
DFT Studies on the Stereoselectivity of α-Silyloxy Diazoalkane Cycloadditions

Abstract
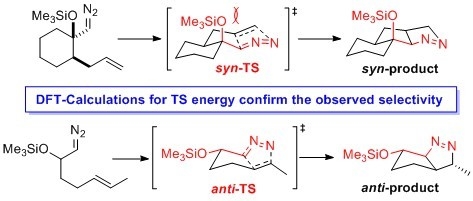
:
{kind=link}
{kind=link}
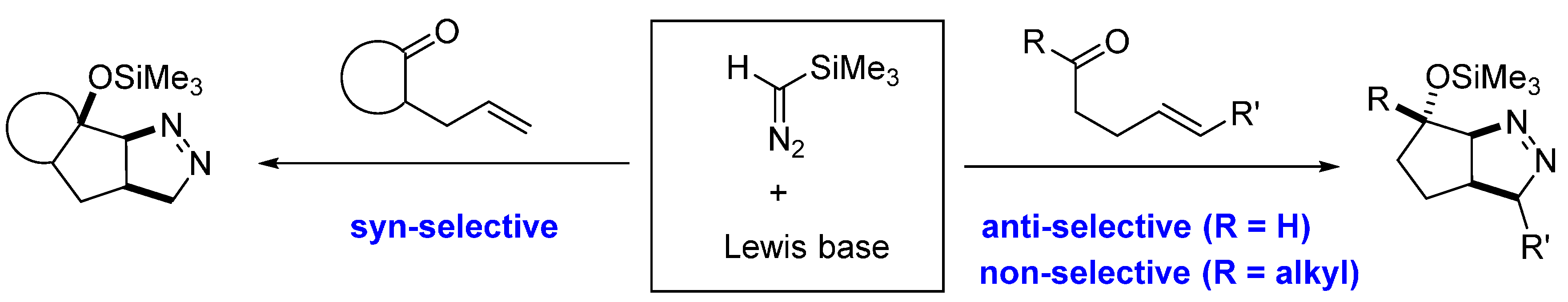{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
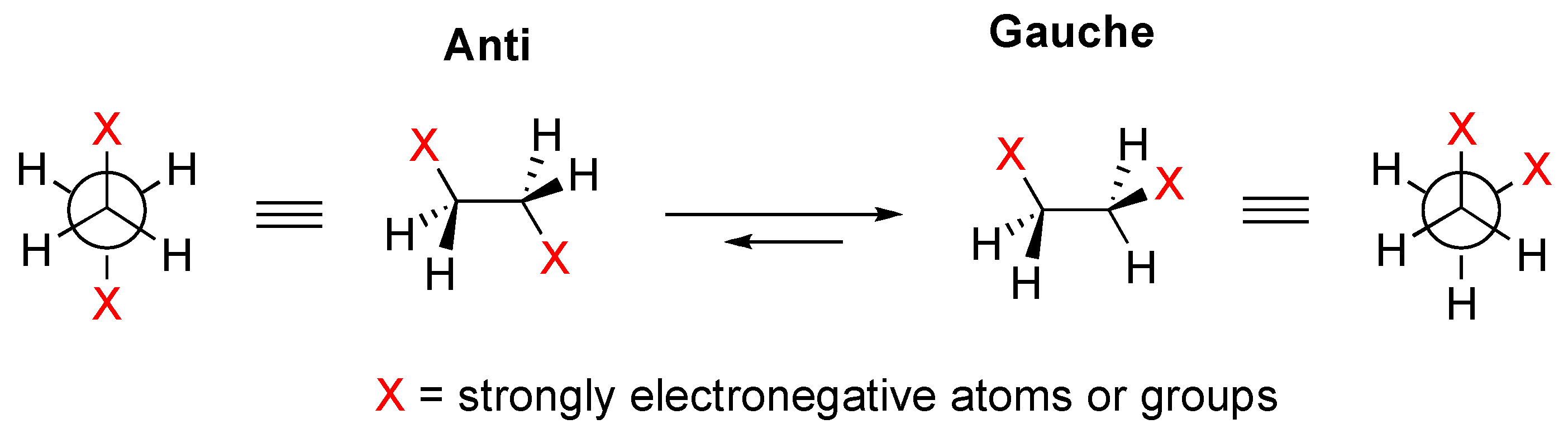
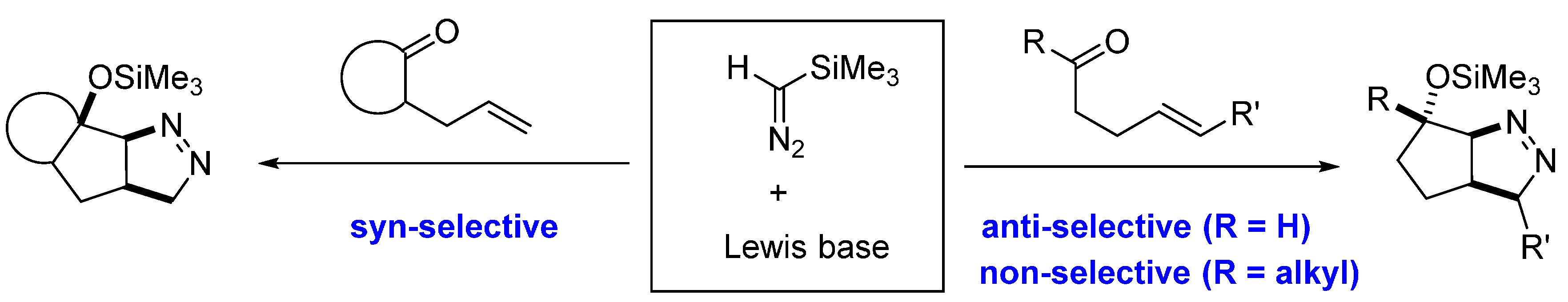
2. Results and Discussion
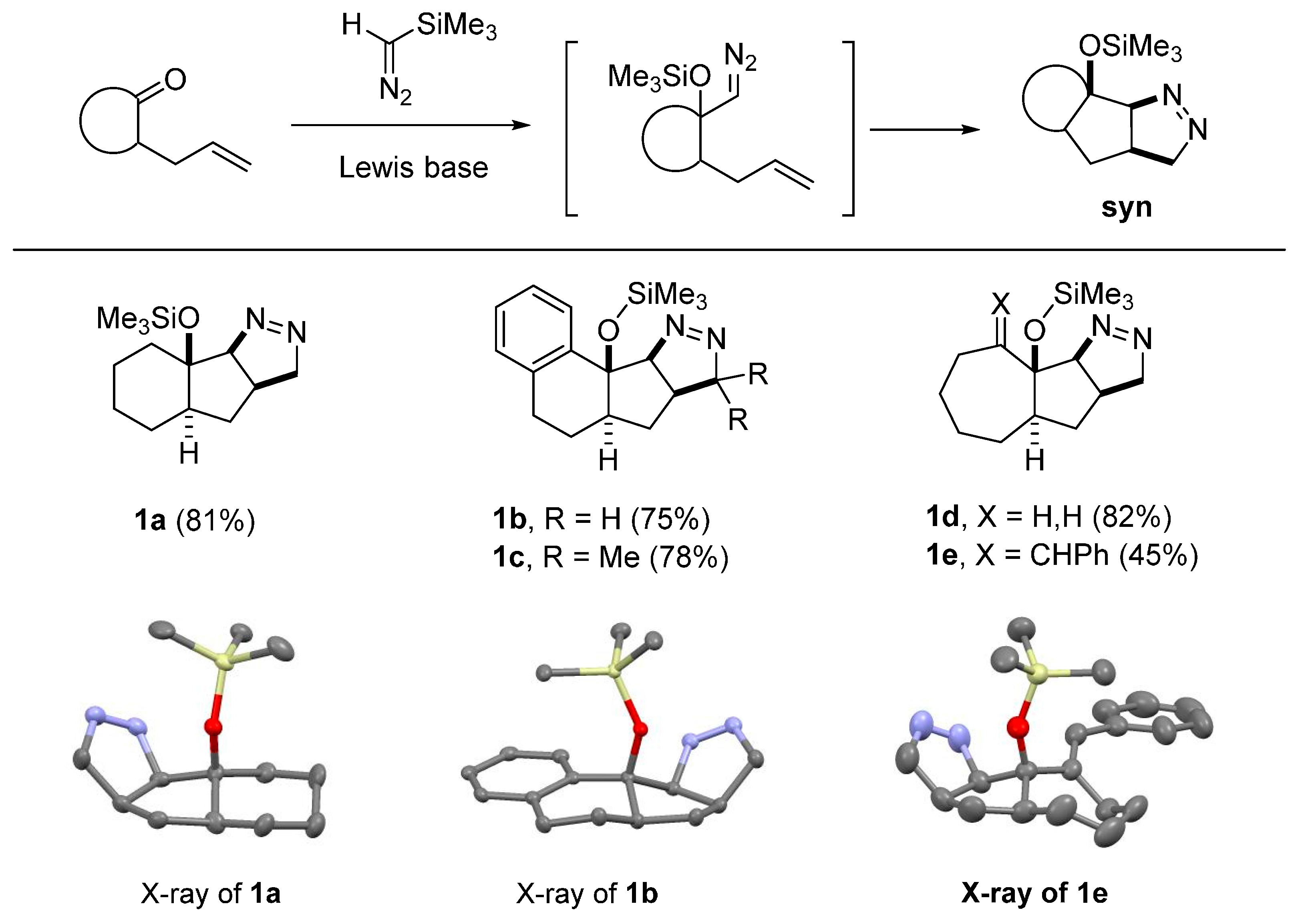
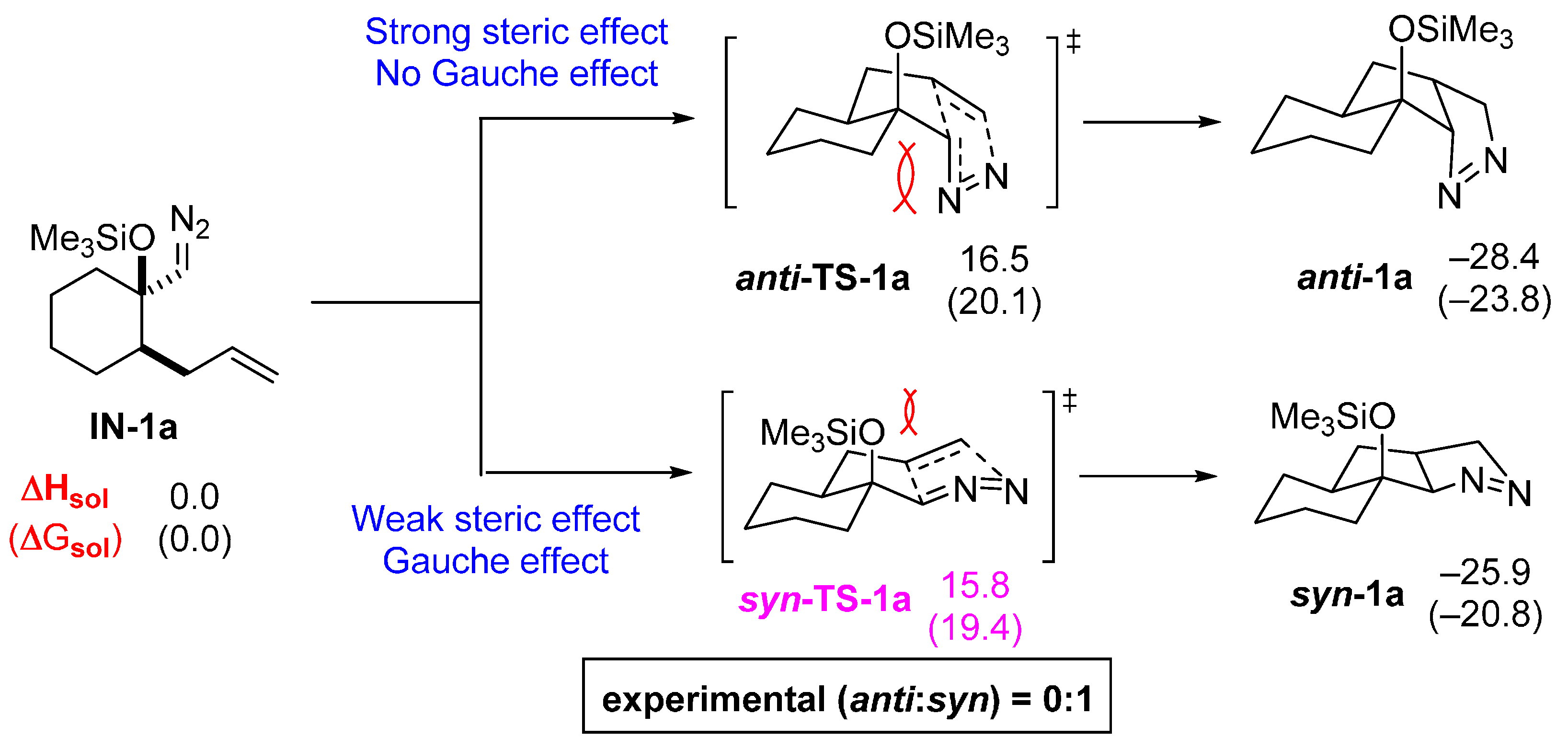
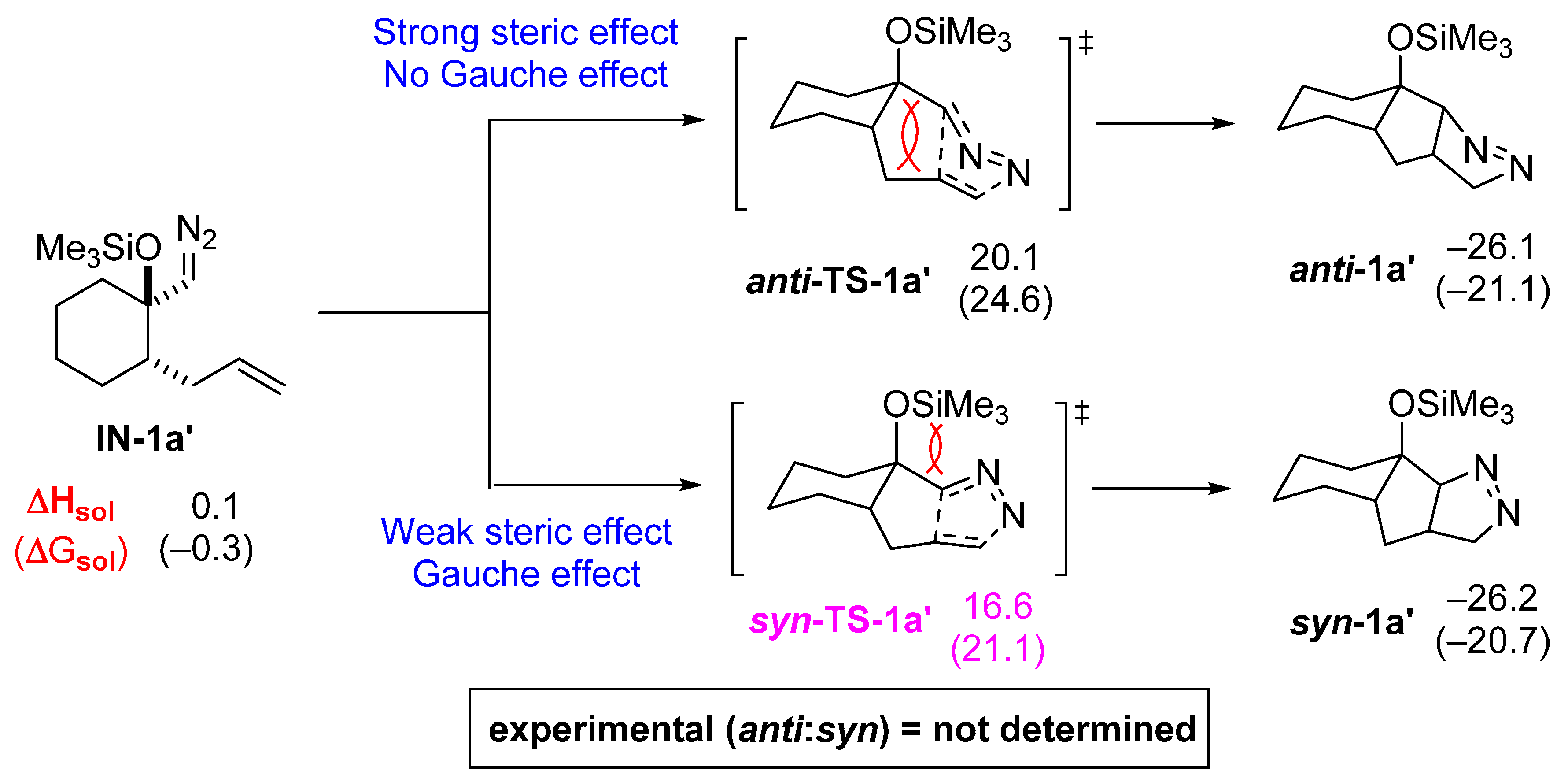
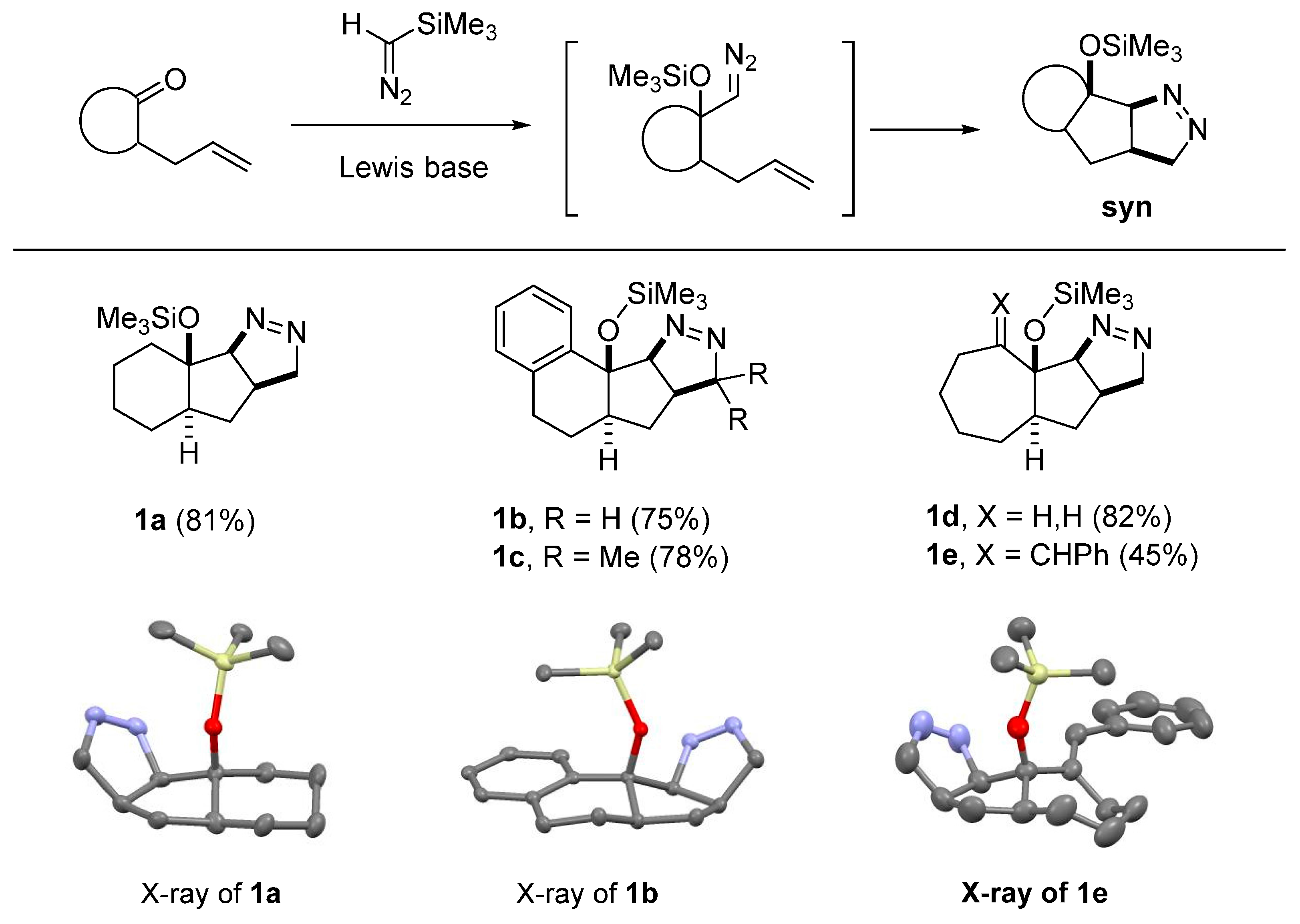
2.1. DFT Calculations on Pyrazolines Formed with High syn-Diastereoselectivity
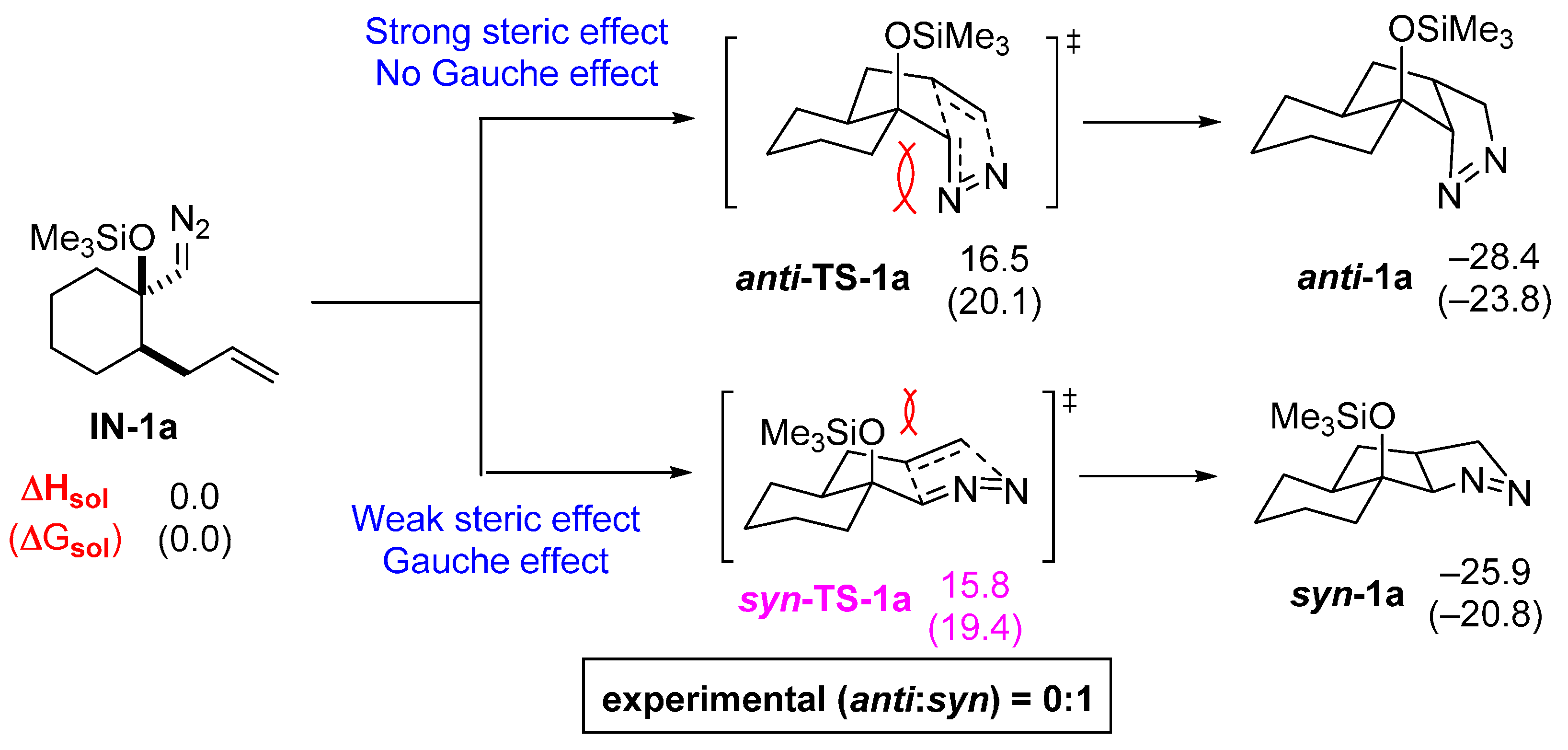
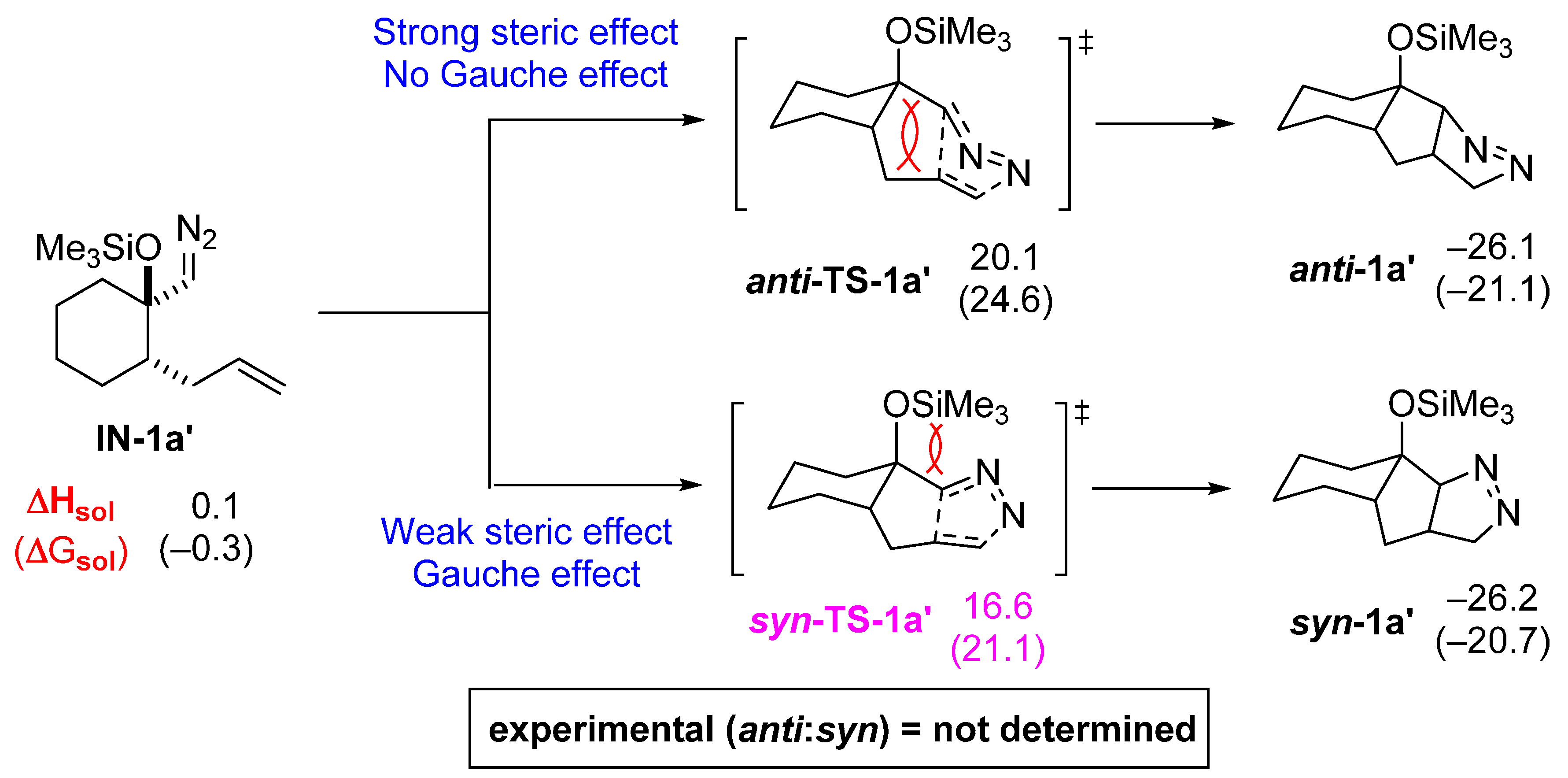
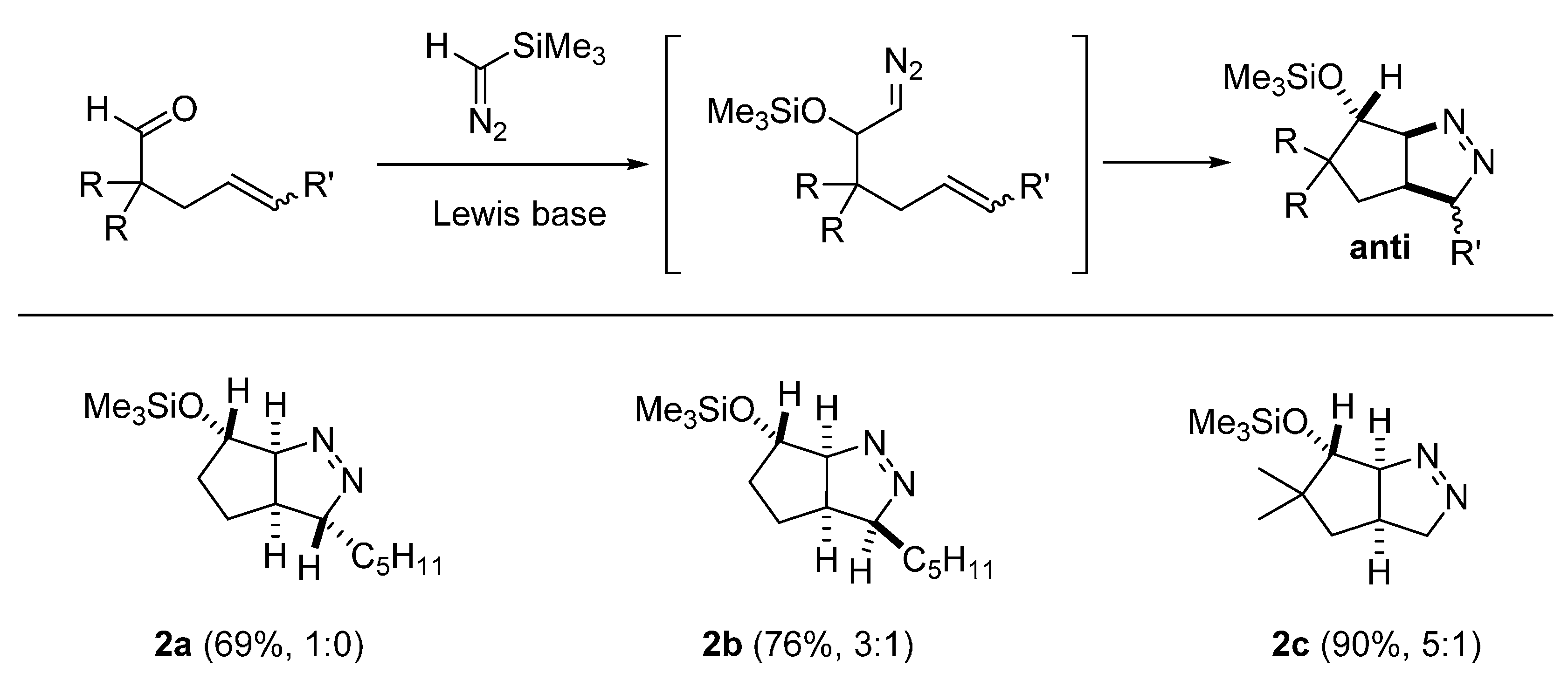
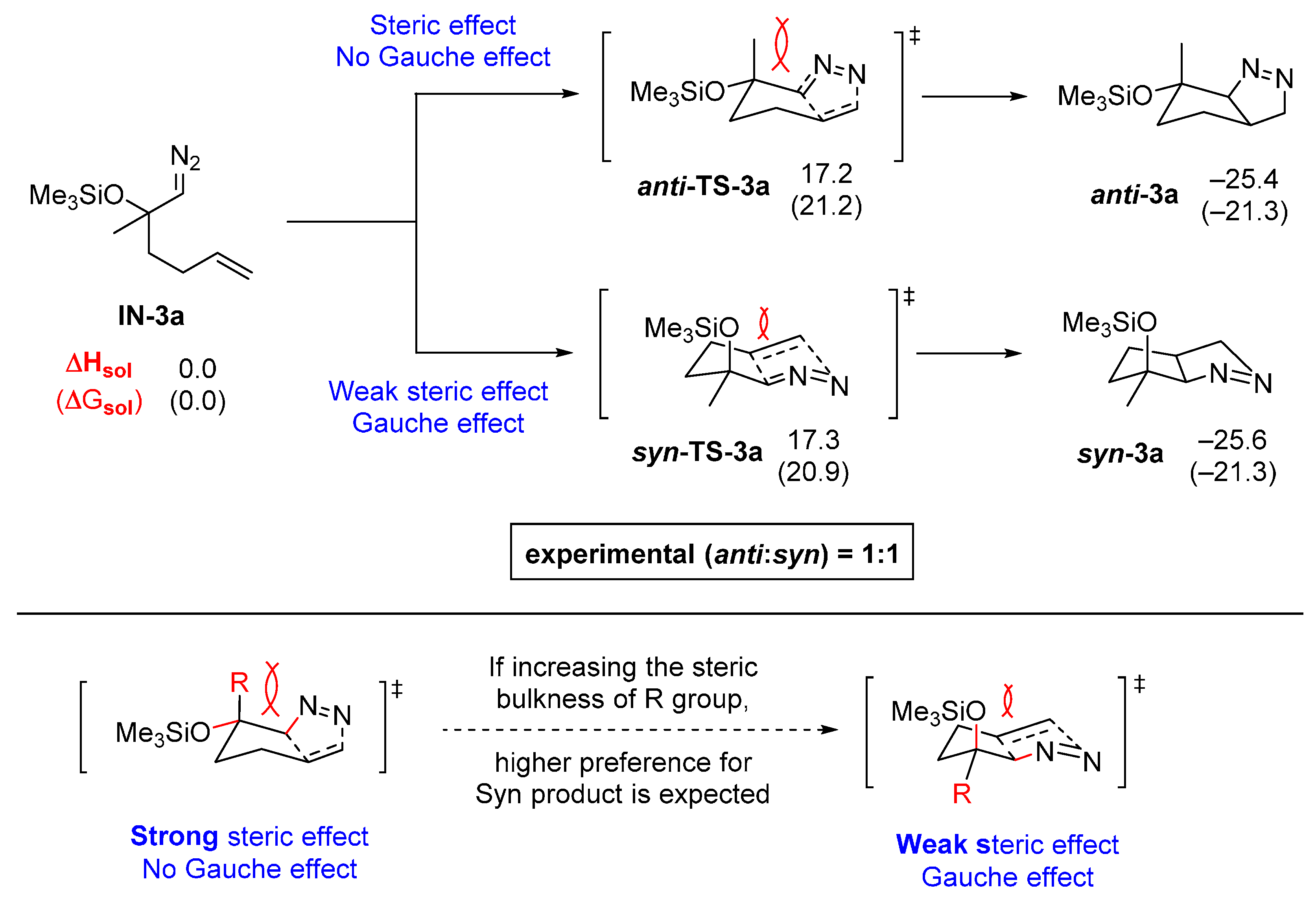
2.2. DFT Calculations on Pyrazolines Formed with High Anti-Diastereoselectivity
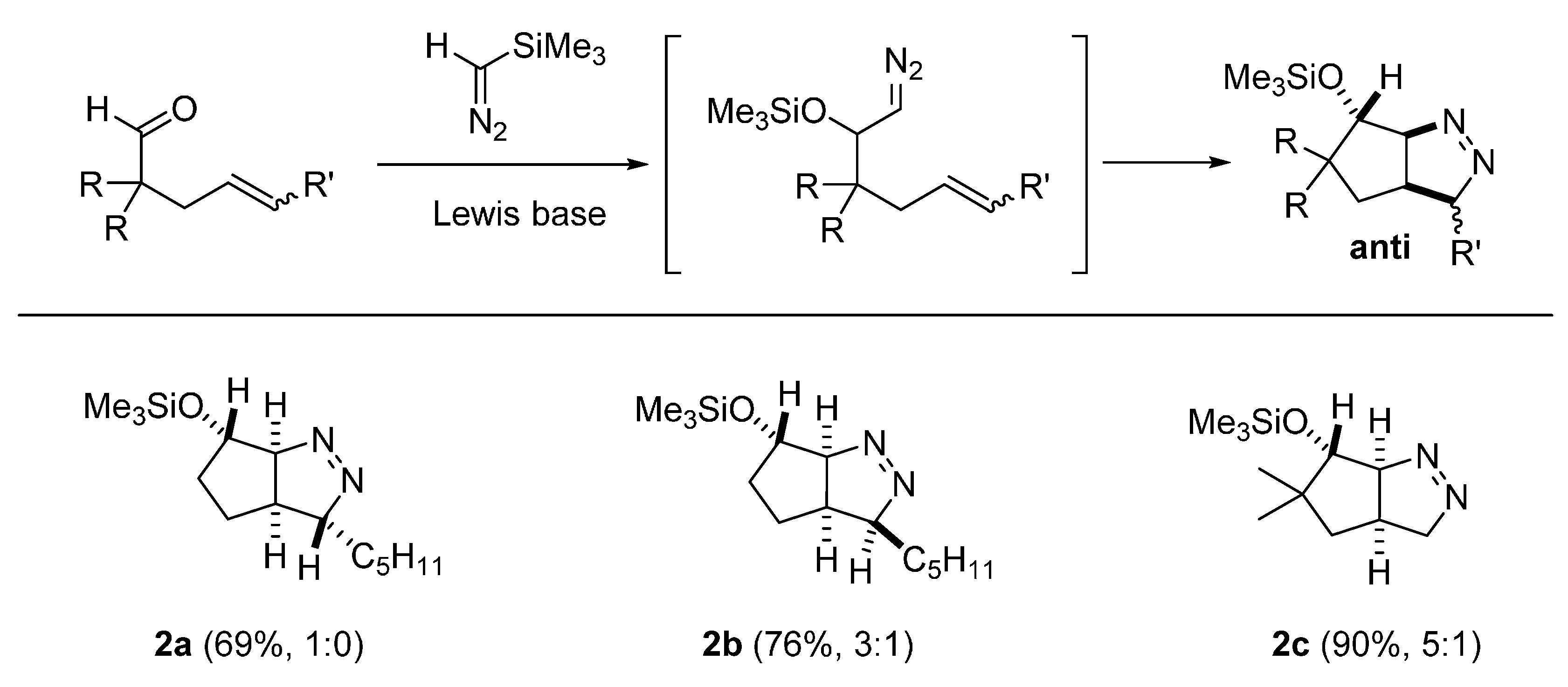

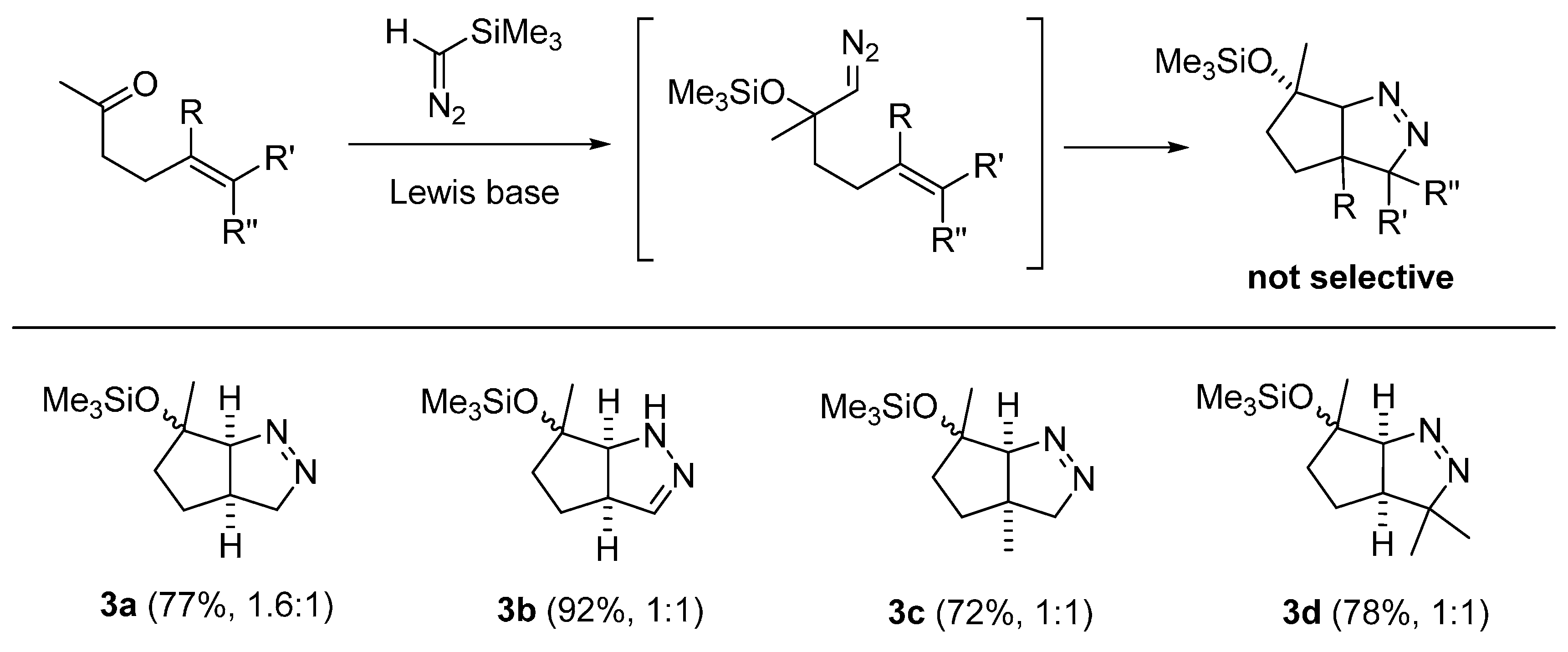
2.3. DFT Calculations on Pyrazolines Formed with Negligible Diastereoselectivity
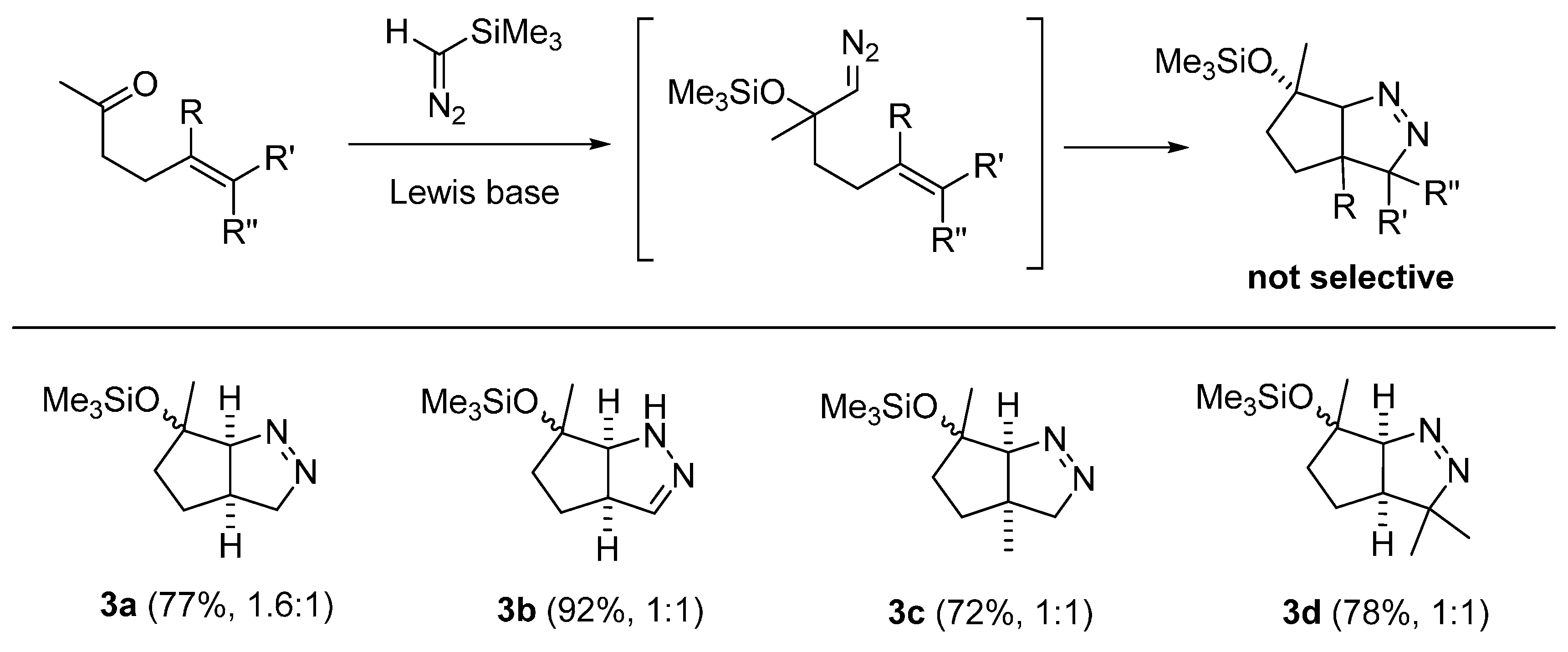
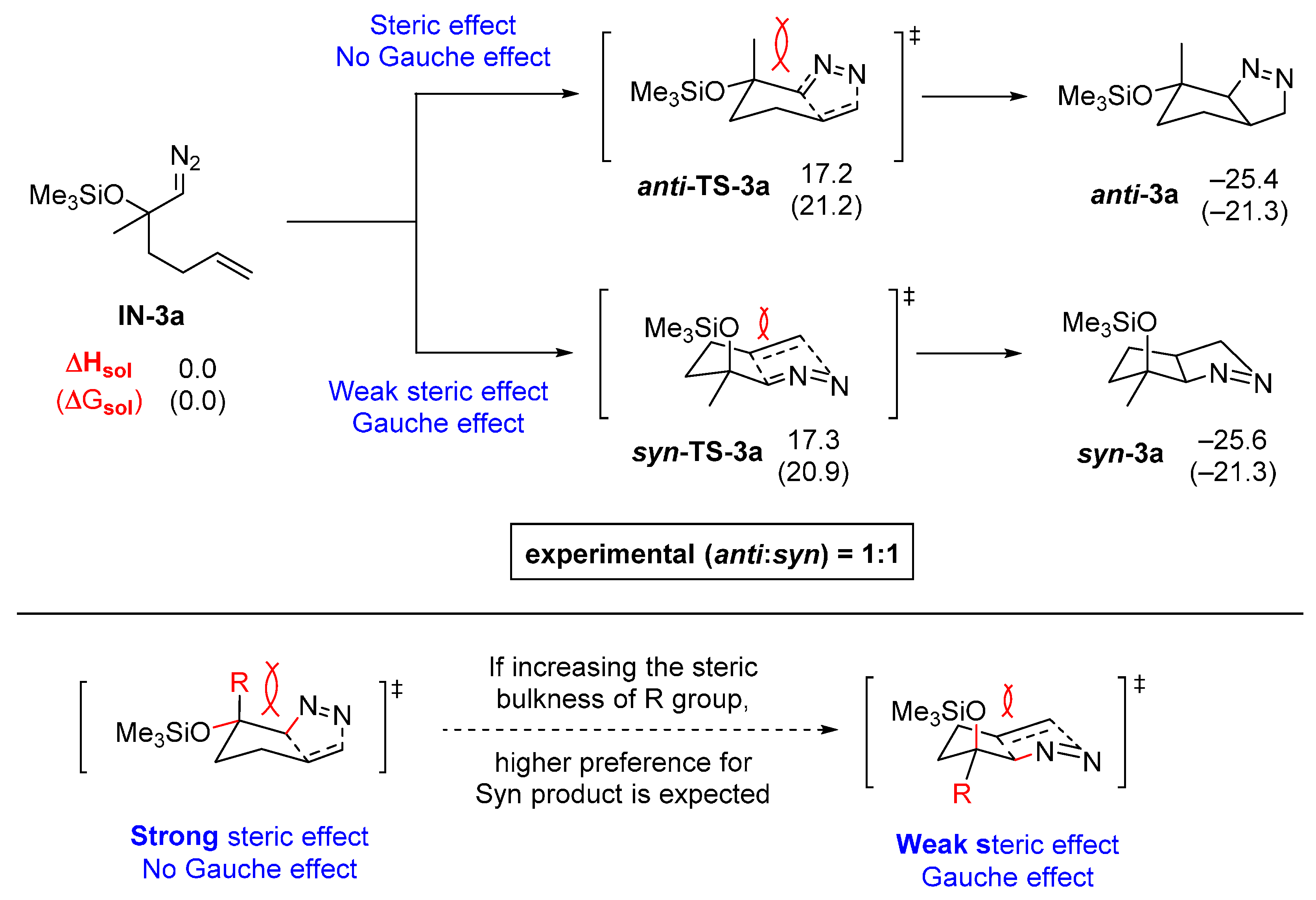
3. Experimental Section
3.1. Calculation Details
3.2. General Procedure for the Preparation of Pyrazolines
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
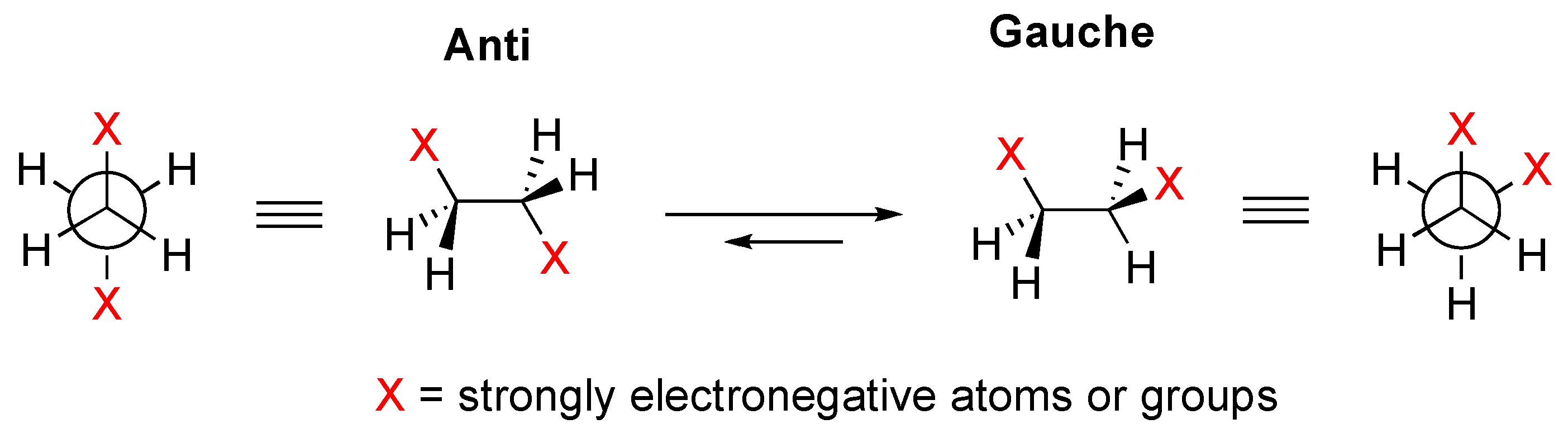
- Wolfe, S. Gauche effect. Stereochemical consequences of adjacent electron pairs and polar bonds. Acc. Chem. Res. 1972, 5, 102–111. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Murcko, M.A.; Laidig, K.E.; MacDougall, P.J. Origin of the Gauche Effect in substituted ethanes and ethenes. J. Phys. Chem. 1990, 94, 6956–6959. [Google Scholar] [CrossRef]
- Durig, J.R.; Liu, J.; Little, T.S.; Kalaskinsky, V.F. Conformational Analysls, Barriers to Internal Rotation, Vibrational Assignment, and ab Initio Calculations of 1,2-Difluoroethane. J. Phys. Chem. 1992, 96, 8224–8233. [Google Scholar] [CrossRef]
- Wiberg, K.B. Bent Bonds in Organic Compounds. Acc. Chem. Res. 1996, 29, 229–234. [Google Scholar] [CrossRef]
- Rablen, P.R.; Hoffmann, R.W.; Hrovat, D.A.; Borden, W.T. Is hyperconjugation responsible for the “gauche effect” in 1-fluoropropane and other 2-substituted-1-fluoroethanes? J. Chem. Soc. Perkin Trans. 2 1999, 1719–1726. [Google Scholar] [CrossRef]
- Buissonneaud, D.Y.; van Mourik, T.; O’Hagan, D. A DFT study on the origin of the fluorine gauche effect in substituted fluoroethanes. Tetrahedron 2010, 66, 2196–2202. [Google Scholar] [CrossRef]
- Craig, N.C.; Chen, A.; Suh, K.W.; Klee, S.; Mellau, G.C.; Winnewisser, B.P.; Winnewisser, M. Contribution to the Study of the Gauche Effect. The Complete Structure of the Anti Rotamer of 1,2-Difluoroethane. J. Am. Chem. Soc. 1997, 119, 4789–4790. [Google Scholar] [CrossRef]
- Engkvist, O.; Karlstrom, G.; Widmark, P.O. On the origin of the gauche effect. A quantum chemical study of 1,2-difluoroethane. Chem. Phys. Lett. 1997, 265, 19–23. [Google Scholar] [CrossRef]
- Goodman, L.; Gu, H.; Pophristic, V. Gauche Effect in 1,2-Difluoroethane. Hyperconjugation, Bent Bonds, Steric Repulsion. J. Phys. Chem. A 2005, 109, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Rey, Y.P.; Zimmer, L.E.; Sparr, C.; Tanzer, E.M.; Schweizer, W.B.; Senn, H.M.; Lakhdar, S.; Gilmour, R. Molecular Design Exploiting a Fluorine gauche Effect as a Stereoelectronic Trigger. Eur. J. Org. Chem. 2014, 2014, 1202–1211. [Google Scholar] [CrossRef]
- Zimmer, L.E.; Sparr, C.; Gilmour, R. Fluorine Conformational Effects in Organocatalysis: An Emerging Strategy for Molecular Design. Angew. Chem. Int. Ed. 2011, 50, 11860–11871. [Google Scholar] [CrossRef] [PubMed]
- Briggs, C.R.S.; Allen, M.J.; O’Hagan, D.; Tozer, D.J.; Slawin, A.M.Z.; Goeta, A.E.; Howard, J.A.K. The observation of a large gauche preference when 2-fluoroethylamine and 2-fluoroethanol become protonated. Org. Biomol. Chem. 2004, 2, 732–740. [Google Scholar] [CrossRef] [PubMed]
- DeRider, M.L.; Wilkens, S.J.; Waddell, M.J.; Bretscher, L.E.; Weinhold, F.; Raines, R.T.; Markley, J.L. Collagen Stability: Insights from NMR Spectroscopic and Hybrid Density Functional Computational Investigations of the Effect of Electronegative Substituents on Prolyl Ring Conformations. J. Am. Chem. Soc. 2002, 124, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, K.N.; Woods, R.J. Solvent interactions determine carbohydrate conformation. Proc. Natl. Acad. Sci. USA 2001, 98, 10541–10545. [Google Scholar] [CrossRef] [PubMed]
- Plavec, J.; Tong, W.; Chattopadhyaya, J. How do the gauche and anomeric effects drive the pseudorotational equilibrium of the pentofuranose moiety of nucleosides? J. Am. Chem. Soc. 1993, 115, 9734–9746. [Google Scholar] [CrossRef]
- Hodges, J.A.; Raines, R.T. Stereoelectronic Effects on Collagen Stability: The Dichotomy of 4-Fluoroproline Diastereomers. J. Am. Chem. Soc. 2003, 125, 9262–9263. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; O’Connor, M.J.; Sun, C.; Wink, D.J.; Lee, D. Sequential Reactions of Trimethylsilyldiazomethane with Carbonyls & Tethered Alkenes Catalyzed by Lewis Bases. Org. Lett. 2013, 15, 2974–2977. [Google Scholar] [PubMed]
- CCDC-1430957 (1a), CCDC-962222 (1b), CCDC-962219 (1e), and CCDC-1430960 (PNB-2a) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Connor, M.J.; Liu, H.; Lee, D.; Zhou, T.; Xia, Y. DFT Studies on the Stereoselectivity of α-Silyloxy Diazoalkane Cycloadditions. Molecules 2015, 20, 21433-21441. https://doi.org/10.3390/molecules201219783
O’Connor MJ, Liu H, Lee D, Zhou T, Xia Y. DFT Studies on the Stereoselectivity of α-Silyloxy Diazoalkane Cycloadditions. Molecules. 2015; 20(12):21433-21441. https://doi.org/10.3390/molecules201219783
Chicago/Turabian StyleO’Connor, Matthew J., Huaqing Liu, Daesung Lee, Tao Zhou, and Yuanzhi Xia. 2015. "DFT Studies on the Stereoselectivity of α-Silyloxy Diazoalkane Cycloadditions" Molecules 20, no. 12: 21433-21441. https://doi.org/10.3390/molecules201219783
APA StyleO’Connor, M. J., Liu, H., Lee, D., Zhou, T., & Xia, Y. (2015). DFT Studies on the Stereoselectivity of α-Silyloxy Diazoalkane Cycloadditions. Molecules, 20(12), 21433-21441. https://doi.org/10.3390/molecules201219783