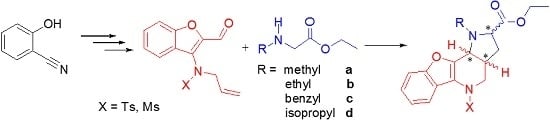
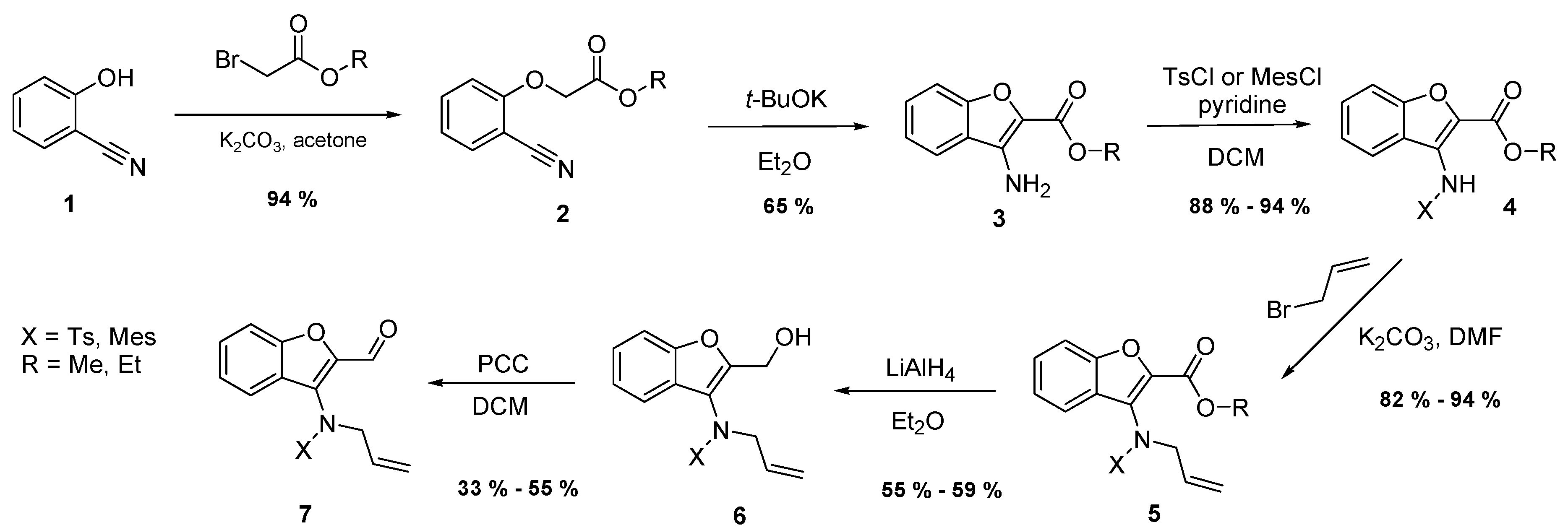
3.2. Chemistry
Methyl-(2-cyanophenoxy)acetate (2). Potassium carbonate (2.78 g, 20.2 mmol, 1.20 eq.) and methyl bromoacetate (3.08 g, 20.2 mmol, 1.20 eq.) were added to a solution of 2-hydroxybenzonitrile (1, 2.00 g, 16.8 mmol, 1.00 eq.) dissolved in dried acetone (85 mL). The reaction mixture was refluxed for 7 h. After cooling down to room temperature, the mixture was filtered and the acetone solution evaporated. The reaction afforded light yellow crystals with a yield of 94%; mp 69 °C. 1H-NMR: δ = 7.59 (dd, J = 7.7, 1.6, 1H, H1), 7.52 (td, J = 9.2, 7.7, 1.7, 1H, H4), 7.07 (td, J = 7.6, 0.7, 1H, H2), 6.86 (d, J = 8.5, 1H, H3), 4.78 (s, 2H, CH2), 3.81 (s, 3H, COOCH3). 13C-NMR: δ = 168.6 (COOCH3), 159.8 (C6), 134.3 and 134.5 (C2 and C3), 122.2 (C1), 116.3 (C5), 112.7 (C4), 103.0 (CN), 66.0 (CH2), 52.7 (COOCH3). MS (EI, 70 eV) Found: 191 (M calculated for: C10H9NO3 191.19); m/z (%) = 191 (M+, 50), 162 (50), 146 (10), 132 (70), 117 (15), 102 (70), 85 (60), 83 (100), 75 (35), 45 (60).
Methyl-3-aminobenzo[b]furan-2-carboxylate (3). Compound 2 (2.62 g, 13.9 mmol, 1.00 eq.) was dissolved in dried diethyl ether and poured into a flask in which an argon atmosphere was maintained. Then, potassium t-butoxide (0.78 g, 6.9 mmol, 0.50 eq.) was carefully added and the mixture stirred at room temperature. After 25 min, several drops of water were added and the solvent evaporated. The remainder was mixed with water (30 mL) and the mixture extracted three times with 30 mL of diethyl ether. After drying by sodium sulfate, filtration, and the evaporation of ether, light yellow crystals were obtained with a yield of 65%; mp 94 °C. 1H-NMR: δ = 7.55 (d, J = 7.9, 1H, H4), 7.48–7.41 (m, 2H, H7 and H6), 7.19–7.30 (m, 1H, H5), 5.00 (bs, 2H, NH2), 3.96 (s, 3H, COOCH3). 13C-NMR: δ = 162.1 (COOCH3), 154.3 (C7a), 138.9 (C3), 129.1 (C4), 125.6 (C3a), 122.5 (C5), 121.8 (C2), 119.8 (C6), 112.8 (C7), 51.7 (COOCH3). MS (EI, 70 eV) Found: 191 (calculated for C10H9NO3: 191.19); m/z (%) = 191 (M+, 100), 159 (50), 133 (25), 103 (90), 83 (20), 77 (50), 51 (25).
General procedure for the preparation of compounds 4A and 4B. Either tosyl chloride (1.29 g, 6.78 mmol, 1.20 eq.) or methane sulfonylchloride (1.29 g, 11.3 mmol, 2.00 eq.) were added to a stirred solution of 3 (1.16 g, 5.65 mmol, 1.00 eq.) and pyridine (1.35 g, 16.9 mmol, 3.00 eq.) in dichloromethane (12 mL), and the mixture was stirred under an argon atmosphere at room temperature overnight. The progress of the reaction was monitored by TLC. After removal of the solvent, the crude product was dissolved in ethyl acetate (40 mL) and subsequently extracted by 3 × 20 mL water, 1 × 15 mL HCl, and 1 × 15 mL brine. The extract was dried by sodium sulfate, filtered, and then thickened to crystallization. It was then subsequently recrystallized either from ethyl acetate or acetone.
Methyl-3-tosylaminobenzo[b]furan-2-carboxylate (4A), colourless crystals, yield 88%, mp 116 °C. 1H-NMR: δ = 8.36 (bs, 1H, NH), 8.31 (d, 1H, J = 8.1, H4), 7.60 (d, J = 8.2, 2H, HTs), 7.41–7.54 (m, 2H, H7 and H6), 7.37 (t, J = 7.4, 1H, H5), 7.17 (d, J = 8.2, 2H, HTs), 3.86 (s, 3H, COOCH3), 2.35 (s, 3H, SO2C6H4CH3). 13C-NMR: δ = 161.0 (COOCH3), 154.3 (C7a), 144.5 (C3), 135.3 (C tosyl), 132.7 (C tosyl), 130.4 (C2), 129.8 (2C tosyl), 129.4 (C6), 127.6 (2C tosyl), 124.4 (C4), 124.3 (C5), 122.4 (C3a), 112.5 (C7), 52.5 (COOCH3), 21.7 (NHSO2C6H4CH3). MS (EI, 70 eV) Found: 345 (calculated for C17H15NO5S: 345.37); m/z (%) = 345 (M+, 50), 222 (10), 190 (45), 162 (40), 134 (55), 119 (20), 103 (20), 91 (100), 76 (30), 65 (45).
Methyl-3-mesylaminobenzo[b]furan-2-carboxylate (4B) ochre crystals, yield 94%, mp 173 °C. 1H-NMR: δ = 8.24 (bs, 1H, NH), 8.19 (d, J = 8.2, 1H, H4), 7.53 (d, J = 3.6, 2H, H7 and H6), 7.34–7.41 (m, 1H, H5), 4.03 (s, 3H, COOCH3), 3.09 (s, 3H, SO2CH3). 13C-NMR: δ = 161.5 (COOCH3), 154.5 (C7a), 132.2 (C3), 129.6 (C6), 124.4 (C4), 124.1 (C5), 122.5 (C2), 122.0 (C3a), 112.6 (C7), 52.8 (COOCH3), 40.3 (NHSO2CH3). MS (EI, 70 eV) found: 269 (calculated for C11H11NO5S: 269.27); m/z (%) = 269 (M+, 70), 237 (15), 190 (85), 162 (50), 148 (15), 134 (100), 119 (30), 103 (50), 91 (35), 76 (70), 59 (20), 50 (25).
General procedure for the preparation of compounds 5A and 5B. A solution of compound 4A or 4B (3.24 mmol, 1.00 eq.) in DMF (10 mL) was mixed with anhydrous potassium carbonate (672 mg, 4.86 mmol, 1.50 eq.). While the mixture was stirred, allyl bromide (590 mg, 4.86 mmol, 1.50 eq.) was added dropwise. The stirring continued for 20 h (for compound 5A) and 6 h (for 5B). The progress of the reaction was monitored by TLC (EtOAc/PE = 1:2). The process was finished by removing the solvent and the remainder was mixed with water (20 mL); the final product was extracted by dichloromethane and the extract washed with brine (15 mL). After drying by Na2SO4, filtration, and removal of the solvent by distillation, the product was crystallized from methanol.
Methyl-3-(N-allyl-N-tosylamino)benzo[b]furan-2-carboxylate (5A) white crystals, yield 94%, mp 86 °C. 1H-NMR: δ = 7.64 (d, J = 8.2, 2H, HTs), 7.53 (d, 1H, J = 8.3, H4), 7.43–7.50 (m, 2H, H7 and H6), 7.27–7.36 (m, 1H, H5), 7.27 (d, J = 8.2, 2H, HTs), 5.75–5.92 (m, 1H, CH2CH=CH2), 5.05 (dd, J = 17.1, 1.2, 1H, CH2CH=CH2), 4.99 (dd, J = 10.1, 0.9, 1H, CH2CH=CH2), 4.10–4.56 (bs, 2H, CH2CH=CH2), 3.67 (s, 3H, COOCH3), 2.43 (s, 3H, SO2C6H4CH3). 13C-NMR: δ = 158.8 (COOCH3), 154.0 (C7a), 144.0 (C3), 141.3 (C tosyl), 136.7 (C tosyl), 133.3 (CH2CH=CH2), 129.8 (2C tosyl), 128.7 (C6), 128.0 (2C tosyl), 127.7 (C2), 127.4 (C3a), 124.6 (C4), 122.6 (C5), 119.5 (CH2CH=CH2), 112.7 (C7), 54.3 (CH2CH=CH2), 52.5 (COOCH3), 21.9 (SO2C6H4CH3). MS (EI, 70 eV) Found: 230 [M − Ts] (calculated for C20H19NO5S: 385.434); m/z (%) = 230 (100), 198 (85), 170 (45), 115 (25), 91 (45), 65 (20), 45 (30).
Methyl-3-(N-allyl-N-mesylamino)benzo[b]furan-2-carboxylate (5B) orange crystals, yield 82%, mp 91 °C. 1H-NMR: δ = 7.80 (d, J = 7.8, 1H, H4), 7.44–7.56 (m, 2H, H7 and H6), 7.33–7.40 (m, 1H, H5), 5.72–5.90 (m, 1H, CH2CH=CH2), 5.11 (dd, J = 17.1, 1.3, 1H, CH2CH=CH2), 5.02 (dd, J = 10.1, 1.2, 1H, CH2CH=CH2), 4.40 (bs, 2H, CH2CH=CH2), 4.02 (s, 3H, COOCH3), 3.06 (s, 3H, SO2CH3). 13C-NMR: δ = 159.5 (COOCH3), 154.0 (C7a), 140.4 (C3), 133.2 (CH2CH=CH2), 129.1 (C6), 128.9 (C2), 127.7 (C3a), 124.9 (C4), 122.6 (C5), 119.7 (CH2CH=CH2), 112.7 (C7), 54.1 (CH2CH=CH2), 52.9 (COOCH3), 40.1 (SO2CH3). MS (EI 70 eV) Found: 230 [M − Ms] (calculated for C14H15NO5S: 309.338); m/z (%) = 230 (80), 198 (100), 170 (70), 156 (15), 115 (30), 102 (15), 83 (70), 59 (15), 45 (45).
Ester group reduction—transformation of compound 5 to compound 6. A solution of compound 5A or 5B (1.27 mmol, 1.00 eq.) in dry diethyl ether (13 mL) was slowly added to an intensively stirred suspension of LiAlH4 (58 mg, 1.5 mmol, 1.2 eq.) in dry diethyl ether (2 mL) cooled down to 0 °C under an argon atmosphere. After addition, the temperature was raised to room temperature and the reaction mixture was kept at this temperature for a further 30 min. Then, the reaction was stopped by the addition of diethyl ether containing several drops of water. Subsequently, the reaction mixture was extracted by diethyl ether (3 × 10 mL). The collected extracts were dried by Na2SO4 and filtered, and the solvent was further evaporated. The liquid product was purified by column chromatography (EtOAc/PE = 1:3).
N-(2-Hydroxymethylbenzo[b]furan-3-yl)-N-allyl-4-methylbenzenesulfonamide (6A) light yellow oil, yield 59%. 1H-NMR: δ = 7.54 (d, J = 8.2, 2H, HTs), 7.42 (d, J = 8.3, 1H, H4), 7.12–7.24 (m, 3H, H6 and 2HTs), 6.87–6.98 (m, 1H, H5), 6.41 (d, J = 7.8, 1H, H7), 5.72–5.91 (m, 1H, CH2CH=CH2), 5.03 (dd, J = 16.9, 1.2, 1H in CH2CH=CH2), 5.05 (dd, J = 1.0, J = 10.1, 1H in CH2CH=CH2), 4.19–4.88 (bs, 4H, 2H in CH2CH=CH2 and 2H in CH2OH), 2.41 (s, 3H, SO2C6H4CH3). 13C-NMR: δ = 157.1 (C7a), 153.7 (C2), 144.4 (C3), 135.9 (C tosyl), 132.6 (CH2CH=CH2), 130.0 (2C tosyl), 127.8 (2C tosyl), 125.1 (C6), 124.2 (C tosyl), 122.9 (C5), 119.9 (C4), 119.1 (CH2CH=CH2), 117.4 (C3a), 112.5 (C7), 55.3 (CH2OH), 52.9 (CH2CH=CH2), 21.8 (SO2C6H4CH3). HRMS (ESI) (m/z): [M + H]+ calculated for C19H20NO4S+: 358.1108. Found: 385.1112.
N-(2-Hydroxymethylbenzo[b]furan-3-yl)-N-allyl-methanesulfonamide (6B) light yellow oil, yield 55%. 1H-NMR: δ = 7.54 (d, J = 7.9, 1H, H4), 7.44–7.49 (m, 1H, H7), 7.33–7.41 (m, 1H, H5), 7.31 (dd, J = 1.2, 7.4, 1H, H6), 5.78–5.96 (m, 1H, CH2CH=CH2), 5.06–5.18 (m, 2H, CH2CH=CH2), 4.70 (s, 2H, CH2OH), 4.37 (bs, 2H, CH2CH=CH2), 2.99 (s, 3H, NHSO2CH3). 13C-NMR: δ = 157.3 (C7a), 154.0 (C2), 132.6 (CH2CH=CH2), 125.7 (C6), 124.5 (C3), 123.9 (C5), 120.3 (C4), 118.6 (CH2CH=CH2), 116.9 (C3a), 113.1 (C7), 55.3 (CH2OH), 53.2 (CH2CH=CH2), 39.5 (NHSO2CH3). HRMS (ESI) (m/z): [M + H]+ calculated for C13H16NO4S+: 282.0795. Found: 282.0795.
Procedure for the preparation of compound 7. Pyridinium chlorochromate (400 mg, 1.86 mmol, 2.50 eq.) was added to a solution of alcohol 6 (0.74 mmol, 1.00 eq.) in dry dichloromethane (5 mL). The resulting black suspension was stirred under argon at room temperature for 26 h. Subsequently, diethyl ether (15 mL) was added and the suspension stirred again for 10 min. Then, the upper layer was decanted and filtered through a short column (5 cm) of Florisil. The obtained light yellow solution was concentrated to crystallization and the product recrystallized from ethanol.
N-Allyl-N-(2-formylbenzo[b]furan-3-yl)-4-toluenesulfonamide (7A) yellow crystals, yield 55%, mp 107 °C. 1H-NMR: δ = 9.72 (s, 1H, CH=O), 7.58 (d, J = 8.2, 2H, HTs), 7.56 (d, J = 7.6, 1H, H4), 7.43–7.51 (m, 1H, H6), 7.20–7.29 (m, 2H, HTs), 7.18 (t, J = 7.5, 1H, H5), 7.08 (d, J = 7.8, 1H, H7), 5.73–5.89 (m, 1H, CH2CH=CH2), 5.06 (dd, J = 9.4, 0.9, 1H in CH2CH=CH2), 5.04 (dd, J = 17.0, 0.9, 1H in CH2CH=CH2), 4.35 (bs, 2H, CH2CH=CH2), 2.43 (s, 3H, SO2C6H4CH3). 13C-NMR: 179.0 (CH=O), 161.1 (C7a), 155.0 (C3), 149.4 (C-tosyl), 144.8 (C2), 135.2 (C-tosyl), 132.0 (CH2CH=CH2), 130.1 (2C tosyl), 129.6 (C6), 127.9 (2C tosyl), 124.7 (C3a), 124.8 (C5), 122.0 (C4), 120.6 (CH2CH=CH2), 113.5 (C7), 53.8 (CH2CH=CH2), 21.8 (SO2C6H4CH3). MS (EI 70 eV): Found: 200 [M − Ts] (calculated for C19H17NO4S: 355.408); m/z (%) = 200 (100), 172 (60), 158 (30), 144 (25), 115 (20), 103 (15), 91 (95), 77 (15), 65 (50).
N-Allyl-N-(2-formylbenzo[b]furan-3-yl)-methanesulfonamide (7B) yellow oil, yield 33%. 1H-NMR: δ = 10.02 (s, 1H, CH=O), 7.80 (d, J = 8.0, 1H, H4), 7.52–7.62 (m, 2H, H7 and H6), 7.37–7.44 (m, 1H, H5), 5.72–5.89 (m, 1H, CH2CH=CH2), 5.11 (dd, J = 17.1, 1.2, 1H, CH2CH=CH2), 5.08 (dd, J = 10.1, 0.9, 1H, CH2CH=CH2), 4.43 (d, J = 0.7, 2H, CH2CH=CH2), 3.02 (s, 3H, NSO2CH3). 13C-NMR: 180.5 (CH=O), 155.0 (C7a), 148.1 (C3), 133.7 (C2), 132.3 (CH2CH=CH2), 130.0 (C6), 125.1 (C5), 122.4 (C4), 120.4 (CH2CH=CH2), 118.1 (C3a), 113.4 (C7), 53.6 (CH2CH=CH2), 39.4 (NSO2CH3). HRMS (APCI): m/z [M + H]+ calculated for C13H13NO4S+: 280.0638. Found: 280.0638.
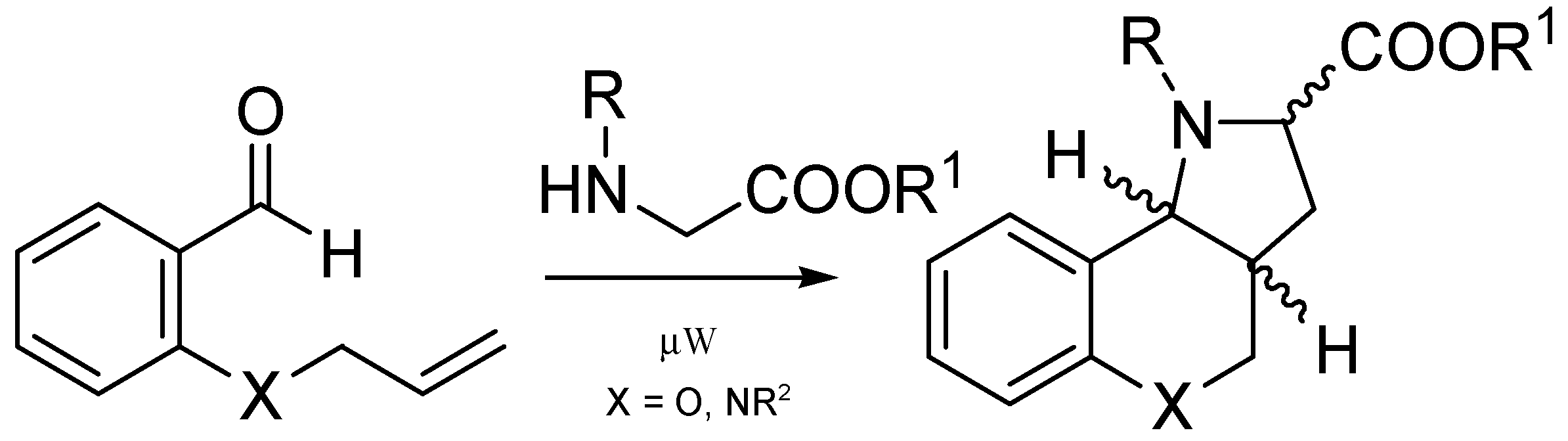
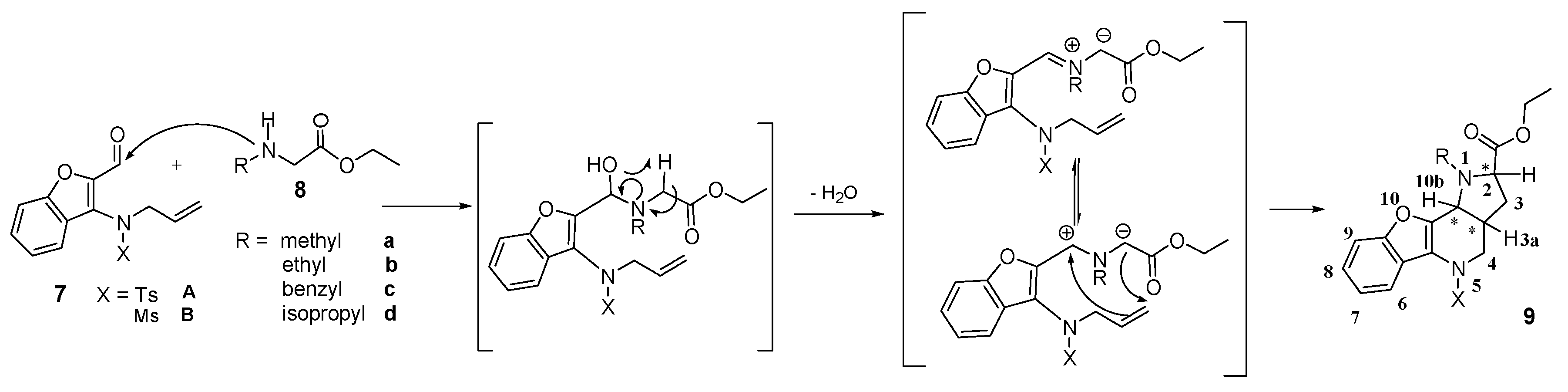
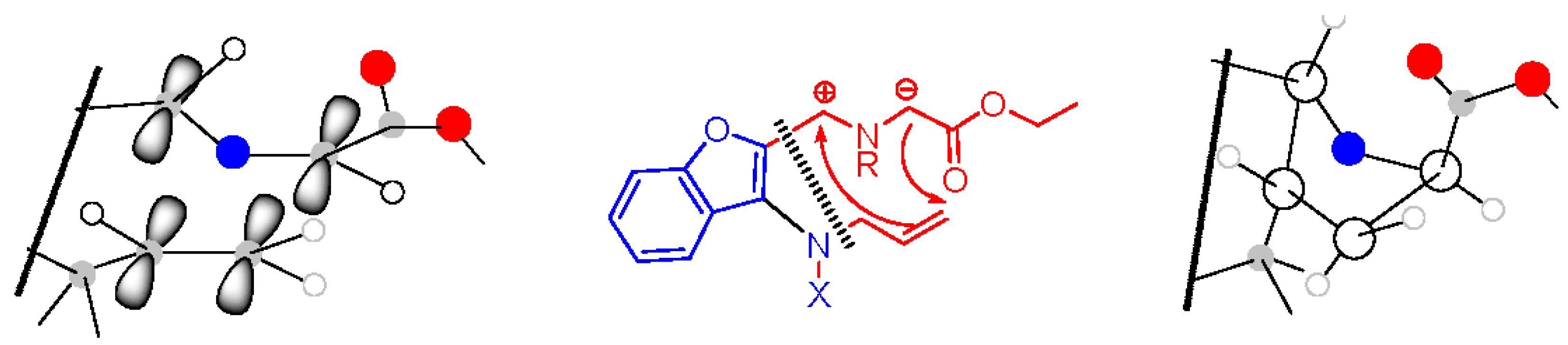
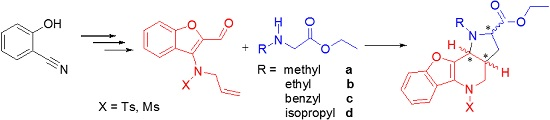
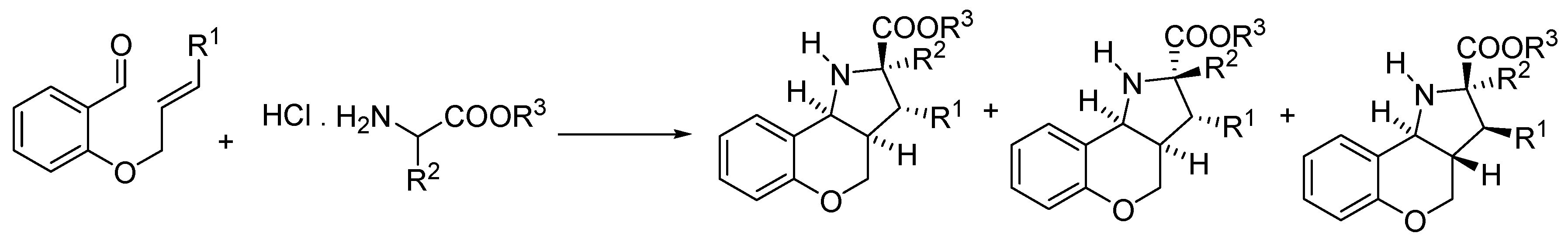
General procedure for the formation of azomethine ylide and its following intramolecular 1,3-dipolar cycloaddition—preparation of compound 9. Compound 7 (0.14 mmol, 1.00 eq.) and a two-fold excess of secondary amine 8 (0.28 mmol, 2.00 eq.) were mixed in a test tube filled with an argon atmosphere and stopped with a balloon. Then, the tube was immersed in a preheated oil bath (130–140 °C) and stirred for about 30 min until the mixture became darker in color. After cooling down, the mixture was separated by HPFC (AcOEt/PE = 1:3). The products were crystallized from methanol.
![Molecules 21 00187 i001]()
(2R,3aR,10bR) and (2S,3aS,10bS)-Ethyl-1-methyl-5-tosyl-2,3,3a,4,5,10b-hexahydro-1H-benzo[b] furo[3,2-b]pyrrolo[2,3-d]pyridine-2-carboxylate (9Aa). Yellow crystals, yield 80%, mp 64 °C. 1H-NMR: δ = 8.12–8.14 (m, 1H, H6), 7.49 (d, J = 8.3, 2H, HTs), 7.44 (dd, J = 1.7, 7.1, 1H, H9), 7.28–7.35 (m, 2H, H8 and H7), 7.19 (d, J = 8.0, 2H, HTs), 4.10–4.18 (m, 2H, COOCH2CH3), 4.07 (dd, J = 14.1, 4.8, 1H, H4), 3.73 (d, J = 6.7, 1H, H10b), 3.64 (dd, J = 2.1, 8.6, 1H, H2), 3.17 (dd, J = 14.0, 12.0, 1H, H4), 2.63 (s, 3H, NCH3), 2.39 (s, 3H, SO2C6H4CH3), 2.03–2.09 (m, 1H, H3), 1.95–2.02 (m, 1H, H3a), 1.62–1.68 (m, 1H, H3), 1.25 (t, J = 7.1, 3H, COOCH2CH3). 13C-NMR: δ = 173.3 (COOCH2CH3), 153.9 (C9a), 148.1 (C10a), 144.1 (C5a), 135.3 (C-SO2C6H4CH3), 129.8 (2CH SO2C6H4CH3), 127.4 (2CH-SO2C6H4CH3), 124.9 (C8), 123.2 (C SO2C6H4CH3), 123.1 (C6), 122.8 (C7), 119.1 (C5b), 111.3 (C9), 65.0 (C2), 60.3 (COOCH2CH3), 56.0 (C10b), 51.0 (C4), 36.4 (NCH3), 33.5 (C3a), 31.7 (C3), 21.6 (SO2C6H4CH3), 14.3 (COOCH2CH3). MS (EI, 70 eV) found 454 (calculated for C24H26N2O5S: 454.54); m/z (%) = 454 (M+, <5), 381 (65), 299 (20), 225 (100), 211 (15), 196 (10), 184 (10), 170 (30), 155 (10), 91 (55), 65 (20), 57 (45). HRMS (ESI) m/z [M + H]+ calculated for C24H27N2O5S+: 455.1635). Found: 455.1636.
![Molecules 21 00187 i002]()
(2R,3aR,10bR) and (2S,3aS,10bS)-Ethyl-1-ethyl-5-tosyl-2,3,3a,4,5,10b-hexahydro-1H-benzo[b] furo[3,2-b]pyrrolo[2,3-d]pyridine-2-carboxylate (9Ab). Light yellow crystals, yield 94%, mp 105 °C. 1H-NMR: δ = 8.13–8.16 (m, 1H, H6), 7.48 (d, J = 8.3, 2H, HTs), 7.44 (dd, J = 1.7, 7.1, 1H, H9), 7.28–7.34 (m, 2H, H8 and H7), 7.19 (d, J = 8.1, 2H, HTs), 4.10–4.17 (m, 2H, COOCH2CH3), 4.05 (dd, J = 14.1, 4.7, 1H, H4), 3.86 (d, J = 6.7, 1H, H10b), 3.81 (dd, J = 1.9, 8.7, 1H, H2), 3.30–3.38 (m, 1H, NCH2CH3), 3.21 (dd, J = 14.0, 11.8, 1H, H4), 2.64–2.72 (m, 1H, NCH2CH3), 2.38 (s, 3H, SO2C6H4CH3), 2.03–2.08 (m, 1H, H3), 1.92–2.00 (m, 1H, H3a), 1.61–1.67 (m, 1H, H3), 1.25 (t, J = 7.1, 3H, COOCH2CH3), 1.04 (t, J = 7.3, 3H, NCH2CH3). 13C-NMR: δ = 173.6 (COOCH2CH3), 153.9 (C9a), 148.2 (C10a), 144.1 (C5a), 135.4 (C in SO2C6H4CH3), 129.8 (2CH in SO2C6H4CH3), 127.4 (2CH in SO2C6H4CH3), 124.8 (C8), 123.2 (C in SO2C6H4CH3), 123.1 (C6), 122.8 (C7), 119.1 (C5b), 112.3 (C9), 60.7 (C2), 60.2 (COOCH2CH3), 55.4 (C4), 51.0 (NCH2CH3), 43.3 (C10b), 32.9 (C3a), 31.5 (C3), 21.6 (SO2C6H4CH3), 14.3 (COOCH2CH3), 13.6 (NCH2CH3). MS (EI, 70 eV) Found: 395 [M − COOEt] (calculated for C25H28N2O5S: 468.57); m/z (%) = 395 (80), 313 (20), 239 (100), 211 (25), 184 (15), 170 (20), 91 (40), 71 (25), 56 (30). HRMS (ESI) m/z [M + H]+ calculated for C25H29N2O5S+: 469.1792). Found: 469.1791.
![Molecules 21 00187 i003]()
(2R,3aR,10bR) and (2S,3aS,10bS)-Ethyl-1-benzyl-5-tosyl-2,3,3a,4,5,10b-hexahydro-1H-benzo[b] furo[3,2-b]pyrrolo[2,3-d]pyridine-2-carboxylate (9Ac). Yellow crystals, yield 96%, mp 130 °C. 1H-NMR: δ = 8.17–8.21 (m, 1H, H6), 7.50 (d, J = 8.3, 2H, HTs), 7.37–7.42 (m, 1H, H9), 7.30–7.33 (m, 2H, H8 and H7), 7.20 (d, J = 8.6, 2H, HTs), 7.15–7.18 (m, 5H, CH2C6H5), 4.58 (d, J = 13.7, 1H, H4), 4.06–4.14 (m, 3H, 2H in COOCH2CH3 and 1H in CH2C6H5), 4.05 (d, J = 3.6, 1H, H10b), 3.48–3.51 (m, 1H, H2), 3.82 (d, J = 13.7, 1H, H4), 3.30 (dd, J = 14.0, 11.6, 1H, CH2C6H5), 2.40 (s, 3H, SO2C6H4CH3), 1.98–2.03 (m, 1H, H3), 1.92–1.97 (m, 1H, H3a), 1.60–1.67 (m, 1H, H3), 1.20 (t, J = 7.1, 3H, COOCH2CH3). 13C-NMR: δ = 173.8 (COOCH2CH3), 153.9 (C9a), 148.0 (C10a), 144.1 (C5a), 138.8 (C in CH2C6H5), 135.3 (C in SO2C6H4CH3), 129.8 (2CH in SO2C6H4CH3), 128.4 (2CH in CH2C6H5), 128.1 (2CH in CH2C6H5), 127.4 (2CH in SO2C6H4CH3), 127.0 (CH in CH2C6H5), 123.1 (C7), 124.9 (C8), 123.1 (C in SO2C6H4CH3), 122.9 (C6), 119.2 (C5b), 111.3 (C9), 60.5 (C2), 60.2 (COOCH2CH3), 55.1 (C10b), 52.8 (CH2C6H5), 51.0 (C3), 32.9 (C3a), 31.6 (C4), 21.6 (SO2C6H4CH3), 14.3 (COOCH2CH3). HRMS (APCI) m/z [M + H]+ calculated for C30H31N2O5S+: 531.1948). Found: 531.1947.
![Molecules 21 00187 i004]()
(2R,3aR,10bR) and (2S,3aS,10bS)-Ethyl-1-isopropyl-5-tosyl-2,3,3a,4,5,10b-hexahydro-1H-benzo [b]furo[3,2-b]pyrrolo[2,3-d]pyridine-2-carboxylate (9Ad). Colourless crystals, yield 44%, mp 102 °C. 1H-NMR: δ = 8.13–8.17 (m, 1H, H8), 7.43 (dd, J = 7.3, 1.4, 1H, H9), 7.27–7.34 (m, 2H, H8 and H7), 7.20 (d, J = 8.0, 2H, HTs), 4.24 (d, J = 6.6, 1H, H10b), 4.05–4.13 (m, 2H, COOCH2CH3), 3.98 (dd, J = 14.0, 4.4, 1H, H4), 3.74 (dd, J = 8.5, 2.0, 1H, H2), 3.49–3.56 (m, 1H, CH(CH3)2), 3.27 (dd, J = 14.0, 11.1, 1H, H4), 2.39 (s, 3H, SO2C6H4CH3), 1.97–2.08 (m, 1H, H3a), 1.89–1.96 (m, 1H, H3), 1.56–1.64 (m, 1H, H3), 1.25 (t, J = 7.2, 3H, COOCH2CH3), 1.12 (d, J = 6.8, 3H, CH(CH3)2), 1.06 (d, J = 6.6, 3H, CH(CH3)2). 13C-NMR: δ = 176.6 (COOCH2CH3), 153.8 (C9a), 147.6 (C10a), 144.0 (C5a), 135.6 (C in SO2C6H4CH3), 129.8 (2CH in SO2C6H4CH3), 127.4 (2CH in SO2C6H4CH3), 124.8 (C8), 123.1 (C in SO2C6H4CH3), 123.0 (C6), 122.9 (C7), 119.3 (C5b), 111.3 (C9), 60.5 (COOCH2CH3), 58.0 (C2), 51.9 (C10b), 50.9 (C4), 47.8 (CH(CH3)2), 33.0 (C3a), 31.8 (C3), 21.6 (SO2C6H4CH3), 21.1 (CH(CH3)2), 17.8 (CH(CH3)2), 14.3 (COOCH2CH3). MS (EI 70 eV) found 482 (calculated for C26H30N2O5S: 482.59); m/z (%) = 482 (M+, <5), 409 (95), 327 (15), 253 (100), 211 (75), 184 (25), 170 (25), 155 (15), 91 (65), 84 (25), 70 (55), 65 (20), 43 (30). HRMS (ESI) m/z [M + H]+ calculated for C26H31N2O5S+: 483.1948). Found: 483.1948.
![Molecules 21 00187 i005]()
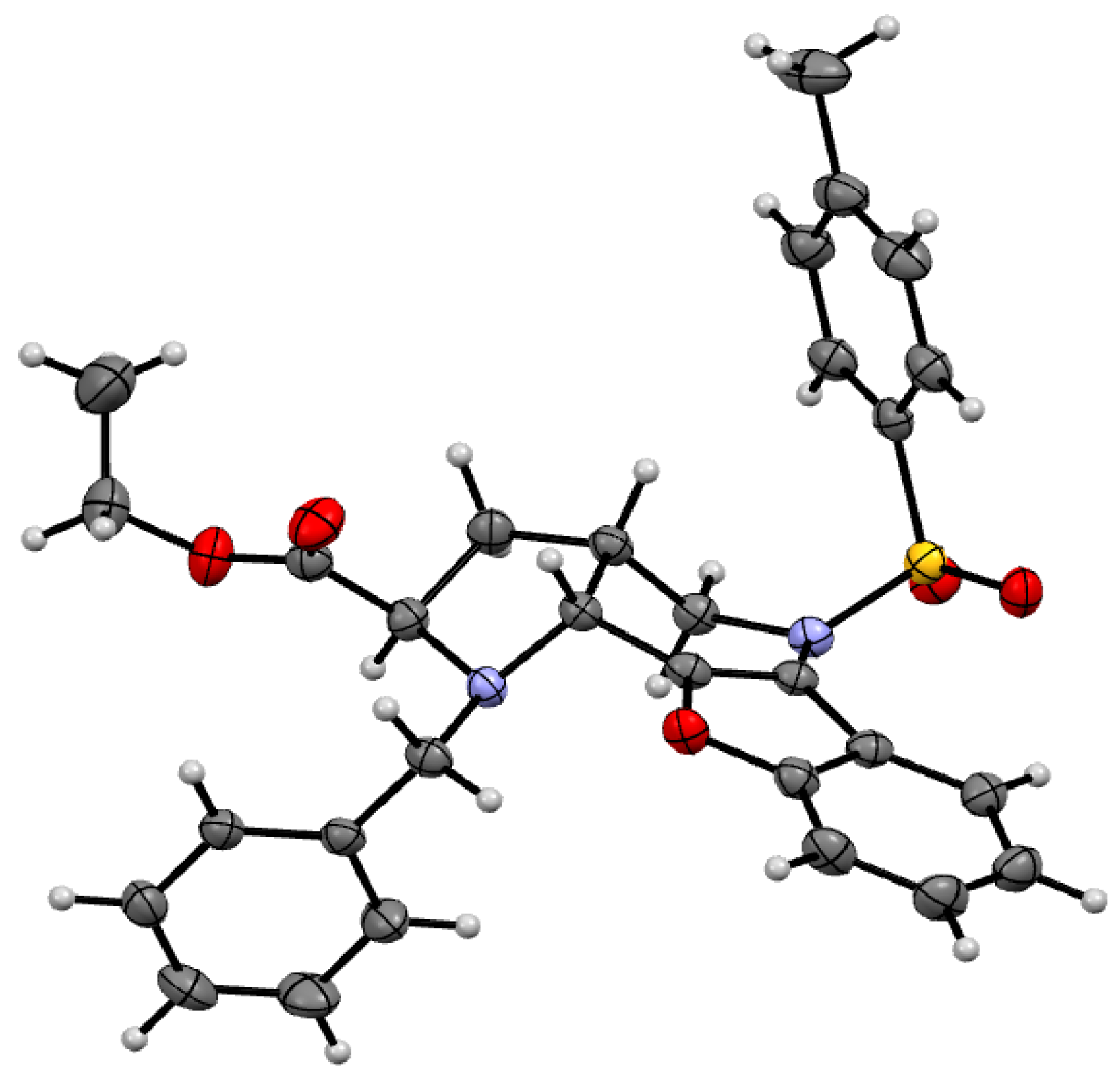
(2R,3aR,10bR) and (2S,3aS,10bS)-Ethyl-1-benzyl-5-mesyl-2,3,3a,4,5,10b-hexahydro-1H-benzo[b] furo[3,2-b]pyrrolo[2,3-d]pyridine-2-carboxylate (9Bc). Light yellow crystals, yield 29%, mp 106 °C. 1H-NMR: δ = 8.02 (d, J = 7.5, 1H, H6), 7.41 (d, J = 7.8, 1H, H9), 7.17–7.33 (m, 7H, 5H in C6H5, H8 and H7), 4.63 (d, J = 13.7, 1H, CH2C6H5), 4.58 (d, J = 6.8, 1H, H10b), 4.11–4.21 (m, 2H, COOCH2CH3), 4.07 (dd, J = 13.6, 4.6, 1H, H4), 3.96 (d, J = 13.7, 1H, CH2C6H5), 3.61 (dd, J = 8.9, 1H, H2), 3.41 (dd, J = 13.4, 11.2, 1H, H4), 2.98 (s, 3H, NSO2CH3), 2.79–2.88 (m, 1H, H3a), 2.21–2.29 (m, 1H, H3), 1.84–1.92 (m, 1H, H3), 1.27 (t, J = 7.1, 3H, COOCH2CH3). 13C-NMR: δ = 173.7 (COOCH2CH3), 154.0 (C9a), 147.0 (C10a), 145.1 (C5a), 138.7 (C C6H5), 128.5 (2CH C6H5), 128.2 (2CH C6H5), 127.1 (CH C6H5), 125.0 (C8), 123.2 (C7), 122.2 (C6), 119.1 (C5b), 111.5 (C9), 60.8 (C2), 60.4 (COCH2CH3), 55.4 (C10b), 53.0 (CH2C6H5), 50.8 (C4), 39.3 (SO2CH3), 34.9 (C3a), 31.7 (C3), 14.3 (COOCH2CH3). MS (EI 70 eV) Found 381[M − COOEt] (calculated for C24H26N2O5S: 454.539); m/z (%) = 381 (40), 301 (15), 211 (20), 91 (100). HRMS (ESI) m/z [M + H]+ calculated for C24H27N2O5S+: 455.1641). Found: 455.1642.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}