Feasibility of Treatment with Rivaroxaban for Non-Canonical Indications: A Case Series Analysis. Clin. Drug Investig. 2016, 36, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Lip, G.Y.H.; Pan, X.; Kamble, S.; Kawabata, H.; Mardekian, J.; Masseria, C.; Bruno, A.; Phatak, H. Major bleeding risk among non-valvular atrial fibrillation patients initiated on apixaban, dabigatran, rivaroxaban or warfarin: A “real-world” observational study in the United States. Int. J. Clin. Pract. 2016, 70, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Vene, N.; Mavri, A.; Gubenšek, M.; Tratar, G.; Vižintin Cuderman, T.; Pohar Perme, M.; Blinc, A. Risk of Thromboembolic Events in Patients with Non-Valvular Atrial Fibrillation After Dabigatran or Rivaroxaban Discontinuation—Data from the Ljubljana Registry. PLoS ONE 2016, 11, e0156943. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, L. Serine Protease Mechanism and Specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef] [PubMed]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- The Protein Databank. Available online: http://www.rcsb.org/pdb (accessed on 28 August 2017).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Forli, S. Charting a Path to Success in Virtual Screening. Molecules 2015, 20, 18732–18758. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Di Giovanni, C. Virtual screening strategies in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef] [PubMed]
- Krovat, E.M.; Fruhwirth, K.H.; Langer, T. Pharmacophore identification, in silico screening, and virtual library design for inhibitors of the human factor Xa. J. Chem. Inf. Model. 2005, 45, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Markt, P.; Petersen, R.K.; Flindt, E.N.; Kristiansen, K.; Kirchmair, J.; Spitzer, G.; Distinto, S.; Schuster, D.; Wolber, G.; Laggner, C.; et al. Discovery of Novel PPAR Ligands by a Virtual Screening Approach Based on Pharmacophore Modeling, 3D Shape, and Electrostatic Similarity Screening. J. Med. Chem. 2008, 51, 6303–6317. [Google Scholar] [CrossRef] [PubMed]
- Muchmore, S.W.; Souers, A.J.; Akritopoulou-Zanze, I. The Use of Three-Dimensional Shape and Electrostatic Similarity Searching in the Identification of a Melanin-Concentrating Hormone Receptor 1 Antagonist. Chem. Biol. Drug Des. 2006, 67, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Murcia, M.; Ortiz, A.R. Virtual screening with flexible docking and COMBINE-based models. Application to a series of factor Xa inhibitors. J. Med. Chem. 2004, 47, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Svensson, F.; Karlén, A.; Sköld, C. Virtual Screening Data Fusion Using Both Structure- and Ligand-Based Methods. J. Chem. Inf. Model. 2012, 52, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.S.S.; Burkart, T. Advances in oral anticoagulation therapy—What’s in the pipeline? Blood Rev. 2017, 31, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Kohrt, J.T.; Bigge, C.F.; Bryant, J.W.; Casimiro-Garcia, A.; Chi, L.; Cody, W.L.; Dahring, T.; Dudley, D.A.; Filipski, K.J.; Haarer, S.; et al. The Discovery of (2R,4R)-N-(4-chlorophenyl)-N-(2-fluoro-4-(2-oxopyridin-1(2H)-yl)phenyl)-4-methoxypyrrolidine-1,2-dicarboxamide (PD 0348292), an Orally Efficacious Factor Xa Inhibitor. Chem. Biol. Drug Des. 2007, 70, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Roehrig, S.; Straub, A.; Pohlmann, J.; Lampe, T.; Pernerstorfer, J.; Schlemmer, K.-H.; Reinemer, P.; Perzborn, E. Discovery of the Novel Antithrombotic Agent 5-Chloro-N-({(5S)-2-oxo-3-[4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene- 2-carboxamide (BAY 59-7939): An Oral, Direct Factor Xa Inhibitor. J. Med. Chem. 2005, 48, 5900–5908. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Brown, R.D.; Varma-O’brien, S.; Rogers, D. Cheminformatics analysis and learning in a data pipelining environment. Mol. Divers. 2006, 10, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Zbinden, K.G.; Anselm, L.; Banner, D.W.; Benz, J.; Blasco, F.; Decoret, G.; Himber, J.; Kuhn, B.; Panday, N.; Ricklin, F.; et al. Design of novel aminopyrrolidine factor Xa inhibitors from a screening hit. Eur. J. Med. Chem. 2009, 44, 2787–2795. [Google Scholar] [CrossRef] [PubMed]
- Anselm, L.; Banner, D.W.; Benz, J.; Groebke Zbinden, K.; Himber, J.; Hilpert, H.; Huber, W.; Kuhn, B.; Mary, J.-L.; Otteneder, M.B.; et al. Discovery of a factor Xa inhibitor (3R,4R)-1-(2,2-difluoro-ethyl)-pyrrolidine-3,4-dicarboxylic acid 3-[(5-chloro-pyridin-2-yl)-amide] 4-{[2-fluoro-4-(2-oxo-2H-pyridin-1-yl)-phenyl]-amide} as a clinical candidate. Bioorg. Med. Chem. Lett. 2010, 20, 5313–5319. [Google Scholar] [CrossRef] [PubMed]
- Van Huis, C.A.; Casimiro-Garcia, A.; Bigge, C.F.; Cody, W.L.; Dudley, D.A.; Filipski, K.J.; Heemstra, R.J.; Kohrt, J.T.; Leadley, R.J., Jr.; Narasimhan, L.S.; et al. Exploration of 4,4-disubstituted pyrrolidine-1,2-dicarboxamides as potent, orally active Factor Xa inhibitors with extended duration of action. Bioorg. Med. Chem. 2009, 17, 2501–2511. [Google Scholar] [CrossRef] [PubMed]
- Meneyrol, J.; Follmann, M.; Lassalle, G.; Wehner, V.; Barre, G.; Rousseaux, T.; Altenburger, J.-M.; Petit, F.; Bocskei, Z.; Schreuder, H.; et al. 5-Chlorothiophene-2-carboxylic Acid [(S)-2-[2-Methyl-3-(2-oxopyrrolidin-1-yl)benzenesulfonylamino]-3-(4-methylpiperazin-1-yl)-3-oxopropyl]amide (SAR107375), a Selective and Potent Orally Active Dual Thrombin and Factor Xa Inhibitor. J. Med. Chem. 2013, 56, 9441–9456. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.J.P.; Orwat, M.J.; Koch, S.; Rossi, K.A.; Alexander, R.S.; Smallwood, A.; Wong, P.C.; Rendina, A.R.; Luettgen, J.M.; Knabb, R.M.; et al. Discovery of 1-(4-Methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (Apixaban, BMS-562247), a Highly Potent, Selective, Efficacious, and Orally Bioavailable Inhibitor of Blood Coagulation Factor Xa. J. Med. Chem. 2007, 50, 5339–5356. [Google Scholar] [PubMed]
- NCI/CAAD Group Website. Available online: http://cactus.nci.nih.gov/download/nci/ (accessed on 13 January 2017).
- Hawkins, P.C.; Nicholls, A. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef] [PubMed]
- OMEGA, v2.5.1.4: OpenEye Scientific Software, Santa Fe, NM, USA. 2013. Available online: http://www.eyesopen.com/ (accessed on 13 January 2017).
- Grant, J.A.; Gallardo, M.A.; Pickup, B.T. A fast method of molecular shape comparison: A simple application of a Gaussian description of molecular shape. J. Comput. Chem. 1996, 17, 1653–1666. [Google Scholar] [CrossRef]
- Grant, J.A.; Pickup, B.T. A Gaussian Description of Molecular Shape. J. Phys. Chem. 1995, 99, 3503–3510. [Google Scholar] [CrossRef]
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef] [PubMed]
- FRED, v3.2.0.2: OpenEye Scientific Software, Santa Fe, NM, USA. 2015. Available online: http://www.eyesopen.com/ (accessed on 13 January 2017).
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput. Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. Docking Screens for Novel Ligands Conferring New Biology. J. Med. Chem. 2016, 59, 4103–4120. [Google Scholar] [CrossRef] [PubMed]
- McGann, M.R.; Almond, H.R.; Nicholls, A.; Grant, J.A.; Brown, F.K. Gaussian docking functions. Biopolymers 2003, 68, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Developmental Therapeutic Program at NCI-NIH. Available online: http://dtp.nci.nih.gov/ (accessed on 13 January 2017).
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL web services: Streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015, 43, W612–W620. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrian-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.L.; Nissink, J.W.M.; Strasser, J.M.; Francis, S.; Higgins, L.; Zhou, H.; Zhang, Z.; Walters, M.A. PAINS in the Assay: Chemical Mechanisms of Assay Interference and Promiscuous Enzymatic Inhibition Observed during a Sulfhydryl-Scavenging HTS. J. Med. Chem. 2015, 58, 2091–2113. [Google Scholar] [CrossRef] [PubMed]
- Capuzzi, S.J.; Muratov, E.N.; Tropsha, A. Phantom PAINS: Problems with the Utility of Alerts for Pan-Assay INterference CompoundS. J. Chem. Inf. Model. 2017, 57, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the Developmental Therapeutics Program (DTP) at the National Cancer Institute (NCI). |
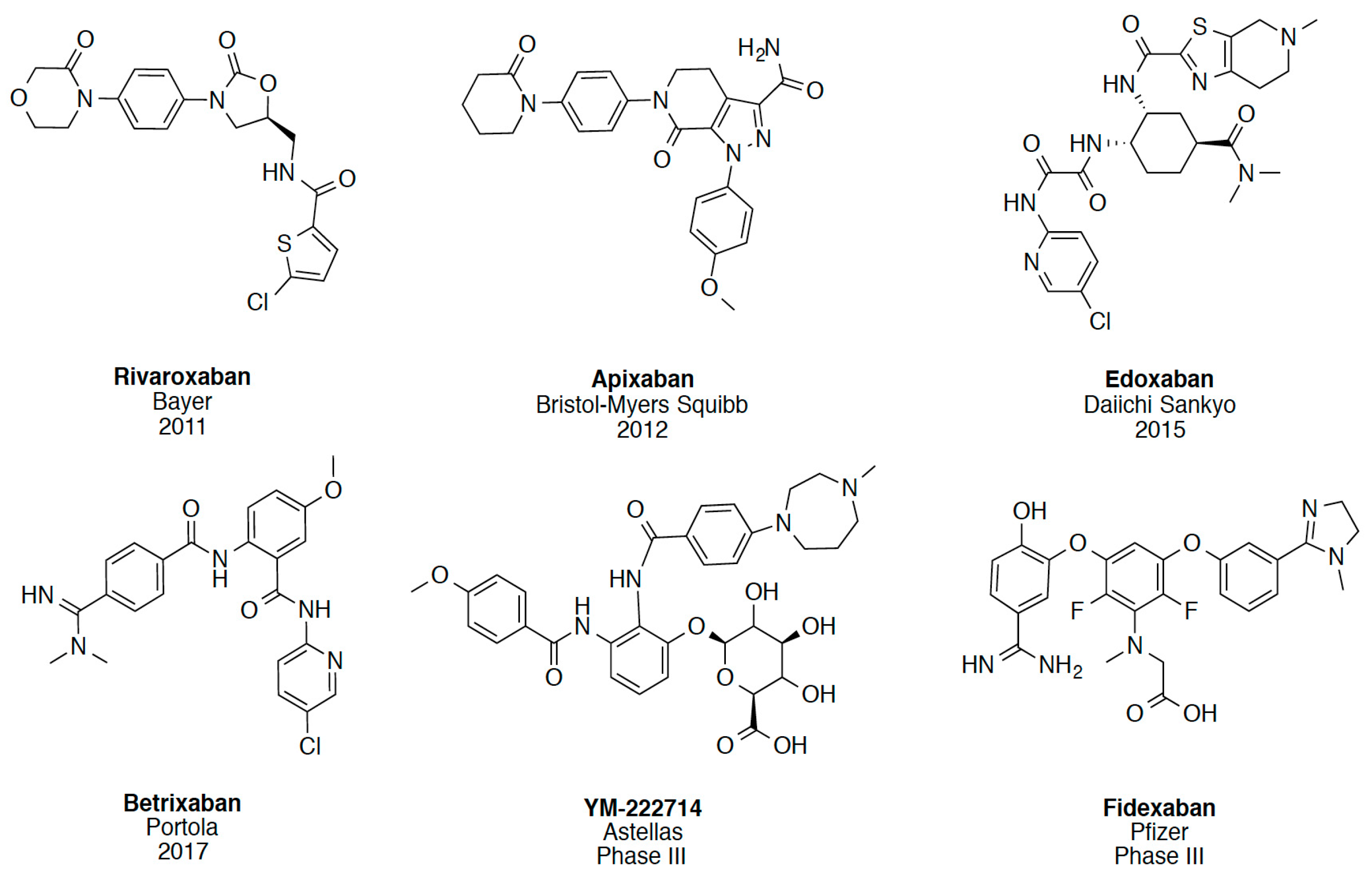


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID (Reference) | Resolution (Å) | Ligand Structure | RMSD Cluster Center (Å) | RMSD Binding Site (6 Å) a | Cluster ID | Tanimoto Distance b |
|---|---|---|---|---|---|---|
| 2VWN [30] | 1.61 |  | ----- | ----- | I | 0.000 |
| 2VWO [30] | 1.60 |  | 0.435 | 0.269 | I | 0.091 |
| 2VVC [30] | 1.95 |  | 0.320 | 0.711 | I | 0.109 |
| 2VVU [30] | 2.30 |  | 0.327 | 0.911 | I | 0.125 |
| 2VWL [30] | 1.80 |  | 0.137 | 0.732 | I | 0.155 |
| 2VWM [30] | 1.96 |  | 0.338 | 0.695 | I | 0.213 |
| 2VVV [30] | 1.73 |  | 0.301 | 0.977 | I | 0.371 |
| 2XBX [31] | 1.85 |  | ----- | ----- | II | 0.000 |
| 2XC5 [31] | 1.70 |  | 0.121 | 0.620 | II | 0.045 |
| 2XBW [31] | 1.72 |  | 0.194 | 0.659 | II | 0.111 |
| 2XC0 [31] | 2.05 |  | 0.120 | 0.767 | II | 0.128 |
| 2XC4 [31] | 1.67 |  | 0.358 | 0.866 | II | 0.130 |
| 2PHB [26] | 2.30 |  | 0.416 | 0.956 | II | 0.365 |
| 2XBV [31] | 1.66 |  | 0.193 | 0.417 | II | 0.378 |
| 2W3K [32] | 2.05 |  | 0.377 | 1.019 | II | 0.411 |
| 4BTI [33] | 2.30 |  | ----- | ----- | III | 0.000 |
| 4BTU [33] | 2.37 |  | 0.440 | 0.605 | III | 0.426 |
| 4BTT [33] | 2.59 |  | 0.530 | 0.479 | III | 0.582 |
| 2W26 Rivaroxaban [27] | 2.08 |  | 0.276 | 0.774 | III | 0.618 |
| 2P16 Apixaban [34] | 2.30 |  | 0.290 | 0.928 | III | 0.689 |
| Number | Cluster | NSC Code | FXa Activity Inhibition >50% at 10 μM | FXa Activity Inhibition >50% at 1 μM | ChemGauss4 Score | ΔG Ludi3 (kcal/mol) | Consensus Score | EON ET Combo | EON Rank | ROCS Tanimoto Combo | ROCS Rank |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 647716 | (+) | (−) | −11.325 | −11.06 | 10 | 0.733 | 34 | 0.541 | 202 |
| 3 | 2 | 635553 | (+) | (−) | −10.852 | −12.26 | 10 | 0.919 | 234 | 0.541 | 202 |
| 5 | 2 | 635393 | (+) | (+) | −10.487 | −11.25 | 9 | 1.095 | 27 | 0.632 | 6 |
| 7 | 1 | 141296 | (+) | (−) | −10.025 | −9.42 | 7 | 1.274 | 34 | 0.686 | 117 |
| 9 | 1 | 634395 | (+) | (−) | −9.972 | −10.12 | 9 | 1.164 | 78 | 0.593 | 1187 |
| 10 | 1 | 351149 | (+) | (−) | −9.830 | −10.65 | 7 | 1.289 | 29 | 0.630 | 500 |
| 12 | 3 | 371867 | (+) | (−) | −9.594 | −12.49 | 6 | 0.700 | 72 | 0.463 | 2404 |
| 9 | 2 | 635550 | (+) | (−) | −8.880 | −12.41 | 7 | 0.895 | 284 | 0.567 | 76 |
| 22 | 2 | 634416 | (+) | (−) | −8.737 | −9.63 | 7 | 1.026 | 77 | 0.485 | 1180 |
| 23 | 2 | 635142 | (+) | (−) | −8.729 | −9.87 | 8 | 0.989 | 112 | 0.481 | 1329 |
| 24 | 2 | 646798 | (+) | (−) | −7.480 | −12.00 | 7 | 0.910 | 250 | 0.504 | 617 |
| - | - | Rivaroxaban | (+) | (+) | −13.715 | −13.21 | 10 | 1.552 | 1 | 0.768 | 7 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lagos, C.F.; Segovia, G.F.; Nuñez-Navarro, N.; Faúndez, M.A.; Zacconi, F.C. Novel FXa Inhibitor Identification through Integration of Ligand- and Structure-Based Approaches. Molecules 2017, 22, 1588. https://doi.org/10.3390/molecules22101588
Lagos CF, Segovia GF, Nuñez-Navarro N, Faúndez MA, Zacconi FC. Novel FXa Inhibitor Identification through Integration of Ligand- and Structure-Based Approaches. Molecules. 2017; 22(10):1588. https://doi.org/10.3390/molecules22101588
Chicago/Turabian StyleLagos, Carlos F., Gerardine F. Segovia, Nicolás Nuñez-Navarro, Mario A. Faúndez, and Flavia C. Zacconi. 2017. "Novel FXa Inhibitor Identification through Integration of Ligand- and Structure-Based Approaches" Molecules 22, no. 10: 1588. https://doi.org/10.3390/molecules22101588