
Chemical Constituents of Malaysian U. cordata var. ferruginea and Their in Vitro α-Glucosidase Inhibitory Activities

Abstract
:1. Introduction
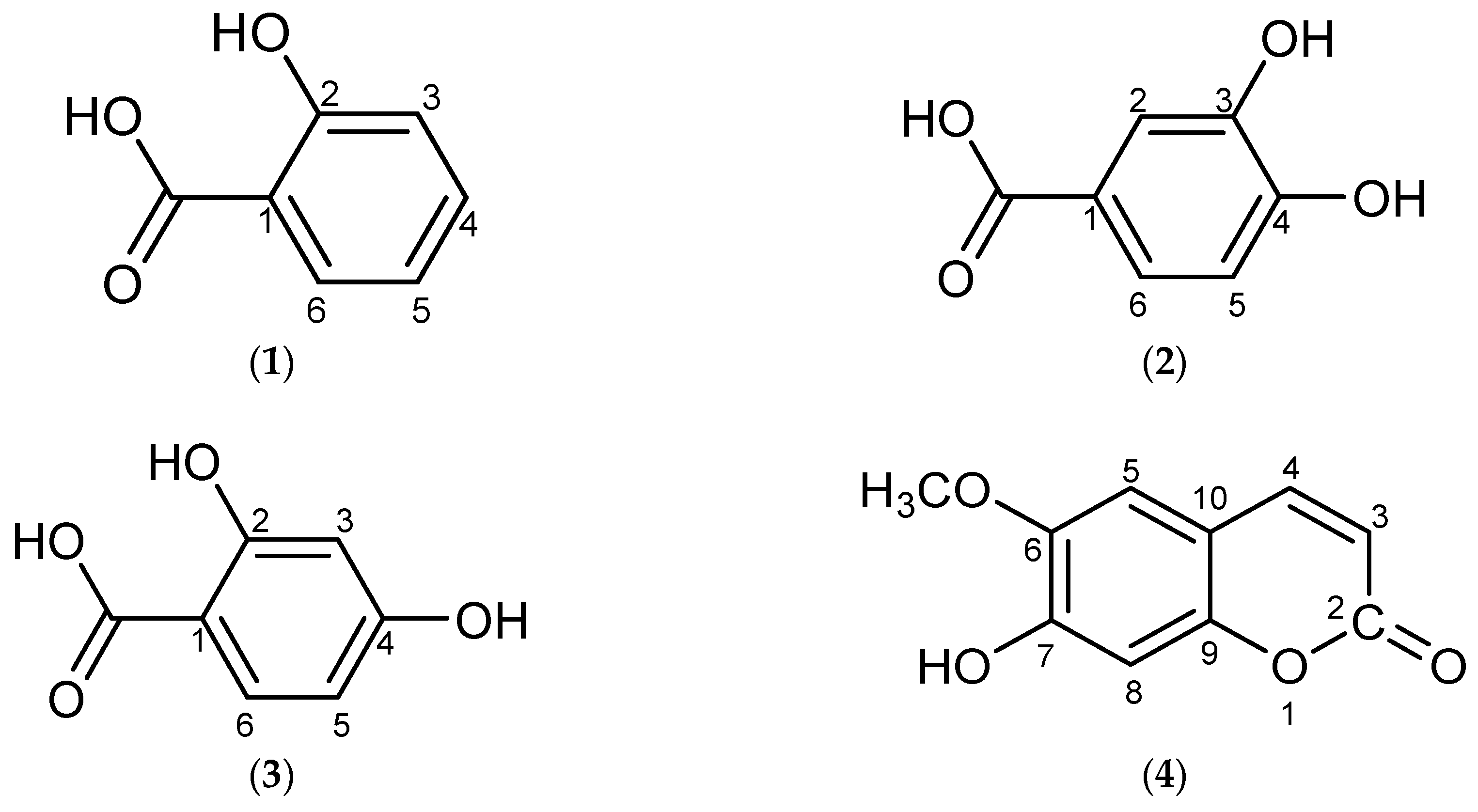
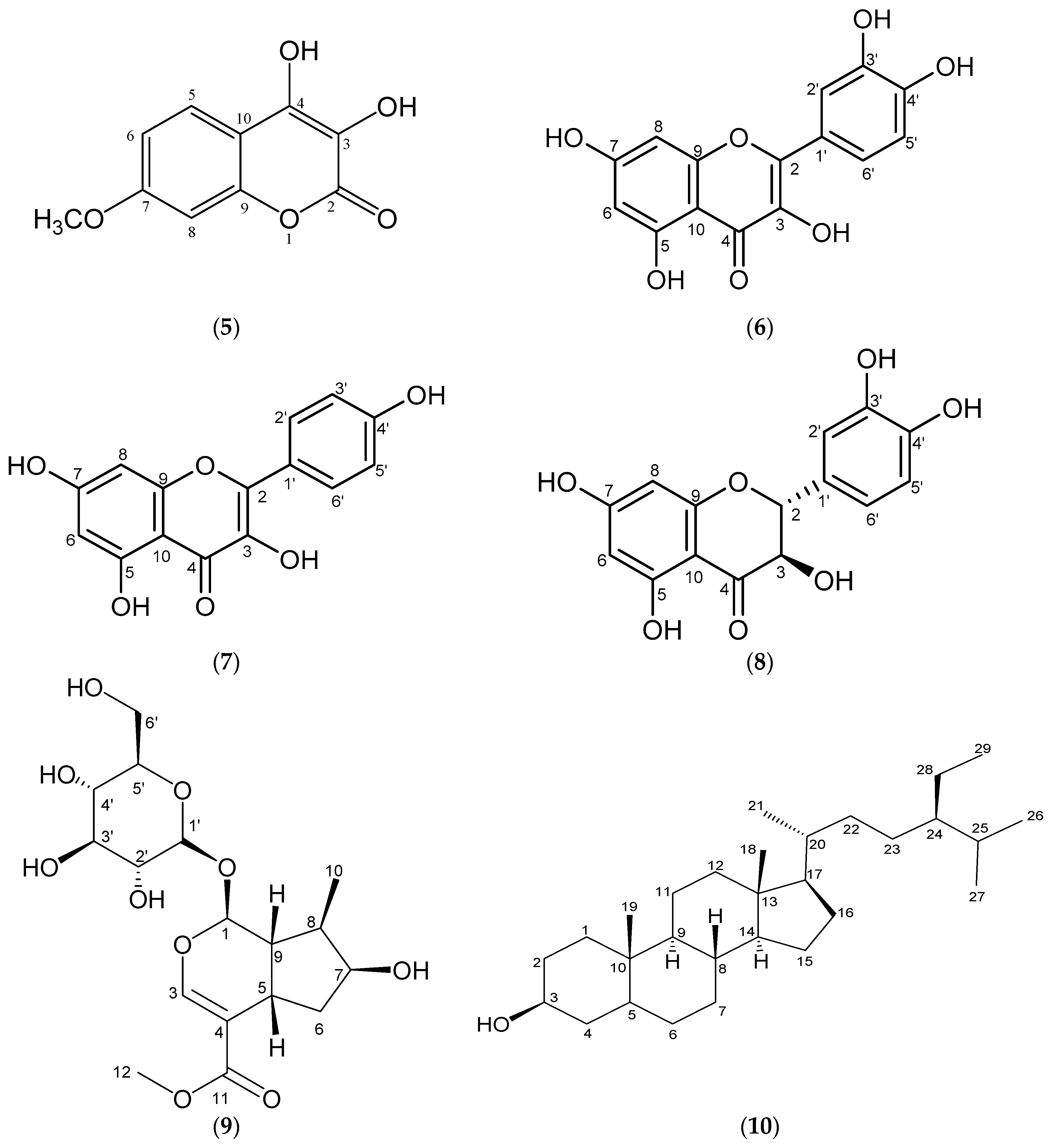
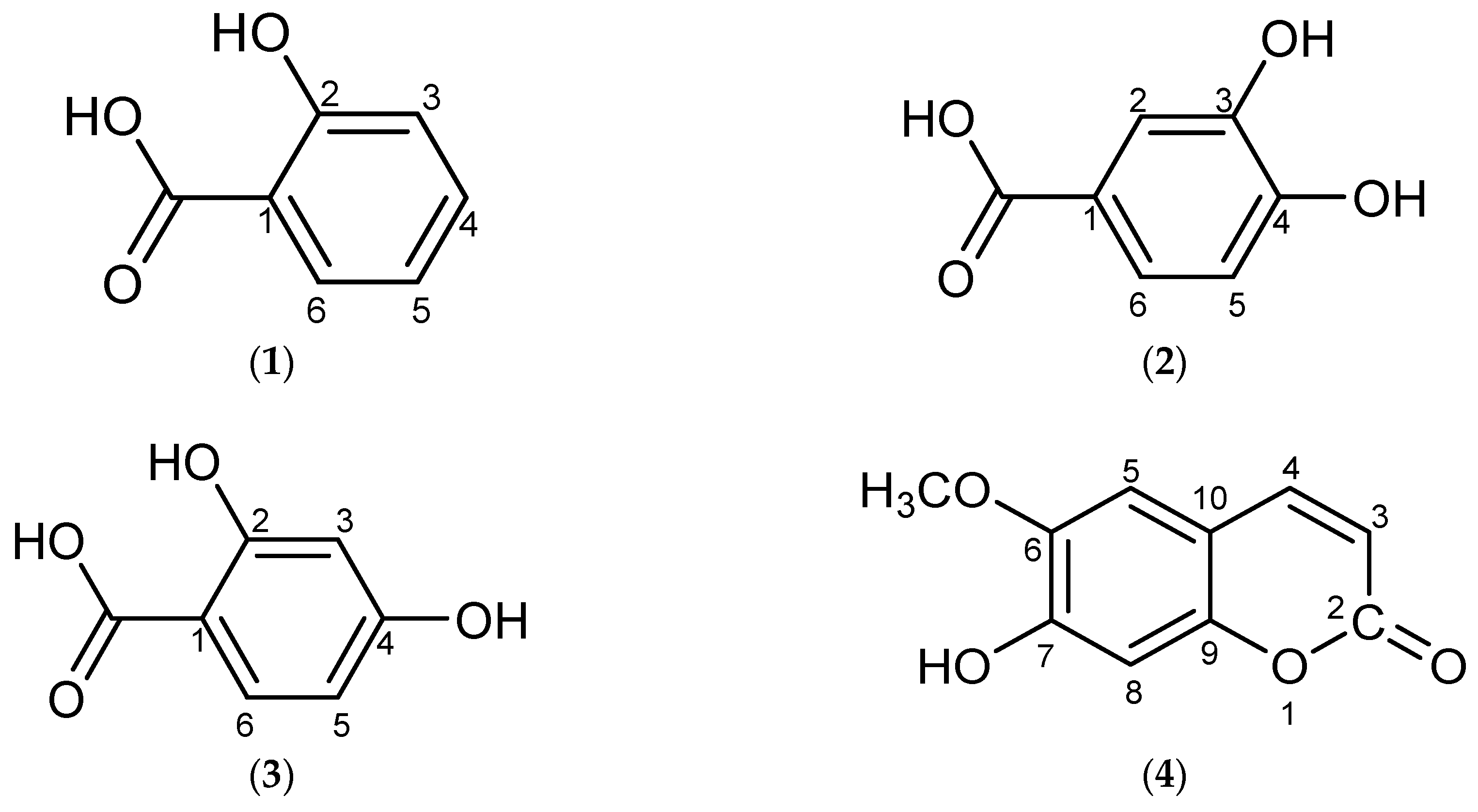
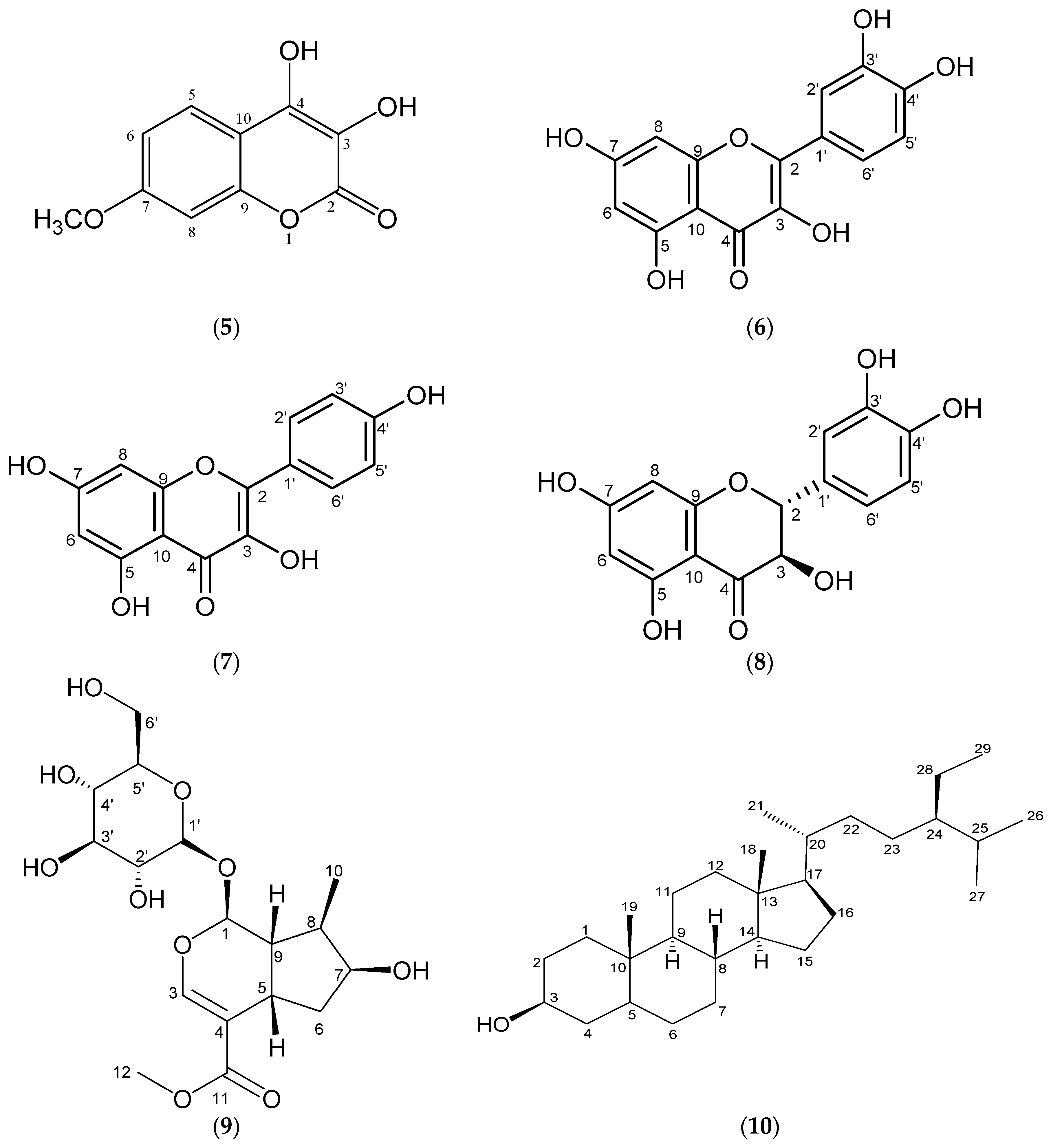
2. Results and Discussion
α-Glucosidase Inhibitory Activities
3. Experimental Section
3.1. General Information
3.2. Plant Material
3.2.1. First Batch of Plant
3.2.2. Second Batch of Plant
3.3. Extraction and Isolation of Compounds
3.3.1. First Batch of Plant
Stem Extract
Flower Extract
3.3.2. Second Batch of Plant
3.4. Physical and Spectral Data of Isolated Compounds
3.5. α-Glucosidase Inhibitory Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Heitzman, M.E.; Neto, C.C.; Winiarz, E.; Vaisberg, A.J.; Hammond, G.B. Ethnobotany, phytochemistry and pharmacology of Uncaria (Rubiaceae). Phytochemistry 2005, 66, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Salim, F.; Ahmad, R. Isopteropodic acid from Malaysian Uncaria longiflora var. pteropoda. World Appl. Sci. J. 2010, 10, 1334–1337. [Google Scholar]
- Salim, F.; Ahmad, R. Alkaloids from Malaysian Uncaria longiflora var. pteropoda. Biochem. Syst. Ecol. 2011, 39, 151–152. [Google Scholar] [CrossRef]
- Salim, F.; Ismail, N.H.; Awang, K.; Ahmad, R. Rauniticine-allo-oxindole B and rauniticinic-allo acid B, new heteroyohimbine-type oxindole alkaloids from the stems of Malaysian Uncaria longiflora var. pteropoda. Molecules 2011, 16, 6541–6548. [Google Scholar] [CrossRef] [PubMed]
- Andre, N.; Wang, X.M.; He, Y.; Pan, G.; Kojo, A.; Liu, Y. A review of the occurrence of non-alkaloid constituents in Uncaria species and their structure-activity relationships. Am. J. Biomed. Life Sci. 2013, 1, 79–98. [Google Scholar] [CrossRef]
- Salim, F.; Zain, M.M.; Ridzuan, M.S.M.; Langat, M.; Mulholland, D.; Ahmad, R. Flavan-3-ols from the leaves of Malaysian Uncaria longiflora var. pteropoda (Miq.) Ridsd. Phytochem. Lett. 2013, 6, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Kam, T.S.; Lee, K.H.; Goh, S.W. Alkaloid distribution in Malaysian Uncaria. Phytochemistry 1992, 31, 2031–2034. [Google Scholar] [CrossRef]
- Pavia, D.L.; Lampman, G.M.; Kriz, G.S.; Vyvyan, J.R. Introduction to Spectroscopy, 4th ed.; Brooks/Cole: Belmont, CA, USA, 2009; Volume 26. [Google Scholar]
- Banday, J.A.; Mir, F.A.; Farooq, S.; Qurishi, M.A.; Koul, S.; Razdan, T.K. Salicylic acid and Methyl gallate from the roots of Conyza canedensis. Int. J. Chem. Anal. Sci. 2012, 3, 1305–1308. [Google Scholar]
- Gurial, M.; Mitra, P.; Ghosh, T.; Gupta, S.; Basu, B.; Mitra, P.K. 3,4-dihydroxybenzoic acid Isolated from the Leaves of Ageratum conyzoides L. Eur. J. Biotechnol. Biosci. 2013, 1, 25–28. [Google Scholar]
- Lee, I.C.; Bae, J.S.; Kim, T.; Kwon, O.J.; Kim, T.H. Polyphenolic constituents from the aerial parts of Thymus quiquecostatus var. japonica collected on Ulleung Island. J. Korean Soc. Appl. Biol. Chem. 2011, 54, 811–816. [Google Scholar] [CrossRef]
- Ahmad, R.; Hashim, H.M.; Noor, Z.M.; Ismail, N.H.; Salim, F.; Lajis, N.H.; Shaari, K. Antioxidant and antidiabetic potential of Malaysian Uncaria. Res. J. Med. Plants 2011, 5, 587–595. [Google Scholar] [CrossRef]
- Hartati, S.; Dewi, R.T.; Darmawan, A.M. Development of antidiabetic active compounds from ethyl acetate extract of Acorus calamus L. Rhizom. Asian Trans. Basic Appl. Sci. 2012, 2, 27–30. [Google Scholar]
- Nair, S.S.; Kavrekar, V.; Mishra, A. In vitro studies on alpha amylase and alpha glucosidase inhibitory activities of selected plant extracts. Eur. J. Exp. Biol. 2013, 3, 128–132. [Google Scholar]
- Wei, J.F.; Zhang, Y.B.; Kang, W.Y. Antioxidant and a-glucosidase inhibitory compounds in Lysimachia clethroide. Afr. J. Pharm. Pharmacol. 2012, 6, 3230–3234. [Google Scholar] [CrossRef]
- Yin, Z.; Zhang, W.; Feng, F.; Zhang, Y.; Kang, W. α-Glucosidase inhibitors isolated from medicinal plants. Food Sci. Hum. Wellness 2014, 3, 136–174. [Google Scholar] [CrossRef]
- Wansi, J.D.; Lallemand, M.C.; Chiozem, D.C.; A.Toze, F.A.; Mbaze, L.M.; Khan, S.N.; Iqbal, C.M.; Wandji, J.; Tillequin, F.; Tanee, Z.F. α-Glucosidase inhibitory constituents from stem bark of Terminalia superba (Combretaceae). Phytochemistry 2007, 68, 2096–2100. [Google Scholar] [CrossRef] [PubMed]
- Mbaze, L.C.; Poumale, H.M.P.; Wansi, J.D.; Lado, J.A.; Khan, S.N.; Iqbal, C.M.; Ngadjui, B.T.; Laatsch, H. α-Glucosidase inhibitory pentacyclic triterpenes from the stem bark of Fagara tessmannii (Rutaceae). Phytochemistry 2007, 68, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Yuan, T.; Cirello, A.L.; Seeram, N.P. Antioxidant and α-glucosidase inhibitory phenolics isolated from highbush blueberry flowers. Food Chem. 2012, 135, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Jabeen, B.; Riaz, N.; Saleem, M.; Naveed, M.A.; Ashraf, M.; Alam, U.; Rafiq, H.M.; Tareen, R.B.; Jabbar, A. Isolation of natural compounds from Phlomis stewartii showing a-glucosidase inhibitory activity. Phytochemistry 2013, 96, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Qi, J.; Yu, B.Y. Iridoid and phenylpropanoid glycosides from Scrophularia ningpoensis Hemsl. and their α-glucosidase inhibitory activities. Fitoterapia 2014, 93, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Minoshima, Y.; Yamamoto, J.; Adachi, I.; Watson, A.; Nash, R.J. Protective effects of dietary chamomile tea on diabetic complication. J. Agric. Food Chem. 2008, 56, 8206–8211. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Gao, H.; Tang, Z.; Song, X.; Wu, L. Several phenolic acids from the fruit of Capparis spinosa. Asian J. Tradit. Med. 2006, 1, 3–4. [Google Scholar]
- Valizadeh, H.; Kordib, F.M.; Kouhkana, R.; Bahadoria, M.B.; Farimanic, M.M. Isolation and structure elucidation of coumarin and cinamate derivatives from Lycium ruthenicum. Iran. Chem. Commun. 2014, 2, 277–282. [Google Scholar]
- Salim, F. Phytochemicals from Malaysian Uncaria longiflora var. pteropoda (Miq) Ridsd. and their Activities Against the Human Neuroblastoma SH-SY5Y Cell Line. Ph.D. Thesis, Universiti Teknologi MARA, Shah Alam, Selangor, Malaysia, 2013. [Google Scholar]
- Kim, S.Y.; Park, J.Y.; Park, P.S.; Bang, S.H.; Lee, K.M.; Lee, Y.R.; Jang, Y.H.; Kim, M.J.; Chun, W.; Heo, M.Y.; et al. Flavonoid glycosides as acetylcholinesterase inhibitors from the whole plants of Persicaria thunbergii. Nat. Prod. Sci. 2014, 20, 191–195. [Google Scholar]
- Kavitha, H.P.; Venkatraman, B.R. Isolation, characterization and anti-inflammatory property of Thevetia peruvian. E-J. Chem. 2010, 7, 1584–1590. [Google Scholar]
- Li, Y.L.; Li, J.; Wang, N.L.; Yao, X.S. Flavonoids and a new polyacetylene from Bidens parviflora Willd. Molecules 2008, 13, 1931–1941. [Google Scholar] [CrossRef] [PubMed]
- Morel, L.J.D.F.; Baratto, D.M.; Pereira, P.S.; Contini, S.H.T.; Momm, H.G.; Bertoni, B.W.; Franca, S.D.C.; Pereira, A.M.S. Loganin production in Palicourea rigida H.B.K. (Rubiaceae) from populations native to Brazilian Cerrado. J. Med. Plants Res. 2011, 5, 2559–2565. [Google Scholar]
- Yinusa, I.; Ndukwe, I.G.; Rufai, Y.; Ayo, R.G. Characterization and microbial activities of β-sitosterol and β-sitostenone mixture isolated from the stem bark of methanol fraction of Sarcocephalus latifolius (Smith Bruce). Int. Res. J. Nat. Sci. 2014, 2, 1–13. [Google Scholar]
- Sample Availability: Samples of the compounds are not available from the authors.

{kind=link}
{kind=link}
| Samples | α-Glucosidase Inhibitory * (%) | IC50 (μg/mL) | IC50 (mM) |
|---|---|---|---|
| Stems | 87.7 ± 2.3 | 102 | - |
| Fractions | |||
| DCM | 75.3 ± 2.4 | 360 | - |
| Acetone | 89.2 ± 3.8 | 200 | - |
| Acarbose | 90.9 ± 2.7 | 580 | 0.89 |
| Compounds | |||
| 2,4-DHBA | 78.8 ± 0.4 | 549 | 3.56 |
| Quercetin | 84.5 ± 0.6 | 556 | 1.84 |
| Scopoletin | 34.5 ± 0.4 | NA | - |
| Loganin | 44.9 ± 0.7 | NA | - |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdullah, N.H.; Salim, F.; Ahmad, R. Chemical Constituents of Malaysian U. cordata var. ferruginea and Their in Vitro α-Glucosidase Inhibitory Activities. Molecules 2016, 21, 525. https://doi.org/10.3390/molecules21050525
Abdullah NH, Salim F, Ahmad R. Chemical Constituents of Malaysian U. cordata var. ferruginea and Their in Vitro α-Glucosidase Inhibitory Activities. Molecules. 2016; 21(5):525. https://doi.org/10.3390/molecules21050525
Chicago/Turabian StyleAbdullah, Nur Hakimah, Fatimah Salim, and Rohaya Ahmad. 2016. "Chemical Constituents of Malaysian U. cordata var. ferruginea and Their in Vitro α-Glucosidase Inhibitory Activities" Molecules 21, no. 5: 525. https://doi.org/10.3390/molecules21050525
APA StyleAbdullah, N. H., Salim, F., & Ahmad, R. (2016). Chemical Constituents of Malaysian U. cordata var. ferruginea and Their in Vitro α-Glucosidase Inhibitory Activities. Molecules, 21(5), 525. https://doi.org/10.3390/molecules21050525