3.2. Chemistry
3.2.1. Synthesis of 2,2-Dimethyl-δ-valerolactone (2h)
To a magnetically stirred solution of diisopropylamine (23.27 g, 230 mmol) in dried THF (250 mL) cooled at −78 °C under N2 was added dropwise 1.6 M n-BuLi in n-hexane (131 mL, 210 mmol) via syringe. After addition, the resulting mixture was stirred at this temperature for 10 min, followed by dropwise addition of δ-valerolactone 2b (10.01 g, 100 mmol). The stirring was continued at this temperature for another 0.5 h, followed by successive additions of MeI (28.39 g, 200 mmol) and DMPU (14.10 g, 110 mmol) in a dropwise manner via syringe. After addition, the reaction mixture was stirred at room temperature overnight.
The reaction mixture was concentrated on a rotary evaporator to about 100 mL and then poured into ice-water (500 mL). The resulting aqueous mixture was extracted with CH
2Cl
2 (100 mL × 3), and the combined extracts were washed successively with 1 M hydrochloric acid (100 mL × 3) and 5% brine (100 mL), dried over anhydrous Na
2SO
4, and evaporated on a rotary evaporator to afford a residue, which was purified by column chromatography to yield a colorless oil. The colorless oil was subjected to the identical procedure described above once again to yield
2h after column chromatography. Colorless oil, 9.10 g (71%).
1H-NMR (DMSO-
d6, 400 MHz) δ: 4.27 (t,
J = 5.8 Hz, 2H), 1.78–1.84 (m, 2H), 1.68–1.71 (m, 2H), 1.17 (s, 6H). The
1H-NMR data were in good agreement with those reported [
35].
3.2.2. General Procedure for the Synthesis of Lactones 2i and 2j
To a stirred suspension of NaBH4 (5.67 g, 150 mmol) in dried THF (50 mL) cooled in an ice-water bath was added dropwise a solution of 18 or 19 (100 mmol) in dried THF (50 mL). The resulting mixture was stirred at room temperature for 5 h and then re-cooled in an ice-water bath, followed by addition of 6 M hydrochloric acid (50 mL). The mixture thus obtained was stirred for another 5 min and poured into ice-water (300 mL). The resulting mixture was extracted with CH2Cl2 (100 mL × 3), and the combined extracts were washed with 5% brine (100 mL), dried over anhydrous Na2SO4, and evaporated on a rotary evaporator to afford a residue, which was purified by column chromatography to yield 2i or 2j.
3,3-Dimethyl-δ-valerolactone (
2i): Colorless oil; 9.36 g (73%).
1H-NMR (DMSO-
d6, 400 MHz) δ: 4.28 (t,
J = 6.2 Hz, 2H), 2.27 (s, 2H), 1.62 (t,
J = 6.0 Hz, 2H), 0.99 (s, 6H). The
1H-NMR data were in good agreement with those reported [
36].
8-Oxaspiro[4,5]decan-7-one (2j): Colorless oil; 11.41 g (74%). 1H-NMR (DMSO-d6, 400 MHz) δ: 4.28 (t, J = 6.0 Hz, 2H), 2.37 (s, 2H), 1.70 (t, J = 6.2 Hz, 2H), 1.45–1.62 (m, 4H), 1.37–1.42 (m, 2H).
3.2.3. General Procedure for the Synthesis of Acyl Hydrazides 3a–3k
To a stirred solution of esters 2a–2j (70 mmol) in MeOH (30 mL) cooled in an ice-water bath was added dropwise 80% aqueous hydrazine hydrate (6.26 g, 100 mmol). The resulting solution was stirred at room temperature (2a–2b or 2d–2j), or reflux (2c), until the completion of reaction as indicated by TLC analysis (typically within 5 h).
The reaction mixture was evaporated on a rotary evaporator to give a residue, which was purified by column chromatography through a short silica gel column to yield 3a–3k after trituration with n-hexane if possible.
4-Pentenoyl hydrazide (
3a): White solid; 6.47 g (81%); m.p. 44.5–45.5 °C (literature value, 45 °C [
37]).
1H-NMR (DMSO-
d6, 400 MHz) δ: 8.92 (brs, 1H), 5.72–5.82 (m, 1H), 4.98–5.03 (m, 1H), 4.92–4.95 (m, 1H), 4.13 (brs, 2H), 2.20–2.25 (m, 2H), 2.08 (t,
J = 7.6 Hz, 2H).
5-Hydroxypentanoyl hydrazide (
3b): White solid; 7.59 g (82%); m.p. 108–109.5 °C (literature value, 107–108 °C [
38]).
1H-NMR (DMSO-
d6, 400 MHz) δ: 8.89 (brs, 1H), 4.34 (brs, 1H), 4.12 (brs, 2H), 3.35 (t,
J = 6.4 Hz, 2H), 1.99 (t,
J = 7.4 Hz, 2H), 1.45–1.53 (m, 2H), 1.33–1.40 (m, 2H).
6-Hydroxyhexanoyl hydrazide (
3c): White solid; 8.39 g (82%); m.p. 116.5–118 °C (literature value, 114.5–116 °C [
39]).
1H-NMR (DMSO-
d6, 400 MHz) δ: 8.88 (brs, 1H), 4.31 (t,
J = 5.0 Hz, 1H), 4.11 (brs, 2H), 3.35 (q,
J = 6.0 Hz, 2H), 1.98 (t,
J = 7.6 Hz, 2H), 1.42–1.50 (m, 2H), 1.35–1.40 (m, 2H), 1.19–1.26 (m, 2H).
2-Hydroxyacetyl hydrazide (
3d): White solid; 5.36 g (85%); m.p. 91.5–93 °C (literature value, 93 °C [
40]).
1H-NMR (DMSO-
d6, 400 MHz) δ: 8.81 (brs, 1H), 5.28 (brs, 1H), 4.20 (brs, 2H), 3.82 (s, 2H).
5-Methylpyrazolidin-3-one (
3e): Colorless thick oil; 0.56 g (8%);
1H-NMR (DMSO-
d6, 400 MHz) δ: 8.89 (brs, 1H), 5.06 (brs, 1H), 3.46-3.51 (m, 1H), 2.29 (dd,
J = 7.0 Hz and 15.4 Hz, 1H), 1.91 (dd,
J = 8.2 Hz and 15.8 Hz, 1H), 1.09 (d,
J = 6.4 Hz, 3H). The
1H-NMR data were in good agreement with those reported [
29].
3-Butenoyl hydrazide (
3f): White solid; 5.19 g (74%); m.p. 47.5–48.5 °C.
1H-NMR (DMSO-
d6, 400 MHz) δ: 8.97 (brs, 1H), 5.79–5.89 (m, 1H), 5.03–5.10 (m, 2H), 4.15 (brs, 2H), 2.81 (d,
J = 6.8 Hz, 2H). The
1H-NMR data were in good agreement with those reported [
41].
3-Hydroxypropionyl hydrazide (
3g): White solid; 5.76 g (79%); m.p. 102–103.5 °C (literature value, 103–104 °C [
42]).
1H-NMR (DMSO-
d6, 400 MHz) δ: 8.93 (brs, 1H), 4.57 (brs, 1H), 4.15 (brs, 2H), 3.58 (t,
J = 6.6 Hz, 2H), 2.16 (t,
J = 6.6 Hz, 2H).
3-Hydroxybutanoyl hydrazide (
3h): White solid; 6.86 g (83%); m.p. 124–126 °C (literature value, 126–128 °C [
43]).
1H-NMR (DMSO-
d6, 400 MHz) δ: 8.90 (brs, 1H), 4.58 (brs, 1H), 4.12 (brs, 2H), 3.91–3.98 (m, 1H), 2.14 (dd,
J = 7.0 Hz and 13.8 Hz, 1H), 2.03 (dd,
J = 6.0 Hz and 13.6 Hz, 1H), 1.03 (d,
J = 6.0 Hz, 3H).
5-Hydroxy-2,2-dimethylpentanoyl hydrazide (3i): Colorless oil; 9.76 g (87%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.71 (brs, 1H), 4.32 (t, J = 5.2 Hz, 1H), 4.13 (brs, 2H), 3.28–3.33 (m, 2H), 1.37–1.41 (m, 2H), 1.23–1.33 (m, 2H), 1.03 (s, 6H).
5-Hydroxy-3,3-dimethylpentanoyl hydrazide (3j): White solid; 9.20 g (82%); m.p. 57.5–59 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.87 (brs, 1H), 4.33 (brs, 1H), 4.16 (brs, 2H), 3.44–3.47 (m, 2H), 1.91 (s, 2H), 1.44 (t, J = 7.4 Hz, 2H), 0.91 (s, 6H).
2-(1-(2-Hydroxyethyl)cyclopentyl)acetyl hydrazide (3k): Colorless oil; 11.21 g (86%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.90 (brs, 1H), 4.38 (brs, 1H), 4.16 (brs, 2H), 3.47 (t, J = 7.2 Hz, 2H), 1.99 (s, 2H), 1.49–1.57 (m, 8H), 1.30–1.37 (m, 2H).
3.2.4. Synthesis of (4-Bromonaphth-1-yl)methylamine 4j
A suspension of
11 (35.37 g, 160 mmol), BPO (0.78 g, 3.2 mmol), and NBS (34.17 g, 192 mmol) in
n-hexane (400 mL) was refluxed under N
2 until the completion of reaction as indicated by TLC analysis (typically 36 h; once the reaction commenced, 0.78 g of BPO was added every 8 h until the reaction completed). The reaction mixture was cooled to room temperature while stirring, and the precipitates were collected via vacuum filtration. The precipitates were triturated successively with saturated aqueous NaHCO
3 (500 mL × 2), water (800 mL × 2), and
n-hexane (800 mL) to give rise to (4-bromonaphth-1-yl)methyl bromide
12. White solid; 35.04 g (73%); m.p. 104.5–106 °C (literature value, 102–104 °C [
44]).
1H-NMR (DMSO-
d6, 400 MHz) δ: 8.20–8.26 (m, 2H), 7.85 (d,
J = 7.6 Hz, 1H), 7.71–7.77 (m, 2H), 7.62 (d,
J = 7.6 Hz, 1H), 5.21 (s, 2H).
A mixture of 12 (33.00 g, 110 mmol) and potassium phthalimide (20.37 g, 110 mmol) in DMF (200 mL) was stirred at 100 °C under N2 until the completion of reaction as indicated by TLC analysis (typically within 12 h). On cooling to room temperature, the reaction mixture was poured into ice-water (600 mL), and the aqueous mixture thus obtained was extracted with CH2Cl2 (150 mL × 3). The combined extracts were washed with 5% brine (100 mL × 5), dried over anhydrous Na2SO4, and evaporated on a rotary evaporator to afford a residue, which was recrystallized from ethanol to produce N-((4-bromonaphth-1-yl)methyl)phthalimide 13. White solid; 36.66 g (91%); m.p. 168.5–170 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.30–8.33 (m, 1H), 8.17–8.21 (m, 1H), 7.83–7.92 (m, 4H), 7.80 (d, J = 8.0 Hz, 1H), 7.69–7.74 (m, 2H), 7.32 (d, J = 7.6 Hz, 1H), 5.23 (s, 2H).
A mixture of 13 (32.96 g, 90 mmol) and 80% aqueous hydrazine hydrate (11.26 g, 180 mmol) in ethanol (600 mL) was refluxed until the completion of reaction as indicated by TLC analysis (typically within 12 h), when a white slurry was formed. On slight cooling, 1 M aqueous NaOH (300 mL) was added to the reaction mixture, which turned to a clear solution and was concentrated on a rotary evaporator to half its original volume. The residue was poured into ice-water (350 mL). The aqueous mixture thus obtained was extracted with CH2Cl2 (100 mL × 3). The combined extracts were washed successively with 5% aqueous NaOH (100 mL × 2) and 5% brine (100 mL), dried over anhydrous Na2SO4, and evaporated on a rotary evaporator to afford a residue, which was purified by column chromatography through a short silica gel column to produce 4j. Colorless oil; 18.27 g (86%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.15–8.18 (m, 2H), 7.83 (d, J = 7.6 Hz, 1H), 7.61–7.70 (m, 2H), 7.49 (d, J = 7.6 Hz, 1H), 4.17 (s, 2H), 1.94 (brs, 2H).
3.2.5. Synthesis of 2-(4-Bromonaphth-1-yl)ethylamine 4k
To a magnetically stirred solution of 1,4-dibromonaphthalene 14 (57.19 g, 200 mmol) in dried THF (600 mL) cooled at −78 °C under N2 was added dropwise 1.6 M n-BuLi in n-hexane (125 mL, 200 mmol) via syringe. After addition, the resulting mixture was stirred at this temperature for another 0.5 h, followed by addition of B(i-PrO)3 (75.23 g, 400 mmol) in a dropwise manner via syringe. The reaction mixture was slowly warmed to room temperature and stirred at room temperature for another 1 h. The reaction mixture was slowly poured into ice-water (600 mL) with concentrated hydrochloric acid (10 mL) while stirring. The precipitates formed were collected via vacuum filtration, washed with cooled water, and triturated with EtOAc/n-hexane to yield 4-bromonaphthalene-1-boronic acid 15. White solid; 46.16 g (92%). A varying amount of boronic anhydride was found to exist in the sample of 15 and therefore 15 was directly used in the next step without further structural characterization.
A mixture of 15 (45.16 g, 180 mmol), aminoacetonitrile hydrochloride (33.31 g, 360 mmol) and NaNO2 (31.05 g, 450 mmol) in toluene (600 mL)/water (30 mL) was stirred at 50 °C until the completion of reaction as indicated by TLC analysis (typically within 12 h). On cooling to room temperature, the reaction mixture was poured into ice-water (300 mL) and the organic phase was separated. The aqueous phase was back-extracted with toluene (100 mL). The combined extracts were washed with 5% brine, dried over anhydrous Na2SO4, and evaporated on a rotary evaporator to afford a residue, which was purified by column chromatography to produce 2-(4-bromonaphth-1-yl)acetonitrile 16. White solid; 35.88 g (81%); m.p. 74.5–75.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.21–8.24 (m, 1H), 8.08–8.11 (m, 1H), 7.92 (d, J = 7.6 Hz, 1H), 7.74–7.79 (m, 2H), 7.53 (d, J = 7.6 Hz, 1H), 4.50 (s, 2H).
To a stirred solution of 16 (34.45 g, 140 mmol) in dried THF (300 mL) cooled in an ice-water bath was added portionwise LiAlH4 (10.63 g, 280 mmol). The resulting mixture was stirred at room temperature under N2 until the completion of reaction as indicated by TLC analysis (typically within 12 h). An appropriate amount of water was carefully added to the stirred reaction mixture to decompose the excess LiAlH4, and the mixture thus obtained was filtered off through celite. The filtrate was poured into ice-water (600 mL), and the resulting mixture was extracted with CH2Cl2 (200 mL × 3). The combined extracts were washed with 5% brine (100 mL), dried over anhydrous Na2SO4, and evaporated on a rotary evaporator to afford a residue, which was purified by column chromatography through a short silica gel column to produce 4k. Colorless oil; 25.56 g (73%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.15–8.19 (m, 2H), 7.79 (d, J = 7.6 Hz, 1H), 7.64–7.69 (m, 2H), 7.29 (d, J = 7.6 Hz, 1H), 3.11 (t, J = 7.4 Hz, 2H), 2.84 (t, J = 7.4 Hz, 2H), 1.73 (brs, 2H).
3.2.6. General Procedure for the Synthesis of 5a–5r and 17
A mixture of 3a–3d or 3f–3k (75 mmol) and DMFDMA (8.94 g, 75 mmol) in MeCN (100 mL) was stirred at 50 °C in an open vessel in a well-ventilated hood until the completion of reaction as indicated by TLC analysis (typically within 2 h). The reaction mixture was evaporated on a rotary evaporator to almost dryness and the residue thus obtained was dissolved in glacial acetic acid (100 mL), followed by addition of 4a–4k (75 mmol). The resulting mixture was refluxed until the completion of reaction as indicated by TLC analysis (typically within 12 h).
On slight cooling, the reaction mixture was concentrated on a rotary evaporator to approximately 50 mL and poured into ice-water (600 mL) while stirring, and the resulting mixture was extracted with CH2Cl2 (200 mL × 3). The combined extracts were washed successively with 1 M hydrochloric acid (100 mL), saturated aqueous NaHCO3 (until the aqueous pH > 7 persistently), and 5% brine (100 mL), dried over anhydrous Na2SO4, and evaporated on a rotary evaporator to afford a residue, which was purified by column chromatography to produce 5a–5r or 17 after trituration with EtOAc/n-hexane if possible.
3-(3-Buten-1-yl)-4-(4-cyclopropylnaphth-1-yl)-4H-1,2,4-triazole (5a): Colorless oil; 16.93 g (78%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.68 (s, 1H), 8.55 (d, J = 8.4 Hz, 1H), 7.69–7.74 (m, 1H), 7.61–7.65 (m, 1H), 7.55 (dd, J = 3.4 Hz and 7.4 Hz, 1H), 7.39 (d, J = 7.6 Hz, 1H), 7.12 (d, J = 8.4 Hz, 1H), 5.61–5.71 (m, 1H), 4.83–4.88 (m, 2H), 2.49–2.55 (m, 1H), 2.20-2.26 (m, 2H), 1.08–1.17 (m, 2H), 0.84–0.89 (m, 2H), 0.76–0.78 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 153.66, 144.97, 141.67, 136.81, 133.32, 129.10, 128.11, 127.78, 127.04, 125.22, 124.91, 122.51, 121.80, 115.60, 30.54, 23.40, 12.81, 7.26, 6.79.
3-(3-Buten-1-yl)-4-(naphth-1-yl)-4H-1,2,4-triazole (5b): Colorless oil; 14.02 g (75%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.73 (s, 1H), 8.16–8.20 (m, 1H), 8.11–8.13 (m, 1H), 7.60–7.71 (m, 4H), 7.15 (d, J = 8.0 Hz, 1H), 5.61–5.71 (m, 1H), 4.86–4.87 (m, 1H), 4.83 (d, J = 0.8 Hz, 1H), 2.54 (t, J = 7.6 Hz, 2H), 2.20–2.25 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 153.59, 144.89, 136.78, 133.66, 130.12, 129.85, 129.11, 128.49, 128.14, 127.13, 125.62, 125.60, 121.25, 115.62, 30.55, 23.44.
3-(3-Buten-1-yl)-4-(4-methylnaphth-1-yl)-4H-1,2,4-triazole (5c): Colorless oil; 15.01 g (76%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.69 (s, 1H), 8.18 (d, J = 8.4 Hz, 1H), 7.69 (t, J = 7.6 Hz, 1H), 7.62 (t, J = 7.6 Hz, 1H), 7.52–7.58 (m, 2H), 7.11 (d, J = 8.4 Hz, 1H), 5.63–5.70 (m, 1H), 4.83–4.87 (m, 2H), 2.75 (s, 3H), 2.52–2.53 (m, 2H), 2.20–2.25 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 153.68, 144.99, 136.93, 136.82, 132.52, 129.15, 128.17, 127.76, 127.02, 126.03, 125.19, 124.98, 121.75, 115.62, 30.57, 23.39, 19.01.
3-(3-Buten-1-yl)-4-(4-ethylnaphth-1-yl)-4H-1,2,4-triazole (5d): Colorless oil; 14.77 g (71%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.69 (s, 1H), 8.24 (d, J = 8.8 Hz, 1H), 7.66–7.70 (m, 1H), 7.58–7.63 (m, 2H), 7.54 (d, J = 7.2 Hz, 1H), 7.12 (d, J = 8.0 Hz, 1H), 5.62–5.72 (m, 1H), 4.86–4.88 (m, 1H), 4.83 (d, J = 1.2 Hz, 1H), 3.11–3.23 (m, 2H), 2.49–2.53 (m, 2H), 2.20–2.26 (m, 2H), 1.34 (t, J = 7.4 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 153.69, 145.00, 142.66, 136.81, 131.74, 129.39, 128.18, 127.64, 127.04, 125.32, 124.50, 124.46, 121.94, 115.59, 30.55, 25.16, 23.42, 14.87.
3-(3-Buten-1-yl)-4-(4-n-propylnaphth-1-yl)-4H-1,2,4-triazole (5e): White solid; 16.61 g (76%); m.p. 92.5–94 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.70 (s, 1H), 8.24 (d, J = 8.4 Hz, 1H), 7.65–7.69 (m, 1H), 7.58–7.62 (m, 2H), 7.52 (d, J = 7.2 Hz, 1H), 7.11 (d, J = 8.0 Hz, 1H), 5.63–5.70 (m, 1H), 4.86–4.87 (m, 1H), 4.83 (d, J = 1.6 Hz, 1H), 3.06–3.18 (m, 2H), 2.49–2.53 (m, 2H), 2.19–2.25 (m, 2H), 1.71–1.77 (m, 2H), 1.00 (t, J = 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 153.68, 145.02, 141.13, 136.84, 131.90, 129.44, 128.21, 127.63, 126.99, 125.51, 125.15, 124.70, 121.92, 115.63, 34.15, 30.56, 23.53, 23.43, 13.91.
3-(3-Buten-1-yl)-4-(4-isopropylnaphth-1-yl)-4H-1,2,4-triazole (5f): Colorless oil; 16.39 g (75%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.70 (s, 1H), 8.34 (d, J = 8.8 Hz, 1H), 7.56–7.72 (m, 4H), 7.12 (d, J = 8.0 Hz, 1H), 5.63–5.71 (m, 1H), 4.87–4.89 (m, 1H), 4.84 (d, J = 1.2 Hz, 1H), 3.82-3.88 (m, 1H), 2.52–2.54 (m, 2H), 2.22–2.27 (m, 2H), 1.41 (d, J = 6.8 Hz, 3H), 1.36 (d, J = 6.8 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 154.39, 147.65, 145.74, 137.55, 132.05, 130.14, 128.75, 128.23, 127.75, 126.03, 124.75, 122.72, 122.22, 116.33, 31.24, 28.76, 24.30, 24.14, 23.66.
3-(3-Buten-1-yl)-4-(4-methoxynaphth-1-yl)-4H-1,2,4-triazole (5g): White solid; 15.29 g (73%); m.p. 92.5–94 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.66 (s, 1H), 8.27–8.30 (m, 1H), 7.60–7.65 (m, 3H), 7.11 (d, J = 8.4 Hz, 1H), 7.03–7.06 (m, 1H), 5.62–5.72 (m, 1H), 4.86–4.88 (m, 1H), 4.84 (d, J = 1.2 Hz, 1H), 4.05 (s, 3H), 2.48–2.53 (m, 2H), 2.20–2.26 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 155.93, 153.89, 145.19, 136.86, 130.00, 128.48, 126.44, 126.25, 125.02, 122.26, 122.22, 121.22, 115.60, 103.81, 56.06, 30.60, 23.37.
3-(3-Buten-1-yl)-4-(4-ethoxynaphth-1-yl)-4H-1,2,4-triazole (5h): White solid; 16.28 g (74%); m.p. 72.5–73.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.65 (s, 1H), 8.29–8.31 (m, 1H), 7.57–7.64 (m, 3H), 7.09 (d, J = 8 Hz, 1H), 7.02–7.05 (m, 1H), 5.61–5.72 (m, 1H), 4.86–4.88 (m, 1H), 4.83 (s, 1H), 4.30 (q, J = 6.9 Hz, 2H), 2.51–2.53 (m, 2H), 2.20–2.25 (m, 2H), 1.49 (t, J = 7.0 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz): 155.17, 153.88, 145.15, 136.82, 130.05, 128.39, 126.29, 126.24, 125.12, 122.30, 122.06, 121.16, 115.52, 104.36, 64.02, 30.58, 23.37, 14.42.
4-(4-Bromonaphth-1-yl)-3-(3-buten-1-yl)-4H-1,2,4-triazole (5i): White solid; 19.20 g (78%); m.p. 97.5–99 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.74 (s, 1H), 8.30 (d, J = 8.4 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.81–7.85 (m, 1H), 7.73 (t, J = 7.6 Hz, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.18 (d, J = 8.4 Hz, 1H), 5.62–5.72 (m, 1H), 4.86–4.88 (m, 1H), 4.84 (s, 1H), 2.52–2.56 (m, 2H), 2.21–2.27 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 153.61, 144.86, 136.75, 131.65, 130.34, 130.00, 129.75, 129.18, 129.03, 127.21, 126.39, 123.84, 122.29, 115.69, 30.52, 23.36.
4-((4-Bromonaphth-1-yl)methyl)-3-(3-buten-1-yl)-4H-1,2,4-triazole (5j): White solid; 19.76 g (77%); m.p. 120.5–122.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.44 (s, 1H), 8.22–8.24 (m, 1H), 8.13–8.16 (m, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.71–7.78 (m, 2H), 6.77 (d, J = 8.0 Hz, 1H), 5.74–5.85 (m, 3H), 4.90–4.99 (m, 2H), 2.72 (t, J = 7.6 Hz, 2H), 2.32–2.38 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 153.21, 144.43, 137.12, 132.41, 131.26, 131.12, 129.66, 128.06, 127.69, 127.17, 125.00, 123.81, 122.13, 115.52, 44.39, 30.45, 23.18.
4-(2-(4-Bromonaphth-1-yl)ethyl)-3-(3-buten-1-yl)-4H-1,2,4-triazole (5k): Colorless oil; 19.77 g (74%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.40 (s, 1H), 8.17-8.19 (m, 2H), 7.76 (d, J = 7.6 Hz, 1H), 7.65–7.73 (m, 2H), 7.12 (d, J = 7.6 Hz, 1H), 5.61–5.72 (m, 1H), 4.89 (s, 1H), 4.85–4.86 (m, 1H), 4.25 (t, J = 7.0 Hz, 2H), 3.49 (t, J = 7.0 Hz, 2H), 2.33–2.37 (m, 2H), 2.14–2.21 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 152.51, 143.66, 137.06, 134.32, 132.61, 131.23, 129.53, 127.75, 127.57, 127.30, 127.12, 124.28, 121.02, 115.27, 43.68, 32.89, 30.30, 22.71.
4-((4-Bromonaphth-1-yl)methyl)-3-(4-hydroxybutyl)-4H-1,2,4-triazole (5l): White solid; 19.72 g (73%); m.p. 126.5–128 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.43 (s, 1H), 8.21–8.24 (m, 1H), 8.13-8.15 (m, 1H), 7.85 (d, J = 7.6 Hz, 1H), 7.71–7.78 (m, 2H), 6.74 (d, J = 8.0 Hz, 1H), 5.74 (s, 2H), 4.35 (t, J = 5.0 Hz, 1H), 3.31–3.35 (m, 2H), 2.64 (t, J = 7.6 Hz, 2H), 1.58–1.66 (m, 2H), 1.38–1.45 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 153.86, 144.34, 132.49, 131.25, 131.13, 129.67, 128.06, 127.70, 127.19, 124.88, 123.78, 122.13, 60.23, 44.40, 31.84, 23.45, 23.17.
4-((4-Bromonaphth-1-yl)methyl)-3-(5-hydroxypentyl)-4H-1,2,4-triazole (5m): White solid; 20.77 g (74%); m.p. 88.5–91 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.43 (s, 1H), 8.22–8.24 (m, 1H), 8.14 (d, J = 8.8 Hz, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.71–7.78 (m, 2H), 6.75 (d, J = 8.0 Hz, 1H), 5.74 (s, 2H), 4.30 (t, J = 5.0 Hz, 1H), 3.29–3.33 (m, 2H), 2.62 (t, J = 7.4 Hz, 2H), 1.52–1.60 (m, 2H), 1.22–1.37 (m, 4H). 13C-NMR (DMSO-d6, 100 MHz) δ: 153.82, 144.35, 132.53, 131.26, 131.13, 129.69, 128.10, 127.72, 127.20, 124.92, 123.81, 122.10, 60.50, 44.39, 32.07, 26.45, 25.07, 23.65.
4-((4-Bromonaphth-1-yl)methyl)-3-(hydroxymethyl)-4H-1,2,4-triazole (5n): White solid; 18.85 g (79%); m.p. 197–200 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.40 (s, 1H), 8.22 (d, J = 8.4 Hz, 1H), 8.17 (d, J = 8.0 Hz, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.69–7.77 (m, 2H), 6.93 (d, J = 7.6 Hz, 1H), 5.82 (s, 2H), 5.75 (t, J = 5.6 Hz, 1H), 4.59 (d, J = 5.6 Hz, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 154.07, 145.58, 132.97, 132.11, 131.84, 130.43, 128.75, 128.49, 127.89, 126.44, 124.53, 122.92, 54.32, 45.53.
4-((4-Bromonaphth-1-yl)methyl)-3-((E)-1-propen-1-yl)-4H-1,2,4-triazole (5o): White solid; 2.95 g (12%); m.p. 157–159 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.54 (s, 1H), 8.21–8.23 (m, 1H), 8.14–8.16 (m, 1H), 7.84 (d, J = 7.6 Hz, 1H), 7.71–7.78 (m, 2H), 6.67 (d, J = 7.6 Hz, 1H), 6.26 (dd, J = 1.6 Hz and 11.6 Hz, 1H), 6.06–6.14 (m, 1H), 5.80 (s, 2H), 2.11 (dd, J = 1.6 Hz and 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 150.79, 143.82, 135.12, 132.61, 131.13, 131.06, 129.66, 128.07, 127.69, 127.15, 124.59, 123.72, 122.02, 112.61, 44.39, 15.69.
3-Allyl-4-((4-bromonaphth-1-yl)methyl)-4H-1,2,4-triazole (5p): White solid; 18.85 g (60%); m.p. 146.5–148 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.43 (s, 1H), 8.22–8.24 (m, 1H), 8.10–8.12 (m, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.71–7.78 (m, 2H), 6.79 (d, J = 7.6 Hz, 1H), 5.85–5.95 (m, 1H), 5.72 (s, 2H), 5.04 (s, 1H), 5.00–5.02 (m, 1H), 3.51 (d, J = 6.4 Hz, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 151.87, 144.57, 132.61, 132.19, 131.27, 131.12, 129.68, 128.05, 127.72, 127.19, 125.20, 123.74, 122.17, 117.42, 44.54, 28.30.
4-((4-Bromonaphth-1-yl)methyl)-3-(2-hydroxyethyl)-4H-1,2,4-triazole (5q): White solid; 19.18 g (77%); m.p. 179–182 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.40 (s, 1H), 8.22–8.24 (m, 1H), 8.15 (d, J = 8.4 Hz, 1H), 7.85 (d, J = 7.6 Hz, 1H), 7.71–7.78 (m, 2H), 6.76 (d, J = 7.6 Hz, 1H), 5.78 (s, 2H), 4.83 (t, J = 5.4 Hz, 1H), 3.68–3.72 (m, 2H), 2.82 (t, J = 6.8 Hz, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 152.36, 144.28, 132.45, 131.28, 131.14, 129.74, 128.12, 127.77, 127.20, 125.02, 123.83, 122.11, 59.11, 44.56, 27.65.
4-((4-Bromonaphth-1-yl)methyl)-3-(2-hydroxypropyl)-4H-1,2,4-triazole (5r): White solid; 18.70 g (72%); m.p. 155.5–157 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.36 (s, 1H), 8.22–8.24 (m, 1H), 8.13–8.15 (m, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.71–7.78 (m, 2H), 6.75 (d, J = 7.6 Hz, 1H), 5.80 (d, J = 17.6 Hz, 1H), 5.76 (d, J = 17.6 Hz, 1H), 4.86 (d, J = 4.4 Hz, 1H), 3.97–4.03 (m, 1H), 2.77 (dd, J = 5.6 Hz and 14.8 Hz, 1H), 2.70 (dd, J = 6.8 Hz and 14.8 Hz, 1H), 1.09 (d, J = 6.4 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 152.34, 144.16, 132.47, 131.28, 131.12, 129.69, 128.06, 127.72, 127.17, 125.05, 123.83, 122.10, 65.16, 44.63, 33.52, 23.17.
4-((4-Bromonaphth-1-yl)methyl)-4H-1,2,4-triazole (17): White solid; 13.83 g (64%); m.p. 201–204 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.61 (s, 2H), 8.22 (d, J = 8.0 Hz, 2H), 7.90 (d, J = 7.6 Hz, 1H), 7.69–7.76 (m, 2H), 7.20 (d, J = 8.0 Hz, 1H), 5.79 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 143.42, 132.61, 131.47, 131.21, 129.77, 128.03, 127.83, 127.23, 126.87, 123.80, 122.57, 45.13.
3.2.7. General Procedure for the Synthesis of 6a–6l
To a stirred solution of 5a–5k or 5o (50 mmol) in THF/H2O (4/1 by v/v, 100 mL in total) at room temperature were added commercially available 50% solution of NMMO in water (23.43 g, 100 mmol) and a 0.16 M stock solution of OsO4 in t-BuOH/H2O (4/1 by v/v; 31.25 mL, 5 mmol). The resulting mixture was stirred at room temperature until the completion of reaction as indicated by TLC analysis (typically within 24 h). The reaction mixture was filtered off through celite via vacuum filtration and the filtrate was poured into ice-water (300 mL). The resulting mixture was extracted with CH2Cl2 (100 mL × 3). The combined extracts were washed successively with 1 M aqueous Na2S2O3 (100 mL) and 5% brine (100 mL), dried over anhydrous Na2SO4, and evaporated on a rotary evaporator to afford a residue, which was purified by column chromatography followed by trituration with EtOAc/n-hexane to produce the pure diol intermediates. These diol intermediates were not structurally characterized and were directly used in the next step.
The diol intermediates (deemed to be 50 mmol) were dissolved in THF/H2O (4/1 by v/v, 200 mL in total), followed by addition of NaIO4 (32.08 g, 150 mmol). The resulting mixture was stirred at room temperature until the completion of reaction as indicated by TLC analysis (typically within 2 h). The reaction mixture was poured into ice-water (400 mL) and the resulting mixture was extracted with CH2Cl2 (100 mL × 3). The combined extracts were washed with 5% brine (100 mL), dried over anhydrous Na2SO4, and evaporated on a rotary evaporator to afford a residue, which was purified by column chromatography to produce 6a–6l after trituration with EtOAc/n-hexane if possible.
3-(4-(4-Cyclopropylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionaldehyde (6a): White solid; 11.36 g (78%); m.p. 126.5–128.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 9.63 (s, 1H), 8.70 (s, 1H), 8.56 (d, J = 8.4 Hz, 1H), 7.71–7.75 (m, 1H), 7.62–7.66 (m, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.40 (d, J = 7.6 Hz, 1H), 7.15 (d, J = 8.4 Hz, 1H), 2.86 (t, J = 7.0 Hz, 2H), 2.63–2.71 (m, 1H), 2.51–2.59 (m, 2H), 1.11–1.14 (m, 2H), 0.84–0.90 (m, 1H), 0.79–0.80 (m, 1H). 13C-NMR (CDCl3, 100 MHz) δ: 199.79, 154.05, 144.70, 142.62, 134.17, 129.47, 127.92, 127.89, 127.12, 125.23, 124.74, 123.07, 121.80, 40.17, 17.15, 13.33, 6.80, 6.74.
3-(4-(Naphth-1-yl)-4H-1,2,4-triazol-3-yl)propionaldehyde (6b): White solid; 9.93 g (79%); m.p. 112–114 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 9.63 (s, 1H), 8.77 (s, 1H), 8.17–8.20 (m, 1H), 8.13 (d, J = 7.6 Hz, 1H), 7.61–7.71 (m, 4H), 7.18 (d, J = 8.4 Hz, 1H), 2.87 (t, J = 7.0 Hz, 2H), 2.66–2.73 (m, 1H), 2.49–2.61 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 201.58, 153.32, 145.03, 133.69, 130.17, 129.72, 129.05, 128.50, 128.20, 127.15, 125.64, 121.29, 39.38, 16.94.
3-(4-(4-Methylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionaldehyde (6c): White solid; 10.08 g (76%); m.p. 101.5–103.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 9.62 (s, 1H), 8.71 (s, 1H), 8.18 (d, J = 8.4 Hz, 1H), 7.68–7.71 (m, 1H), 7.63 (t, J = 7.2 Hz, 1H), 7.53–7.59 (m, 2H), 7.15 (d, J = 8.0 Hz, 1H), 2.85 (t, J = 7.0 Hz, 2H), 2.75 (s, 3H), 2.64–2.73 (m, 1H), 2.52–2.59 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 201.59, 153.43, 145.14, 137.00, 132.57, 129.10, 128.06, 127.83, 127.04, 126.06, 125.23, 125.00, 121.79, 39.40, 19.02, 16.93.
3-(4-(4-Ethylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionaldehyde (6d): White solid; 10.75 g (77%); m.p. 114–115.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 9.63 (s, 1H), 8.71 (s, 1H), 8.25 (d, J = 8.4 Hz, 1H), 7.69 (t, J = 7.6 Hz, 1H), 7.60–7.63 (m, 2H), 7.55 (d, J = 7.2 Hz, 1H), 7.16 (d, J = 8.4 Hz, 1H), 3.12–3.23 (m, 2H), 2.86 (t, J = 7.0 Hz, 2H), 2.64–2.72 (m, 1H), 2.52–2.59 (m, 1H), 1.35 (t, J = 7.6 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 201.63, 153.43, 145.16, 142.74, 131.76, 129.33, 128.06, 127.72, 127.07, 125.36, 124.52, 121.97, 39.38, 25.18, 16.94, 14.93.
3-(4-(4-n-Propylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionaldehyde (6e): White solid; 11.00 g (75%); m.p. 97.5–99.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 9.62 (s, 1H), 8.72 (s, 1H), 8.25 (d, J = 8.4 Hz, 1H), 7.66–7.70 (m, 1H), 7.59–7.63 (m, 2H), 7.53 (d, J = 7.6 Hz, 1H), 7.15 (d, J = 8.4 Hz, 1H), 3.05–3.20 (m, 2H), 2.85 (t, J = 7.0 Hz, 2H), 2.64–2.71 (m, 1H), 2.50–2.59 (m, 1H), 1.70-1.78 (m, 2H), 1.01 (t, J = 7.4 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 201.62, 153.44, 145.16, 141.20, 131.95, 129.39, 128.09, 127.69, 127.01, 125.54, 125.19, 124.71, 121.95, 39.38, 34.18, 23.56, 16.94, 13.95.
3-(4-(4-Isopropylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionaldehyde (6f): Colorless oil; 11.44 g (78%). 1H-NMR (DMSO-d6, 400 MHz) δ: 9.63 (s, 1H), 8.72 (s, 1H), 8.35 (d, J = 8.8 Hz, 1H), 7.60–7.71 (m, 4H), 7.16 (d, J = 8.0 Hz, 1H), 3.82–3.89 (m, 1H), 2.87 (t, J = 7.0 Hz, 2H), 2.64–2.72 (m, 1H), 2.53–2.59 (m, 1H), 1.41 (d, J = 6.8 Hz, 3H), 1.37 (d, J = 6.8 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 201.57, 153.44, 147.00, 145.15, 131.39, 129.39, 127.94, 127.58, 127.05, 125.36, 124.05, 122.06, 121.57, 39.37, 28.08, 23.54, 22.99, 16.97.
3-(4-(4-Methoxynaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionaldehyde (6g): White solid; 10.27 g (73%); m.p. 149–150.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 9.63 (s, 1H), 8.68 (s, 1H), 8.28–8.30 (m, 1H), 7.61–7.65 (m, 3H), 7.13 (d, J = 8.4 Hz, 1H), 7.07–7.10 (m, 1H), 4.06 (s, 3H), 2.85 (t, J = 7.0 Hz, 2H), 2.64–2.72 (m, 1H), 2.54–2.59 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 201.62, 155.98, 153.62, 145.33, 129.94, 128.53, 126.45, 126.28, 125.06, 122.23, 122.14, 121.25, 103.86, 56.08, 39.42, 16.90.
3-(4-(4-Ethoxynaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionaldehyde (6h): White solid; 11.08 g (75%); m.p. 138.5–140 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 9.62 (s, 1H), 8.67 (s, 1H), 8.29–8.32 (m, 1H), 7.60–7.64 (m, 3H), 7.07–7.11 (m, 2H), 4.31 (q, J = 6.9 Hz, 2H), 2.84 (t, J = 7.0 Hz, 2H), 2.64–2.72 (m, 1H), 2.53–2.59 (m, 1H), 1.50 (t, J = 6.8 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 201.61, 155.22, 153.62, 145.32, 129.99, 128.49, 126.35, 126.30, 125.16, 122.33, 121.94, 121.20, 104.47, 64.07, 39.42, 16.89, 14.46.
3-(4-(4-Bromonaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionaldehyde (6i): White solid; 12.88 g (78%); m.p. 139–140.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 9.62 (s, 1H), 8.76 (s, 1H), 8.31 (d, J = 8.4 Hz, 1H), 8.10 (d, J = 8.0 Hz, 1H), 7.82–7.86 (m, 1H), 7.72–7.76 (m, 1H), 7.66 (d, J = 7.6 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 2.86 (t, J = 7.0 Hz, 2H), 2.67–2.74 (m, 1H), 2.56–2.62 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 201.54, 153.35, 144.97, 131.68, 130.30, 129.92, 129.77, 129.21, 129.02, 127.21, 126.41, 123.89, 122.34, 39.39, 16.91.
3-(4-((4-Bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)propionaldehyde (6j): White solid; 13.25 g (77%); m.p. 131.5–133 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 9.69 (s, 1H), 8.46 (s, 1H), 8.22–8.24 (m, 1H), 8.15–8.17 (m, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.72–7.79 (m, 2H), 6.78 (d, J = 7.6 Hz, 1H), 5.77 (s, 2H), 2.85–2.93 (m, 4H). 13C-NMR (DMSO-d6, 100 MHz) δ: 201.77, 152.91, 144.65, 132.19, 131.28, 131.15, 129.73, 128.11, 127.75, 127.20, 125.02, 123.83, 122.16, 44.41, 39.32, 16.88.
3-(4-(2-(4-Bromonaphth-1-yl)ethyl)-4H-1,2,4-triazol-3-yl)propionaldehyde (6k): White solid; 12.90 g (72%); m.p. 42.5–44 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 9.62 (s, 1H), 8.33 (s, 1H), 8.18–8.21 (m, 2H), 7.78 (d, J = 8.0 Hz, 1H), 7.65–7.73 (m, 2H), 7.18 (d, J = 7.6 Hz, 1H), 4.28 (t, J = 7.2 Hz, 2H), 3.50 (t, J = 7.4 Hz, 2H), 2.77 (t, J = 6.8 Hz, 2H), 2.67–2.71 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 201.74, 152.16, 143.94, 134.40, 132.64, 131.22, 129.60, 127.75, 127.66, 127.38, 127.11, 124.38, 120.97, 43.72, 39.39, 32.60, 16.46.
(4-((4-Bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)carboxaldehyde (6l): White solid; 12.01 g (76%); m.p. 211–213 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 10.06 (s, 1H), 8.84 (s, 1H), 8.22–8.25 (m, 1H), 8.13–8.15 (m, 1H), 7.83 (d, J = 7.6 Hz, 1H), 7.72–7.79 (m, 2H), 6.80 (d, J = 7.6 Hz, 1H), 6.04 (s, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 182.52, 150.19, 147.54, 131.96, 131.20, 131.07, 129.69, 128.10, 127.90, 127.23, 125.09, 123.57, 122.29, 46.13.
3.2.8. Synthesis of 1-(4-((4-Bromonaphth-1-yl)methyl)-4H-1,2,4-Triazol-3-yl)acetone 6n
To a stirred solution of DMSO (2.34 g, 30 mmol) in dried CH2Cl2 (40 mL) cooled at −78 °C under N2 was added dropwise a solution of (COCl)2 (1.90 g, 15 mmol) in dried CH2Cl2 (10 mL) via syringe, and the resulting solution was stirred at this temperature for 0.5 h, followed by addition of a solution of 5r (3.46 g, 10 mmol) in dried CH2Cl2 (10 mL) in a dropwise manner via syringe. After addition, the stirring was continued at this temperature for 1 h, and Et3N (6.07 g, 60 mmol) was added in a dropwise manner via syringe. The reaction mixture thus obtained was stirred at room temperature until the completion of reaction as indicated by TLC analysis (typically within 3 h).
The reaction mixture was poured into ice-water (200 mL). The resulting mixture was extracted with CH2Cl2 (100 mL × 3). The combined extracts were washed successively with saturated aqueous NaHCO3 (50 mL), 1 M hydrochloric acid (50 mL), and 5% brine (100 mL), dried over anhydrous Na2SO4, and evaporated on a rotary evaporator to afford a residue, which was purified by column chromatography to produce 6n. White solid; 2.44 g (71%); m.p. 187–190 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.37 (s, 1H), 8.22 (d, J = 8.0 Hz, 1H), 8.08 (d, J = 7.6 Hz, 1H), 7.87 (d, J = 7.6 Hz, 1H), 7.69–7.77 (m, 2H), 6.90 (d, J = 7.6 Hz, 1H), 5.63 (s, 2H), 4.11 (s, 2H), 2.15 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 203.07, 148.91, 144.52, 131.92, 131.43, 131.16, 129.69, 128.05, 127.73, 127.17, 125.90, 123.92, 122.35, 44.89, 39.04, 29.47.
3.2.9. General Procedure for the Synthesis of 7a–7k and 17
To a stirred solution of 6a–6l (35 mmol) in t-BuOH (200 mL) and 2-methyl-2-butene (73.64 g, 1.05 mol) cooled with an ice-water bath was added a suspension of NaClO2 (80%; 11.87 g, 105 mmol) and NaH2PO4 (25.20 g, 210 mmol) in water (50 mL). The resulting mixture was stirred at room temperature until the completion of reaction as indicated by TLC analysis (typically within 6 h).
The reaction mixture was poured into ice-water (500 mL), and the mixture thus obtained was acidified (pH = 1–2) with concentrated hydrochloric acid and extracted with CH2Cl2 (100 mL × 3). The combined extracts were washed with water (100 mL), dried over anhydrous Na2SO4, and evaporated on a rotary evaporator to afford a residue, which was purified by column chromatography to produce 7a–7k or 17 after trituration with EtOAc/n-hexane.
3-(4-(4-Cyclopropylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (7a): White solid; 9.04 g (84%); m.p. 213.5–215 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.22 (brs, 1H), 8.71 (s, 1H), 8.56 (d, J = 8.8 Hz, 1H), 7.70–7.74 (m, 1H), 7.60–7.64 (m, 1H), 7.56 (d, J = 7.6 Hz, 1H), 7.41 (d, J = 7.6 Hz, 1H), 7.16 (d, J = 8.4 Hz, 1H), 2.58–2.67 (m, 3H), 2.45–2.55 (m, 2H), 1.08–1.17 (m, 2H), 0.73-0.91 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.01, 153.49, 145.04, 141.72, 133.37, 129.07, 128.05, 127.81, 127.07, 125.25, 124.92, 122.60, 121.93, 30.44, 19.51, 12.86, 7.18, 6.88.
3-(4-(Naphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (7b): White solid; 7.95 g (85%); m.p. 225–226.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.18 (brs, 1H), 8.74 (s, 1H), 8.18 (dd, J = 2.0 Hz and 7.2 Hz, 1H), 8.13 (d, J = 7.6 Hz, 1H), 7.59–7.72 (m, 4H), 7.19 (d, J = 8.0 Hz, 1H), 2.52–2.67 (m, 4H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.70, 154.11, 145.65, 134.39, 130.85, 130.47, 129.78, 129.18, 128.85, 127.85, 126.34, 122.09, 31.15, 20.22.
3-(4-(4-Methylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (7c): White solid; 8.66 g (88%); m.p. 225 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 12.17 (brs, 1H), 8.70 (s, 1H), 8.18 (d, J = 8.4 Hz, 1H), 7.69 (t, J = 7.6 Hz, 1H), 7.61 (t, J = 7.4 Hz, 1H), 7.53–7.57 (m, 2H), 7.15 (d, J = 8.4 Hz, 1H), 2.75 (s, 3H), 2.58–2.65 (m, 3H), 2.51–2.56 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.01, 153.54, 145.07, 137.02, 132.59, 129.13, 128.10, 127.81, 127.07, 126.08, 125.25, 125.01, 121.89, 30.48, 19.52, 19.05.
3-(4-(4-Ethylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (7d): White solid; 8.48 g (82%); m.p. 206.5 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 12.17 (brs, 1H), 8.70 (s, 1H), 8.25 (d, J = 8.4 Hz, 1H), 7.67–7.71 (m, 1H), 7.58–7.62 (m, 2H), 7.54 (d, J = 7.6 Hz, 1H), 7.16 (d, J = 8.0 Hz, 1H), 3.12–3.23 (m, 2H), 2.59–2.67 (m, 3H), 2.52–2.56 (m, 1H), 1.35 (t, J = 7.4 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.70, 154.20, 145.76, 143.42, 132.46, 130.04, 128.79, 128.36, 127.76, 126.04, 125.20, 122.76, 31.12, 25.88, 20.20, 15.62.
3-(4-(4-n-Propylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (7e): White solid; 9.10 g (84%); m.p. 171–173.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.15 (brs, 1H), 8.71 (s, 1H), 8.24 (d, J = 8.4 Hz, 1H), 7.68 (t, J = 7.6 Hz, 1H), 7.52–7.61 (m, 3H), 7.15 (d, J = 8.4 Hz, 1H), 3.04–3.19 (m, 2H), 2.60–2.64 (m, 2H), 2.49–2.56 (m, 2H), 1.74–1.76 (m, 2H), 1.01 (t, J = 7.2 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.13, 153.53, 145.06, 141.18, 131.94, 129.40, 128.14, 127.64, 127.01, 125.53, 125.18, 124.69, 122.04, 34.19, 30.54, 23.57, 19.55, 13.97.
3-(4-(4-Isopropylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (7f): White solid; 8.88 g (82%); m.p. 178–180.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.16 (brs, 1H), 8.71 (s, 1H), 8.34 (d, J = 8.8 Hz, 1H), 7.69 (t, J = 7.6 Hz, 1H), 7.58–7.64 (m, 3H), 7.16 (d, J = 8.4 Hz, 1H), 3.82–3.89 (m, 1H), 2.59–2.68 (m, 3H), 2.51–2.56 (m, 1H), 1.41 (d, J = 6.8 Hz, 3H), 1.37 (d, J = 6.8 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.03, 153.52, 146.99, 145.08, 131.38, 129.40, 127.97, 127.54, 127.06, 125.36, 124.04, 122.15, 121.57, 30.41, 28.09, 23.57, 23.02, 19.53.
3-(4-(4-Methoxynaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (7g): White solid; 8.74 g (84%); m.p. 243–245 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.15 (brs, 1H), 8.67 (s, 1H), 8.28–8.30 (m, 1H), 7.60–7.65 (m, 3H), 7.07–7.13 (m, 2H), 4.06 (s, 3H), 2.59–2.65 (m, 3H), 2.52–2.57 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.00, 155.98, 153.69, 145.24, 129.96, 128.49, 126.46, 126.28, 125.05, 122.22, 122.18, 121.33, 103.87, 56.09, 30.49, 19.47.
3-(4-(4-Ethoxynaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (7h): White solid; 8.94 g (82%); m.p. 231–233 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.23 (brs, 1H), 8.66 (s, 1H), 8.29–8.32 (m, 1H), 7.57–7.64 (m, 3H), 7.07–7.11 (m, 2H), 4.30 (q, J = 6.9 Hz, 2H), 2.49–2.65 (m, 4H), 1.50 (t, J = 7.0 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.10, 155.21, 153.72, 145.22, 130.01, 128.44, 126.35, 126.28, 125.14, 122.30, 121.99, 121.28, 104.47, 64.07, 30.59, 19.51, 14.47.
3-(4-(4-Bromonaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (7i): White solid; 10.78 g (89%); m.p. 237–240 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.09 (brs, 1H), 8.75 (s, 1H), 8.30 (d, J = 8.4 Hz, 1H), 8.10 (d, J = 8.0 Hz, 1H), 7.83 (t, J = 7.6 Hz, 1H), 7.72 (t, J = 7.6 Hz, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 2.52-2.69 (m, 4H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.01, 153.46, 144.89, 131.68, 130.32, 129.97, 129.78, 129.17, 129.04, 127.21, 126.41, 123.87, 122.44, 30.49, 19.48.
3-(4-((4-Bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)propionic acid (7j): White solid; 10.59 g (84%); m.p. 225–227 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.18 (brs, 1H), 8.43 (s, 1H), 8.23 (d, J = 7.6 Hz, 1H), 8.14–8.16 (m, 1H), 7.85 (d, J = 7.6 Hz, 1H), 7.72–7.75 (m, 2H), 6.78 (d, J = 7.6 Hz, 1H), 5.76 (s, 2H), 2.81–2.85 (m, 2H), 2.68–2.72 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.22, 152.99, 144.50, 132.23, 131.29, 131.14, 129.72, 128.12, 127.76, 127.20, 125.04, 123.85, 122.12, 44.36, 30.45, 19.35.
3-(4-(2-(4-Bromonaphth-1-yl)ethyl)-4H-1,2,4-triazol-3-yl)propionic acid (7k): White solid; 10.61 g (81%); m.p. 210–213 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.18 (brs, 1H), 8.31 (s, 1H), 8.18–8.21 (m, 2H), 7.78 (d, J = 7.6 Hz, 1H), 7.65–7.73 (m, 2H), 7.18 (d, J = 7.6 Hz, 1H), 4.28 (t, J = 7.2 Hz, 2H), 3.49 (t, J = 7.2 Hz, 2H), 2.69 (t, J = 6.6 Hz, 2H), 2.61 (t, J = 6.4 Hz, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.25, 152.34, 143.82, 134.41, 132.66, 131.24, 129.60, 127.74, 127.65, 127.38, 127.13, 124.39, 120.99, 43.72, 32.59, 30.64, 18.96.
4-((4-Bromonaphth-1-yl)methyl)-4H-1,2,4-triazole (17): 4.13 g (41%). The physical properties of the sample 17 obtained by this procedure were in good agreement with those for the sample obtained from 3i–3k described above.
3.2.10. General Procedure for the Synthesis of 7l, 7m and 17
To a stirred solution of 5l–5n (35 mmol) and (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO; 0.55 g, 3.5 mmol) in MeCN (200 mL) and phosphate buffer (150 mL, pH = 6.7; prepared by mixing aqueous 0.67 M Na2HPO4 and 0.67 M NaH2PO4 in a ratio of 1/1) at 35 °C were added aqueous NaClO2 and aqueous NaClO simultaneously over 2 h. The aqueous NaClO2 solution was prepared by dissolving 80% solid NaClO2 (80%; 7.91 g, 70 mmol) in water (40 mL), while the aqueous NaClO solution was prepared by diluting a commercially available aqueous NaClO solution (21% by w/w; 0.35 mL, 1 mmol) with water (20 mL). After addition, the reaction mixture was stirred at 35 °C until the completion of reaction as indicated by TLC analysis (typically within 10 h).
The reaction mixture was poured into ice-water (200 mL), and the aqueous mixture thus obtained was acidified (pH = 1–2) with concentrated hydrochloric acid and extracted with CH2Cl2 (100 mL × 3). The combined extracts were washed successively with 1% aqueous Na2S2O3 (100 mL) and water (100 mL), dried over anhydrous Na2SO4, and evaporated on a rotary evaporator to afford a residue, which was purified by column chromatography through a short silica gel column to produce 7l, 7m or 17 after trituration with EtOAc/n-hexane.
4-(4-((4-Bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)butanoic acid (7l): White solid; 10.74 g (82%); m.p. 175–177.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.03 (brs, 1H), 8.44 (s, 1H), 8.22–8.24 (m, 1H), 8.13–8.15 (m, 1H), 7.85 (d, J = 7.6 Hz, 1H), 7.72–7.78 (m, 2H), 6.72 (d, J = 7.6 Hz, 1H), 5.74 (s, 2H), 2.68 (t, J = 7.6 Hz, 2H), 2.29 (t, J = 7.2 Hz, 2H), 1.81–1.89 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 174.02, 153.43, 144.45, 132.46, 131.24, 131.14, 129.74, 128.11, 127.74, 127.21, 124.74, 123.80, 122.11, 44.41, 32.72, 22.93, 21.93.
5-(4-((4-Bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)pentanoic acid (7m): White solid; 11.01 g (81%); m.p. 179.5–181 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 11.99 (brs, 1H), 8.42 (s, 1H), 8.21–8.24 (m, 1H), 8.13–8.15 (m, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.71–7.78 (m, 2H), 6.76 (d, J = 7.6 Hz, 1H), 5.74 (s, 2H), 2.65 (t, J = 7.4 Hz, 2H), 2.16 (t, J = 7.2 Hz, 2H), 1.58–1.65 (m, 2H), 1.47–1.54 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 174.29, 153.65, 144.38, 132.46, 131.30, 131.17, 129.72, 128.10, 127.74, 127.23, 124.99, 123.82, 122.18, 44.44, 33.28, 25.96, 24.01, 23.36.
4-((4-Bromonaphth-1-yl)methyl)-4H-1,2,4-triazole (17): 3.43 g (34%). The physical properties of sample 17 obtained by this procedure were in good agreement with those for the sample obtained from 3i–3k described above.
3.2.11. General Procedure for the Synthesis of 8a–8m
To a stirred mixture of 7a–7m (28 mmol) in dried CH2Cl2 (100 mL) cooled in an ice-water bath were added EDCI (8.05 g, 42 mmol), DMAP (1.71 g, 14 mmol) and MeOH (8.97 g, 280 mmol), and the resulting mixture was stirred at room temperature under N2 until the completion of reaction as indicated by TLC analysis (typically within 6 h).
The reaction mixture was diluted with CH2Cl2 (200 mL) and washed successively with 1 M hydrochloric acid (100 mL), saturated aqueous Na2CO3 (100 mL), and 5% brine (100 mL). The organic phase was dried over anhydrous Na2SO4 and evaporated on a rotary evaporator to afford a residue, which was purified by column chromatography to produce 8a–8m after trituration with EtOAc/n-hexane if possible.
Methyl 3-(4-(4-cyclopropylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (8a): White solid; 6.93 g (77%); m.p. 88.5–89.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.70 (s, 1H), 8.56 (d, J = 8.4 Hz, 1H), 7.72 (t, J = 7.6 Hz, 1H), 7.64 (t, J = 7.4 Hz, 1H), 7.56 (d, J = 7.6 Hz, 1H), 7.41 (d, J = 7.6 Hz, 1H), 7.15 (d, J = 8.4 Hz, 1H), 3.51 (s, 3H), 2.63–2.75 (m, 3H), 2.51–2.59 (m, 2H), 1.09–1.16 (m, 2H), 0.85–0.88 (m, 1H), 0.76–0.79 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.99, 153.26, 145.09, 141.76, 133.36, 129.05, 127.97, 127.84, 127.09, 125.27, 124.94, 122.59, 121.87, 51.37, 30.11, 19.39, 12.85, 7.20, 6.89.
Methyl 3-(4-(naphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (8b): White solid; 5.99 g (76%); m.p. 129–130.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.75 (s, 1H), 8.17–8.20 (m, 1H), 8.13 (d, J = 7.6 Hz, 1H), 7.61–7.72 (m, 4H), 7.18 (d, J = 8.0 Hz, 1H), 3.51 (s, 3H), 2.65–2.75 (m, 3H), 2.54–2.63 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.97, 153.16, 145.00, 133.69, 130.18, 129.69, 129.05, 128.49, 128.18, 127.16, 125.65, 125.63, 121.32, 51.36, 30.12, 19.40.
Methyl 3-(4-(4-methylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (8c): White solid; 6.37 g (77%); m.p. 138.5–140.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.70 (s, 1H), 8.19 (d, J = 8.0 Hz, 1H), 7.70 (dt, J = 1.2 Hz and 8.4 Hz, 1H), 7.63 (dt, J = 1.2 Hz and 6.8 Hz, 1H), 7.53–7.58 (m, 2H), 7.15 (d, J = 8.0 Hz, 1H), 3.51 (s, 3H), 2.75 (s, 3H), 2.70–2.74 (m, 2H), 2.63–2.69 (m, 1H), 2.52–2.59 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.96, 153.26, 145.10, 137.01, 132.55, 129.09, 128.02, 127.80, 127.04, 126.05, 125.22, 124.99, 121.81, 51.36, 30.13, 19.38, 19.02.
Methyl 3-(4-(4-ethylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (8d): White solid; 6.84 g (79%); m.p. 116.5–118 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.71 (s, 1H), 8.25 (d, J = 8.4 Hz, 1H), 7.67–7.71 (m, 1H), 7.59–7.63 (m, 2H), 7.55 (d, J = 7.2 Hz, 1H), 7.15 (d, J = 8.0 Hz, 1H), 3.51 (s, 3H), 3.14–3.21 (m, 2H), 2.70–2.75 (m, 2H), 2.64–2.67 (m, 1H), 2.54–2.59 (m, 1H), 1.35 (t, J = 7.6 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.97, 153.26, 145.10, 142.74, 131.75, 129.32, 128.03, 127.68, 127.06, 125.34, 124.51, 122.00, 51.35, 30.11, 25.17, 19.39, 14.91.
Methyl 3-(4-(4-n-propylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (8e): White solid; 6.88 g (76%); m.p. 116.5–118 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.72 (s, 1H), 8.25 (d, J = 8.4 Hz, 1H), 7.68 (t, J = 7.6 Hz, 1H), 7.58–7.63 (m, 2H), 7.53 (d, J = 7.6 Hz, 1H), 7.15 (d, J = 8.0 Hz, 1H), 3.51 (s, 3H), 3.06–3.17 (m, 2H), 2.69–2.74 (m, 2H), 2.63–2.67 (m, 1H), 2.54–2.59 (m, 1H), 1.71–1.78 (m, 2H), 1.01 (t, J = 7.4 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.96, 153.25, 145.09, 141.19, 131.93, 129.37, 128.06, 127.64, 126.99, 125.52, 125.16, 124.68, 121.97, 51.34, 34.18, 30.10, 23.55, 19.39, 13.93.
Methyl 3-(4-(4-isopropylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (8f): White solid; 6.88 g (76%); m.p. 80–81.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.71 (s, 1H), 8.34 (d, J = 8.4 Hz, 1H), 7.69 (t, J = 7.6 Hz, 1H), 7.60–7.64 (m, 3H), 7.15 (d, J = 8.4 Hz, 1H), 3.82–3.88 (m, 1H), 3.51 (s, 3H), 2.71–2.76 (m, 2H), 2.64–2.70 (m, 1H), 2.53–2.59 (m, 1H), 1.41 (d, J = 6.8 Hz, 3H), 1.37 (d, J = 6.4 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.97, 153.26, 146.99, 145.10, 131.36, 129.36, 127.89, 127.54, 127.04, 125.34, 124.03, 122.07, 121.55, 51.33, 30.07, 28.05, 23.54, 22.99, 19.39.
Methyl 3-(4-(4-methoxynaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (8g): White solid; 6.80 g (78%); m.p. 134.5–135.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.67 (s, 1H), 8.28–8.30 (m, 1H), 7.60–7.65 (m, 3H), 7.13 (d, J = 8.4 Hz, 1H), 7.06–7.10 (m, 1H), 4.06 (s, 3H), 3.51 (s, 3H), 2.52–2.74 (m, 4H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.99, 156.00, 153.46, 145.29, 129.95, 128.51, 126.45, 126.28, 125.05, 122.23, 122.11, 121.28, 103.86, 56.08, 51.36, 30.15, 19.36.
Methyl 3-(4-(4-ethoxynaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (8h): White solid; 6.92 g (76%); m.p. 117–118.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.67 (s, 1H), 8.29–8.32 (m, 1H), 7.61–7.65 (m, 2H), 7.59 (d, J = 8.0 Hz, 1H), 7.11 (d, J = 8.0 Hz, 1H), 7.06–7.09 (m, 1H), 4.31 (q, J = 6.9 Hz, 2H), 3.51 (s, 3H), 2.64–2.74 (m, 3H), 2.52–2.61 (m, 1H), 1.50 (t, J = 7.0 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.96, 155.22, 153.43, 145.27, 129.98, 128.45, 126.34, 126.27, 125.13, 122.30, 121.90, 121.21, 104.45, 64.06, 51.33, 30.14, 19.34, 14.45.
Methyl 3-(4-(4-bromonaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (8i): White solid; 8.07 g (80%); m.p. 126–127.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.76 (s, 1H), 8.31 (d, J = 8.4 Hz, 1H), 8.10 (d, J = 7.6 Hz, 1H), 7.82–7.86 (m, 1H), 7.72–7.76 (m, 1H), 7.65 (d, J = 8.0 Hz, 1H), 7.22 (d, J = 8.4 Hz, 1H), 3.51 (s, 3H), 2.68–2.75 (m, 3H), 2.53–2.63 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.94, 153.19, 144.93, 131.67, 130.29, 129.88, 129.77, 129.18, 129.02, 127.20, 126.39, 123.89, 122.35, 51.36, 30.12, 19.35.
Methyl 3-(4-((4-bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)propionate (8j): White solid; 8.07 g (77%); m.p. 100–101.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.45 (s, 1H), 8.22–8.25 (m, 1H), 8.14–8.17 (m, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.72–7.79 (m, 2H), 6.77 (d, J = 8.0 Hz, 1H), 5.77 (s, 2H), 3.56 (s, 3H), 2.85–2.88 (m, 2H), 2.75–2.79 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.20, 152.74, 144.60, 132.22, 131.26, 131.13, 129.71, 128.12, 127.75, 127.19, 124.96, 123.84, 122.12, 51.37, 44.35, 30.07, 19.26.
Methyl 3-(4-(2-(4-bromonaphth-1-yl)ethyl)-4H-1,2,4-triazol-3-yl)propionate (8k): White solid; 8.15 g (75%); m.p. 112.5–114 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.33 (s, 1H), 8.18–8.20 (m, 2H), 7.78 (d, J = 7.6 Hz, 1H), 7.65–7.73 (m, 2H), 7.16 (d, J = 7.6 Hz, 1H), 4.28 (t, J = 7.2 Hz, 2H), 3.56 (s, 3H), 3.49 (t, J = 7.2 Hz, 2H), 2.60–2.68 (m, 4H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.17, 152.03, 143.88, 134.38, 132.62, 131.23, 129.58, 127.74, 127.63, 127.36, 127.12, 124.35, 121.00, 51.37, 43.70, 32.64, 30.24, 18.83.
Methyl 4-(4-((4-bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)butanoate (8l): white foam; 8.59 g (79%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.44 (s, 1H), 8.22–8.24 (m, 1H), 8.13–8.15 (m, 1H), 7.85 (d, J = 7.6 Hz, 1H), 7.72–7.79 (m, 2H), 6.71 (d, J = 7.6 Hz, 1H), 5.73 (s, 2H), 3.51 (s, 3H), 2.67 (t, J = 7.4 Hz, 2H), 2.37 (t, J = 7.2 Hz, 2H), 1.83–1.90 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.88, 153.27, 144.46, 132.48, 131.23, 131.13, 129.74, 128.15, 127.76, 127.20, 124.76, 123.82, 122.07, 51.18, 44.37, 32.36, 22.82, 21.81.
Methyl 5-(4-((4-bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)pentanoate (8m): Colorless oil; 9.01 g (80%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.43 (s, 1H), 8.22–8.24 (m, 1H), 8.13–8.15 (m, 1H), 7.85 (d, J = 7.6 Hz, 1H), 7.72–7.78 (m, 2H), 6.74 (d, J = 7.6 Hz, 1H), 5.73 (s, 2H), 3.53 (s, 3H), 2.64 (t, J = 7.2 Hz, 2H), 2.24 (t, J = 7.2 Hz, 2H), 1.50–1.61 (m, 4H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.09, 153.54, 144.38, 132.52, 131.26, 131.13, 129.71, 128.13, 127.73, 127.20, 124.90, 123.83, 122.08, 51.11, 44.36, 32.81, 25.83, 23.86, 23.22.
3.2.12. General Procedure for the Synthesis of 9a–9c and 9f–9q
A mixture of 8a–8m (6 mmol) and N-halosuccinimide (NCS, NBS or NIS; 7.2 mmol) in MeCN (30 mL) was stirred at room temperature (9b, 9d–9l or 9n–9q), 60 °C (for 9m), or reflux (9a and 9c), until the completion of reaction as indicated by TLC analysis (typically within 24 h).
The reaction mixture was poured into ice-water (100 mL) and the aqueous mixture thus obtained was extracted with CH2Cl2 (50 mL × 3). The combined extracts were washed successively with 5% aqueous Na2S2O3 (50 mL), saturated aqueous Na2CO3 (50 mL × 3), and 5% brine (50 mL), dried over anhydrous Na2SO4, and evaporated on a rotary evaporator to afford a residue, which was purified by column chromatography to produce 9a–9c or 9f–9q after trituration with EtOAc/n-hexane if possible.
Methyl 3-(5-chloro-4-(4-cyclopropylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (9a): White solid; 1.26 g (59%); m.p. 104–105.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.58 (d, J = 8.4 Hz, 1H), 7.74 (t, J = 7.6 Hz, 1H), 7.64–7.68 (m, 2H), 7.44 (d, J = 7.6 Hz, 1H), 7.15 (d, J = 8.0 Hz, 1H), 3.52 (s, 3H), 2.62–2.73 (m, 3H), 2.51–2.57 (m, 2H), 1.12–1.16 (m, 2H), 0.84–0.87 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.76, 155.96, 142.81, 141.45, 133.44, 128.71, 128.17, 127.21, 126.48, 126.14, 125.13, 122.67, 121.42, 51.39, 29.38, 20.45, 12.83, 7.23, 7.14.
Methyl 3-(5-bromo-4-(4-cyclopropylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (9b): White solid; 1.61 g (67%); m.p. 122.5–123.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.58 (d, J = 8.4 Hz, 1H), 7.73 (t, J = 7.6 Hz, 1H), 7.62–7.67 (m, 2H), 7.44 (d, J = 7.6 Hz, 1H), 7.10 (d, J = 8.4 Hz, 1H), 3.51 (s, 3H), 2.62–2.72 (m, 3H), 2.51–2.56 (m, 2H), 1.12–1.16 (m, 2H), 0.84–0.87 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.78, 156.41, 142.66, 133.42, 130.48, 128.82, 128.09, 127.18, 127.01, 126.53, 125.11, 122.68, 121.58, 51.40, 29.51, 20.42, 12.84, 7.23, 7.16.
Methyl 3-(4-(4-cyclopropylnaphth-1-yl)-5-iodo-4H-1,2,4-triazol-3-yl)propionate (9c): White solid; 1.37 g (51%); m.p. 138.5–140 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.58 (d, J = 8.4 Hz, 1H), 7.73 (t, J = 7.6 Hz, 1H), 7.64 (t, J = 7.2 Hz, 1H), 7.55 (d, J = 7.6 Hz, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.02 (d, J = 8.4 Hz, 1H), 3.51 (s, 3H), 2.62–2.71 (m, 3H), 2.52–2.57 (m, 2H), 1.12–1.17 (m, 2H), 0.85–0.88 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.83, 156.29, 142.38, 133.41, 129.07, 128.42, 127.91, 127.12, 126.62, 125.06, 122.65, 121.89, 106.22, 51.38, 29.82, 20.33, 12.85, 7.26, 7.18.
Methyl 3-(5-bromo-4-(naphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (9f): White solid; 1.34 g (62%); m.p. 152–154 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.22–8.25 (m, 1H), 8.14–8.16 (m, 1H), 7.72–7.76 (m, 2H), 7.63–7.70 (m, 2H), 7.13 (d, J = 8.0 Hz, 1H), 3.52 (s, 3H), 2.65–2.73 (m, 3H), 2.51–2.58 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.78, 156.34, 133.77, 131.01, 130.29, 128.89, 128.78, 128.68, 128.48, 127.30, 126.92, 125.82, 121.04, 51.42, 29.53, 20.43.
Methyl 3-(5-bromo-4-(4-methylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (9g): White solid; 1.37 g (61%); m.p. 127–128.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.20 (d, J = 8.4 Hz, 1H), 7.68–7.72 (m, 1H), 7.57–7.66 (m, 3H), 7.10 (d, J = 8.4 Hz, 1H), 3.51 (s, 3H), 2.76 (s, 3H), 2.63–2.72 (m, 3H), 2.51–2.56 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.78, 156.43, 138.01, 132.68, 130.52, 128.89, 128.09, 127.17, 127.12, 126.49, 126.28, 125.21, 121.54, 51.42, 29.56, 20.44, 19.08.
Methyl 3-(5-bromo-4-(4-ethylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (9h): White solid; 1.47 g (63%); m.p. 107.5–109 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.27 (d, J = 8.4 Hz, 1H), 7.58–7.72 (m, 4H), 7.10 (d, J = 8.4 Hz, 1H), 3.52 (s, 3H), 3.19 (q, J = 7.6 Hz, 2H), 2.63–2.72 (m, 3H), 2.52–2.57 (m, 1H), 1.37 (t, J = 7.4 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.80, 156.44, 143.57, 131.91, 130.52, 129.09, 127.97, 127.20, 127.12, 126.60, 124.71, 124.61, 121.71, 51.41, 29.53, 25.16, 20.45, 14.68.
Methyl 3-(5-bromo-4-(4-n-propylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (9i): White solid; 1.57 g (65%); m.p. 104.5–106 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.27 (d, J = 8.4 Hz, 1H), 7.57–7.71 (m, 4H), 7.10 (d, J = 8.4 Hz, 1H), 3.51 (s, 3H), 3.13 (t, J = 7.6 Hz, 2H), 2.63–2.72 (m, 3H), 2.52–2.57 (m, 1H), 1.74–1.80 (m, 2H), 1.02 (t, J = 7.4 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.80, 156.43, 142.09, 132.06, 130.52, 129.15, 127.93, 127.14, 126.42, 125.65, 124.88, 121.69, 51.41, 34.19, 29.51, 23.41, 20.44, 13.97.
Methyl 3-(5-bromo-4-(4-isopropylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (9j): White solid; 1.50 g (62%); m.p. 108.5–110 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.36 (d, J = 8.4 Hz, 1H), 7.61–7.72 (m, 4H), 7.11 (d, J = 8.4 Hz, 1H), 3.83–3.90 (m, 1H), 3.52 (s, 3H), 2.69–2.74 (m, 2H), 2.63–2.66 (m, 1H), 2.47–2.54 (m, 1H), 1.40 (d, J = 6.8 Hz, 6H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.81, 156.44, 147.83, 131.48, 130.52, 129.14, 127.82, 127.17, 126.98, 126.59, 124.27, 121.78, 51.40, 29.47, 28.16, 23.28, 23.22, 20.45.
Methyl 3-(5-bromo-4-(4-methoxynaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (9k): White solid; 0.70 g (30%); m.p. 124.5–126.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.29–8.31 (m, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.62–7.66 (m, 2H), 7.17 (d, J = 8.4 Hz, 1H), 7.03–7.05 (m, 1H), 4.08 (s, 3H), 3.52 (s, 3H), 2.66–2.72 (m, 3H), 2.51–2.58 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.80, 156.61, 156.52, 130.90, 129.75, 128.77, 127.66, 126.57, 125.15, 122.41, 121.12, 121.01, 104.11, 56.13, 51.41, 29.59, 20.44.
Methyl 3-(5-bromo-4-(4-ethoxynaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (9l): White solid; 0.75 g (31%); m.p. 120–122.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.31–8.33 (m, 1H), 7.63–7.66 (m, 3H), 7.14 (d, J = 8.4 Hz, 1H), 7.02–7.04 (m, 1H), 4.33 (q, J = 6.9 Hz, 2H), 3.52 (s, 3H), 2.66–2.72 (m, 3H), 2.51–2.58 (m, 1H), 1.51 (t, J = 7.0 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.78, 156.60, 155.77, 130.90, 129.79, 128.72, 127.66, 126.47, 125.22, 122.48, 120.96, 120.90, 104.66, 64.14, 51.40, 29.58, 20.43, 14.46.
Methyl 3-(5-bromo-4-(4-bromonaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (9m): White solid; 1.61 g (61%); m.p. 162–163.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.32 (d, J = 8.4 Hz, 1H), 8.16 (d, J = 8.0 Hz, 1H), 7.85 (t, J = 7.6 Hz, 1H), 7.72–7.78 (m, 2H), 7.19 (d, J = 8.4 Hz, 1H), 3.52 (s, 3H), 2.66–2.73 (m, 3H), 2.52–2.60 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.75, 156.37, 131.82, 130.18, 130.09, 130.02, 129.52, 129.21, 128.94, 127.70, 127.42, 124.94, 122.08, 51.42, 29.57, 20.40.
Methyl 3-(5-bromo-4-((4-bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)propionate (9n): White solid; 1.71 g (63%); m.p. 90.5–91.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.22–8.26 (m, 2H), 7.78–7.83 (m, 3H), 6.36 (d, J = 8.0 Hz, 1H), 5.78 (s, 2H), 3.55 (s, 3H), 2.90 (t, J = 7.0 Hz, 2H), 2.77 (t, J = 7.0 Hz, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.96, 156.06, 131.25, 131.04, 130.82, 130.10, 129.68, 128.29, 127.75, 127.17, 123.78, 122.46, 121.80, 51.42, 44.94, 29.59, 20.13.
Methyl 3-(5-bromo-4-(2-(4-bromonaphth-1-yl)ethyl)-4H-1,2,4-triazol-3-yl)propionate (9o): White foam; 1.82 g (65%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.18–8.21 (m, 1H), 8.14–8.16 (m, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.66–7.74 (m, 2H), 7.09 (d, J = 7.6 Hz, 1H), 4.26 (t, J = 7.0 Hz, 2H), 3.57 (s, 3H), 3.46 (t, J = 7.0 Hz, 2H), 2.60–2.68 (m, 4H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.00, 155.20, 133.79, 132.74, 131.28, 129.59, 128.89, 127.99, 127.69, 127.47, 127.21, 124.05, 121.28, 51.44, 44.66, 31.64, 29.62, 19.78.
Methyl 4-(5-bromo-4-((4-bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)butanoate (9p): White solid; 1.77 g (63%); m.p. 154–155.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.21–8.25 (m, 2H), 7.78–7.82 (m, 3H), 6.31 (d, J = 8.0 Hz, 1H), 5.74 (s, 2H), 3.49 (s, 3H), 2.71 (t, J = 7.4 Hz, 2H), 2.36 (t, J = 7.2 Hz, 2H), 1.81–1.89 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.76, 156.64, 131.43, 131.04, 130.78, 129.98, 129.70, 128.28, 127.73, 127.17, 123.72, 122.31, 121.76, 51.16, 44.99, 32.15, 23.72, 21.52.
Methyl 5-(5-bromo-4-((4-bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)pentanoate (9q): Colorless oil; 1.76 g (61%). 1H-NMR (DMSO-d6, 400 MHz) δ: 8.21–8.25 (m, 2H), 7.77–7.82 (m, 3H), 6.31 (d, J = 8.0 Hz, 1H), 5.74 (s, 2H), 3.50 (s, 3H), 2.68 (t, J = 7.2 Hz, 2H), 2.22 (t, J = 7.2 Hz, 2H), 1.49–1.60 (m, 4H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.04, 156.96, 131.53, 131.06, 130.81, 129.87, 129.69, 128.30, 127.75, 127.18, 123.76, 122.38, 121.76, 51.10, 44.98, 32.73, 25.48, 24.11, 23.68.
3.2.13. General Procedure for the Synthesis of 9d and 9e
To a stirred solution of MeONa (150 mmol) in dried MeOH (for 9d) or EtONa (150 mmol) in dried EtOH (for 9e) prepared by dissolving Na (3.45 g, 150 mmol) in dried MeOH (100 mL) or EtOH (100 mL) was added 9b (12.01 g, 30 mmol). The resulting mixture was stirred at reflux under N2 until the completion of reaction as indicated by TLC analysis (typically within 24 h).
On cooling to room temperature, the reaction mixture was poured into ice-water (200 mL) and the aqueous mixture thus obtained was extracted immediately with CH2Cl2 (50 mL × 3). The combined extracts were washed with 5% brine (50 mL), dried over anhydrous Na2SO4, and evaporated on a rotary evaporator to afford a residue, which was purified by column chromatography to produce 9d or 9e after trituration with EtOAc/n-hexane.
Methyl 3-(4-(4-cyclopropylnaphth-1-yl)-5-methoxy-4H-1,2,4-triazol-3-yl)propionate (9d): White solid; 1.48 g (14%); m.p. 115–116.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.54 (d, J = 8.4 Hz, 1H), 7.71 (t, J = 7.6 Hz, 1H), 7.63 (t, J = 7.6 Hz, 1H), 7.54 (d, J = 7.2 Hz, 1H), 7.38 (d, J = 7.6 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 3.91 (s, 3H), 3.51 (s, 3H), 2.64 (t, J = 7.0 Hz, 2H), 2.51–2.60 (m, 2H), 2.38–2.47 (m, 1H), 1.10–1.13 (m, 2H), 0.77–0.87 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.95, 159.33, 150.97, 141.82, 133.45, 129.14, 127.74, 126.96, 126.39, 126.10, 124.98, 122.73, 121.83, 57.47, 51.34, 29.47, 20.19, 12.82, 7.17, 6.88.
Ethyl 3-(4-(4-cyclopropylnaphth-1-yl)-5-ethoxy-4H-1,2,4-triazol-3-yl)propionate (9e): White solid; 1.48 g (13%); m.p. 94.5–96 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 8.54 (d, J = 8.4 Hz, 1H), 7.71 (t, J = 7.0 Hz, 1H), 7.63 (t, J = 7.2 Hz, 1H), 7.53 (d, J = 7.6 Hz, 1H), 7.38 (d, J = 7.6 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 4.31–4.37 (m, 2H), 3.96 (q, J = 7.1 Hz, 2H), 2.56–2.62 (m, 2H), 2.49–2.54 (m, 2H), 2.38–2.44 (m, 1H), 1.08–1.18 (m, 8H), 0.81–0.84 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 171.48, 158.63, 150.70, 141.72, 133.45, 129.14, 127.65, 126.95, 126.51, 126.03, 124.97, 122.75, 121.87, 66.42, 59.91, 29.63, 20.21, 14.22, 13.95, 12.81, 7.11, 6.95.
3.2.14. General Procedure for the Synthesis of 10a–10q
To a stirred solution of 9a–9q (1.5 mmol) in EtOH (15 mL) was added an aqueous solution of LiOH prepared by dissolving LiOH·H2O (0.19 g, 4.5 mmol) in water (1 mL), and the resulting mixture was stirred at room temperature until the completion of reaction as indicated by TLC analysis (typically within 3 h).
The reaction mixture was poured into ice-water (100 mL) and the mixture thus obtained was acidified (pH = 1–2) by concentrated hydrochloric acid and extracted with CH2Cl2 (50 mL × 3). The combined extracts were washed with water (50 mL), dried over anhydrous Na2SO4, and evaporated on a rotary evaporator to afford a residue, which was purified by column chromatography to produce 10a–10q after trituration with EtOAc/n-hexane.
3-(5-Chloro-4-(4-cyclopropylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (10a): White solid; 0.45 g (88%); m.p. 146–147.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.21 (brs, 1H), 8.58 (d, J = 8.4 Hz, 1H), 7.72–7.76 (m, 1H), 7.63–7.67 (m, 2H), 7.44 (d, J = 7.6 Hz, 1H), 7.16 (d, J = 8.0 Hz, 1H), 2.44–2.65 (m, 5H), 1.13–1.16 (m, 2H), 0.86–0.87 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.81, 156.22, 142.79, 141.38, 133.45, 128.73, 128.16, 127.22, 126.49, 126.21, 125.15, 122.70, 121.50, 29.69, 20.58, 12.86, 7.23, 7.16.
3-(5-Bromo-4-(4-cyclopropylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (10b): White solid; 0.52 g (89%); m.p. 132.5–134.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.21 (brs, 1H), 8.58 (d, J = 8.4 Hz, 1H), 7.73 (t, J = 7.6 Hz, 1H), 7.61–7.66 (m, 2H), 7.44 (d, J = 7.6 Hz, 1H), 7.11 (d, J = 8.4 Hz, 1H), 2.42–2.65 (m, 5H), 1.12–1.16 (m, 2H), 0.84-0.88 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.85, 156.70, 142.66, 133.46, 130.42, 128.87, 128.09, 127.20, 127.11, 126.55, 125.13, 122.72, 121.68, 29.84, 20.58, 12.89, 7.24, 7.19.
3-(4-(4-Cyclopropylnaphth-1-yl)-5-iodo-4H-1,2,4-triazol-3-yl)propionic acid (10c): White solid; 0.55 g (85%); m.p. 151 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 12.18 (brs, 1H), 8.57 (d, J = 8.4 Hz, 1H), 7.72 (t, J = 7.4 Hz, 1H), 7.62 (t, J = 7.6 Hz, 1H), 7.54 (d, J = 7.6 Hz, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.03 (d, J = 8.4 Hz, 1H), 2.46–2.66 (m, 5H), 1.12–1.18 (m, 2H), 0.86–0.90 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.84, 156.52, 142.34, 133.41, 129.08, 128.49, 127.87, 127.11, 126.61, 125.04, 122.66, 121.94, 106.11, 30.13, 20.45, 12.86, 7.26, 7.15.
3-(4-(4-Cyclopropylnaphth-1-yl)-5-methoxy-4H-1,2,4-triazol-3-yl)propionic acid (10d): White solid; 0.46 g (90%); m.p. 167 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 12.18 (brs, 1H), 8.54 (d, J = 8.4 Hz, 1H), 7.70 (t, J = 7.2 Hz, 1H), 7.62 (t, J = 7.4 Hz, 1H), 7.53 (d, J = 7.6 Hz, 1H), 7.38 (d, J = 7.6 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 3.90 (s, 3H), 2.47–2.57 (m, 4H), 2.34–2.43 (m, 1H), 1.08–1.16 (m, 2H), 0.79–0.87 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.02, 159.30, 151.22, 141.77, 133.45, 129.16, 127.71, 126.94, 126.47, 126.09, 124.97, 122.75, 121.88, 57.45, 29.87, 20.35, 12.83, 7.14, 6.88.
3-(4-(4-Cyclopropylnaphth-1-yl)-5-ethoxy-4H-1,2,4-triazol-3-yl)propionic acid (10e): White solid; 0.46 g (87%); m.p. 154.5–155.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.13 (brs, 1H), 8.54 (d, J = 8.4 Hz, 1H), 7.70 (t, J = 7.4 Hz, 1H), 7.62 (t, J = 7.6 Hz, 1H), 7.52 (d, J = 7.6 Hz, 1H), 7.38 (d, J = 7.6 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 4.30–4.38 (m, 2H), 2.46–2.57 (m, 4H), 2.32–2.41 (m, 1H), 1.11–1.18 (m, 5H), 0.82–0.87 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.00, 158.63, 150.92, 141.70, 133.46, 129.15, 127.64, 126.95, 126.57, 126.05, 124.97, 122.77, 121.93, 66.42, 29.77, 20.33, 14.23, 12.83, 7.10, 6.96.
3-(5-Bromo-4-(naphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (10f): White solid; 0.46 g (88%); m.p. 177.5–179 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.22 (brs, 1H), 8.24 (t, J = 4.6 Hz, 1H), 8.15 (d, J = 7.6 Hz, 1H), 7.61–7.74 (m, 4H), 7.14 (d, J = 8.0 Hz, 1H), 2.61–2.68 (m, 3H), 2.46–2.55 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.80, 156.60, 133.78, 130.98, 130.20, 128.92, 128.86, 128.67, 128.44, 127.29, 126.92, 125.82, 121.11, 29.84, 20.56.
3-(5-Bromo-4-(4-methylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (10g): White solid; 0.46 g (86%); m.p. 186 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 12.24 (brs, 1H), 8.20 (d, J = 8.4 Hz, 1H), 7.68–7.72 (m, 1H), 7.57–7.65 (m, 3H), 7.11 (d, J = 8.4 Hz, 1H), 2.76 (s, 3H), 2.59–2.64 (m, 3H), 2.46–2.53 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.96, 156.75, 137.99, 132.68, 130.41, 128.92, 128.08, 127.21, 127.18, 126.50, 126.28, 125.22, 121.60, 30.07, 20.65, 19.10.
3-(5-Bromo-4-(4-ethylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (10h): White solid; 0.48 g (85%); m.p. 167.5–170 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.21 (brs, 1H), 8.27 (d, J = 8.4 Hz, 1H), 7.58–7.72 (m, 4H), 7.11 (d, J = 8.4 Hz, 1H), 3.19 (q, J = 7.5 Hz, 2H), 2.58–2.67 (m, 2H), 2.45–2.53 (m, 2H), 1.37 (t, J = 7.6 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.87, 156.73, 143.57, 131.95, 130.46, 129.15, 127.96, 127.22, 126.62, 124.72, 124.62, 121.82, 29.87, 25.20, 20.62, 14.69.
3-(5-Bromo-4-(4-n-propylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (10i): White solid; 0.52 g (89%); m.p. 139.5–141.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.20 (brs, 1H), 8.26 (d, J = 8.4 Hz, 1H), 7.56–7.71 (m, 4H), 7.10 (d, J = 8.0 Hz, 1H), 3.13 (t, J = 7.6 Hz, 2H), 2.58–2.64 (m, 2H), 2.45–2.52 (m, 2H), 1.72–1.80 (m, 2H), 1.02 (t, J = 7.4 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.52, 157.37, 142.76, 132.76, 131.13, 129.88, 128.61, 127.91, 127.83, 127.12, 126.34, 125.58, 122.46, 34.90, 30.52, 24.11, 21.27, 14.69.
3-(5-Bromo-4-(4-isopropylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (10j): White solid; 0.48 g (83%); m.p. 197 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 12.21 (brs, 1H), 8.36 (d, J = 8.4 Hz, 1H), 7.60–7.72 (m, 4H), 7.11 (d, J = 8.0 Hz, 1H), 3.83–3.90 (m, 1H), 2.59–2.65 (m, 3H), 2.44–2.51 (m, 1H), 1.40 (d, J = 6.8 Hz, 6H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.85, 156.69, 147.82, 131.49, 130.45, 129.17, 127.81, 127.18, 127.06, 126.59, 124.27, 121.86, 121.78, 29.79, 28.17, 23.31, 23.25, 20.58.
3-(5-Bromo-4-(4-methoxynaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (10k): White solid; 0.49 g (87%); m.p. 188 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 12.20 (brs, 1H), 8.29–8.31 (m, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.62–7.65 (m, 2H), 7.17 (d, J = 8.4 Hz, 1H), 7.03–7.06 (m, 1H), 4.08 (s, 3H), 2.59–2.66 (m, 3H), 2.47–2.52 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.83, 156.86, 156.51, 130.82, 129.77, 128.75, 127.66, 126.58, 125.16, 122.40, 121.19, 121.08, 104.13, 56.15, 29.90, 20.56.
3-(5-Bromo-4-(4-ethoxynaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (10l): White solid; 0.52 g (89%); m.p. 108–110.5 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.20 (brs, 1H), 8.30–8.34 (m, 1H), 7.60–7.65 (m, 3H), 7.14 (d, J = 8.0 Hz, 1H), 7.01–7.05 (m, 1H), 4.33 (q, J = 6.9 Hz, 2H), 2.59–2.67 (m, 3H), 2.47–2.55 (m, 1H), 1.51 (t, J = 7.0 Hz, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.81, 156.84, 155.75, 130.82, 129.81, 128.70, 127.66, 126.47, 125.22, 122.47, 121.03, 120.97, 104.66, 64.14, 29.89, 20.55, 14.47.
3-(5-Bromo-4-(4-bromonaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionic acid (10m): White solid; 0.58 g (91%); m.p. 202 °C (dec). 1H-NMR (DMSO-d6, 400 MHz) δ: 12.21 (brs, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.15 (d, J = 8.0 Hz, 1H), 7.85 (t, J = 7.8 Hz, 1H), 7.75 (d, J = 7.6 Hz, 1H), 7.72 (d, J = 8.0 Hz, 1H), 7.19 (d, J = 8.4 Hz, 1H), 2.60–2.69 (m, 3H), 2.49–2.56 (m, 1H). 13C-NMR (DMSO-d6, 100 MHz) δ: 172.80, 156.63, 131.83, 130.11, 130.02, 129.50, 129.22, 129.02, 127.71, 127.42, 124.91, 122.17, 29.87, 20.52.
3-(5-Bromo-4-((4-bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)propionic acid (10n): White solid; 0.56 g (85%); m.p. 173–176 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.22 (brs, 1H), 8.22–8.26 (m, 2H), 7.76–7.82 (m, 3H), 6.36 (d, J = 8.0 Hz, 1H), 5.77 (s, 2H), 2.85 (t, J = 7.0 Hz, 2H), 2.69 (t, J = 7.2 Hz, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.68, 157.03, 131.97, 131.74, 131.52, 130.70, 130.38, 128.99, 128.46, 127.88, 124.48, 123.21, 122.49, 45.63, 30.62, 20.93.
3-(5-Bromo-4-(2-(4-bromonaphth-1-yl)ethyl)-4H-1,2,4-triazol-3-yl)propionic acid (10o): White solid; 0.57 g (84%); m.p. 190–192 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.21 (brs, 1H), 8.15–8.20 (m, 2H), 7.76 (d, J = 7.6 Hz, 1H), 7.66–7.73 (m, 2H), 7.11 (d, J = 8.0 Hz, 1H), 4.26 (t, J = 7.0 Hz, 2H), 3.46 (t, J = 7.0 Hz, 2H), 2.71 (t, J = 6.6 Hz, 2H), 2.62 (t, J = 6.4 Hz, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.10, 155.49, 133.82, 132.78, 131.28, 129.58, 128.82, 127.98, 127.68, 127.47, 127.22, 124.07, 121.26, 44.63, 31.63, 29.96, 19.93.
4-(5-Bromo-4-((4-bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)butanoic acid (10p): White solid; 0.56 g (83%); m.p. 177–179 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 12.02 (brs, 1H), 8.21–8.25 (m, 2H), 7.76–7.82 (m, 3H), 6.30 (d, J = 8.0 Hz, 1H), 5.74 (s, 2H), 2.71 (t, J = 7.4 Hz, 2H), 2.27 (t, J = 7.2 Hz, 2H), 1.78–1.86 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 173.91, 156.81, 131.47, 131.05, 130.79, 129.95, 129.71, 128.30, 127.75, 127.18, 123.74, 122.27, 121.75, 45.01, 32.49, 23.84, 21.67.
5-(5-Bromo-4-((4-bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)pentanoic acid (10q): White solid; 0.57 g (82%); m.p. 166.5–168 °C. 1H-NMR (DMSO-d6, 400 MHz) δ: 11.95 (brs, 1H), 8.21–8.25 (m, 2H), 7.76–7.83 (m, 3H), 6.31 (d, J = 8.0 Hz, 1H), 5.74 (s, 2H), 2.68 (t, J = 7.4 Hz, 2H), 2.14 (t, J = 7.2 Hz, 2H), 1.58–1.64 (m, 2H), 1.45–1.53 (m, 2H). 13C-NMR (DMSO-d6, 100 MHz) δ: 174.21, 157.03, 131.54, 131.07, 130.83, 129.84, 129.70, 128.30, 127.76, 127.19, 123.77, 122.39, 121.78, 44.99, 33.15, 25.57, 24.19, 23.79.
3.2.15. General Procedure for the Synthesis of 1a–1r
To a stirred mixture of 7a or 10a–10q (1 mmol, accurately weighted to four decimal places) in MeOH (5 mL) was added an aqueous solution of NaOH (prepared by dissolving 0.0400 g, 1 mmol of NaOH in a minimal volume of water), and the resulting mixture was stirred at room temperature until a clear solution was obtained (typically within 1 h).
The reaction mixture was filtered off and the filtrate was evaporated on a rotary evaporator to give rise to a residue, which was co-evaporated with CH2Cl2 (10 mL × 3) on a rotary evaporator and further dried in vacuo at room temperature to yield 1a–1r.
Sodium 3-(4-(4-cyclopropylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (1a): White solid; 0.32 g (98%); m.p. 140 °C (dec). 1H-NMR (MeOH-d4, 400 MHz) δ: 8.59 (d, J = 8.4 Hz, 1H), 8.56 (s, 1H), 7.68 (t, J = 7.6 Hz, 1H), 7.60 (t, J = 7.6 Hz, 1H), 7.55 (d, J = 7.6 Hz, 1H), 7.42 (d, J = 7.6 Hz, 1H), 7.19 (d, J = 7.6 Hz, 1H), 2.75–2.80 (m, 2H), 2.43–2.53 (m, 3H), 1.16–1.19 (m, 2H), 0.82–0.85 (m, 2H). 13C-NMR (MeOH-d4, 100 MHz) δ: 179.27, 157.30, 146.36, 144.16, 135.44, 130.90, 129.21, 129.11, 128.23, 126.56, 126.33, 124.00, 122.79, 35.53, 22.43, 14.13, 7.59, 7.56. ESI-HRMS [M − Na]−: (m/z) calcd. for C18H16N3O2: 306.1243, found: 306.1235.
Sodium 3-(5-chloro-4-(4-cyclopropylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (1b): White solid; 0.36 g (99%); m.p. 135 °C (dec). 1H-NMR (MeOH-d4, 400 MHz) δ: 8.61 (d, J = 8.4 Hz, 1H), 7.67–7.72 (m, 1H), 7.60–7.64 (m, 1H), 7.57 (d, J = 7.6 Hz, 1H), 7.45 (dd, J = 0.6 Hz and 7.8 Hz, 1H), 7.17 (d, J = 8.4 Hz, 1H), 2.65–2.78 (m, 2H), 2.40–2.59 (m, 3H), 1.16 (m, 2H), 0.84–0.88 (m, 2H). 13C-NMR (MeOH-d4, 100 MHz) δ: 178.94, 159.53, 144.97, 143.92, 135.58, 130.72, 129.35, 128.33, 127.83, 127.67, 126.50, 124.12, 122.56, 34.80, 23.54, 14.16, 7.63. ESI-HRMS [M − Na]−: (m/z) calcd. for C18H15ClN3O2: 340.0853, found: 340.0854.
Sodium 3-(5-bromo-4-(4-cyclopropylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (1c): White solid; 0.41 g (100%); m.p. 162 °C (dec). 1H-NMR (MeOH-d4, 400 MHz) δ: 8.61 (d, J = 8.8 Hz, 1H), 7.68 (t, J = 7.4 Hz, 1H), 7.60 (t, J = 7.4 Hz, 1H), 7.55 (d, J = 7.6 Hz, 1H), 7.44 (d, J = 7.6 Hz, 1H), 7.13 (d, J = 8.4 Hz, 1H), 2.68–2.77 (m, 2H), 2.38–2.57 (m, 3H), 1.14–1.20 (m, 2H), 0.83–0.89 (m, 2H). 13C-NMR (MeOH-d4, 100 MHz) δ: 178.07, 159.87, 144.91, 135.57, 130.78, 129.67, 129.28, 128.60, 128.32, 127.75, 126.47, 124.10, 122.72, 34.04, 23.17, 14.16, 7.65. ESI-HRMS [M − Na]−: (m/z) calcd. for C18H15BrN3O2: 384.0348 (79Br), found: 384.0339; calcd. for C18H15BrN3O2: 386.0327 (81Br), found: 386.0317.
Sodium 3-(4-(4-cyclopropylnaphth-1-yl)-5-iodo-4H-1,2,4-triazol-3-yl)propionate (1d): White solid; 0.44 g (97%); m.p. 158 °C (dec). 1H-NMR (MeOH-d4, 400 MHz) δ: 8.60 (d, J = 8.8 Hz, 1H), 7.68 (td, J = 0.9 Hz and 7.6 Hz, 1H), 7.57–7.61 (m, 1H), 7.50 (d, J = 7.6 Hz, 1H), 7.44 (d, J = 7.6 Hz, 1H), 7.07 (d, J = 8.4 Hz, 1H), 2.71–2.78 (m, 2H), 2.47–2.56 (m, 2H), 2.37–2.46 (m, 1H), 1.15–1.21 (m, 2H), 0.83-0.90 (m, 2H). 13C-NMR (MeOH-d4, 100 MHz) δ: 179.28, 160.15, 144.70, 135.57, 131.00, 129.99, 129.14, 128.27, 127.95, 126.43, 124.11, 122.99, 105.86, 35.45, 23.54, 14.18, 7.71, 7.60. ESI-HRMS [M − Na]−: (m/z) calcd. for C18H15IN3O2: 432.0209, found: 432.0200.
Sodium 3-(4-(4-cyclopropylnaphth-1-yl)-5-methoxy-4H-1,2,4-triazol-3-yl)propionate (1e): White solid; 0.36 g (99%); m.p. 133 °C (dec). 1H-NMR (MeOH-d4, 400 MHz) δ: 8.57 (d, J = 8.4 Hz, 1H), 7.66 (dt, J = 1.1 Hz and 7.7 Hz, 1H), 7.58 (dt, J = 1.2 Hz and 7.6 Hz, 1H), 7.48 (d, J = 7.6 Hz, 1H), 7.40 (d, J = 7.6 Hz, 1H), 7.25 (d, J = 8.4 Hz, 1H), 3.99 (s, 3H), 2.65 (t, J = 8.0 Hz, 2H), 2.43–2.51 (m, 2H), 2.31–2.39 (m, 1H), 1.14–1.18 (m, 2H), 0.81–0.84 (m, 2H). 13C-NMR (MeOH-d4, 100 MHz) δ: 179.46, 161.32, 155.00, 144.05, 135.55, 131.08, 128.87, 128.03, 127.80, 127.35, 126.32, 124.13, 122.86, 58.56, 35.19, 23.38, 14.12, 7.53, 7.44. ESI-HRMS [M − Na]−: (m/z) calcd. for C19H18N3O3: 336.1348, found: 336.1346.
Sodium 3-(4-(4-cyclopropylnaphth-1-yl)-5-ethoxy-4H-1,2,4-triazol-3-yl)propionate (1f): White solid; 0.37 g (98%); m.p. 101.5 °C (dec). 1H-NMR (MeOH-d4, 400 MHz) δ: 8.57 (d, J = 8.4 Hz, 1H), 7.66 (t, J = 7.0 Hz, 1H), 7.59 (t, J = 7.6 Hz, 1H), 7.48 (d, J = 7.6 Hz, 1H), 7.41 (d, J = 7.6 Hz, 1H), 7.26 (d, J = 8.0 Hz, 1H), 4.39 (q, J = 7.1 Hz, 2H), 2.64 (t, J = 7.6 Hz, 2H), 2.44–2.49 (m, 2H), 2.37–2.38 (m, 1H), 1.22 (t, J = 7.0 Hz, 3H), 1.14–1.19 (m, 2H), 0.82–0.86 (m, 2H). 13C-NMR (MeOH-d4, 100 MHz) δ: 178.85, 160.64, 154.61, 143.99, 135.57, 131.09, 128.82, 128.03, 127.89, 127.33, 126.33, 124.18, 122.93, 68.47, 34.74, 23.20, 14.63, 14.14, 7.51, 7.44. ESI-HRMS [M − Na]−: (m/z) calcd. for C20H20N3O3: 350.1505, found: 350.1497.
Sodium 3-(5-bromo-4-(naphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (1g): White solid; 0.37 g (100%); m.p. 182–184 °C. 1H-NMR (MeOH-d4, 400 MHz) δ: 8.18 (d, J = 7.6 Hz, 1H), 8.06–8.09 (m, 1H), 7.59–7.72 (m, 4H), 7.17–7.19 (m, 1H), 2.68–2.81 (m, 2H), 2.55–2.63 (m, 1H), 2.44–2.52 (m, 1H). 13C-NMR (MeOH-d4, 100 MHz) δ: 178.50, 159.84, 135.80, 132.52, 132.01, 130.82, 130.27, 129.92, 129.67, 128.52, 128.10, 126.72, 122.16, 34.33, 23.29. ESI-HRMS [M − Na]−: (m/z) calcd. for C15H11BrN3O2: 344.0035 (79Br), found: 344.0030; calcd. for C15H11BrN3O2: 346.0014 (81Br), found: 346.0010.
Sodium 3-(5-bromo-4-(4-methylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (1h): White solid; 0.38 g (99%); m.p. 155 °C (dec). 1H-NMR (MeOH-d4, 400 MHz) δ: 8.21 (d, J = 8.4 Hz, 1H), 7.65–7.69 (m, 1H), 7.59–7.63 (m, 1H), 7.54 (s, 2H), 7.13–7.15 (m, 1H), 2.80 (s, 3H), 2.71–2.78 (m, 2H), 2.50–2.58 (m, 1H), 2.38–2.49 (m, 1H). 13C-NMR (MeOH-d4, 100 MHz) δ: 179.26, 160.14, 139.95, 134.69, 132.22, 130.82, 129.24, 128.69, 128.36, 127.74, 127.29, 126.28, 122.72, 35.11, 23.60, 19.64. ESI-HRMS [M − Na]−: (m/z) calcd. for C16H13BrN3O2: 358.0190 (79Br), found: 358.0192; calcd. for C16H13BrN3O2: 360.0171 (81Br), found: 360.0171.
Sodium 3-(5-bromo-4-(4-ethylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (1i): White solid; 0.38 g (97%); m.p. 198 °C (dec). 1H-NMR (MeOH-d4, 400 MHz) δ: 8.27 (d, J = 8.4 Hz, 1H), 7.64–7.69 (m, 2H), 7.57–7.61 (m, 2H), 7.14 (d, J = 8.0 Hz, 1H), 3.23 (q, J = 7.6 Hz, 2H), 2.71–2.76 (m, 2H), 2.52–2.59 (m, 1H), 2.42–2.48 (m, 1H), 1.43 (t, J = 7.4 Hz, 3H). 13C-NMR (MeOH-d4, 100 MHz) δ: 179.13, 160.07, 145.72, 133.80, 132.18, 131.00, 129.11, 128.59, 128.32, 127.81, 125.88, 125.71, 122.82, 34.91, 26.90, 23.52, 15.37. ESI-HRMS [M − Na]−: (m/z) calcd. for C17H15BrN3O2: 372.0348 (79Br), found: 372.0349; calcd. for C17H15BrN3O2: 374.0327 (81Br), found: 374.0333.
Sodium 3-(5-bromo-4-(4-n-propylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (1j): White solid; 0.41 g (99%); m.p. 183 °C (dec). 1H-NMR (MeOH-d4, 400 MHz) δ: 8.25 (d, J = 8.4 Hz, 1H), 7.64–7.68 (m, 1H), 7.53–7.61 (m, 3H), 7.14 (d, J = 8.0 Hz, 1H), 3.17 (t, J = 7.6 Hz, 2H), 2.67–2.76 (m, 2H), 2.54–2.62 (m, 1H), 2.43–2.51 (m, 1H), 1.79–1.88 (m, 2H), 1.08 (t, J = 7.4 Hz, 3H). 13C-NMR (MeOH-d4, 100 MHz) δ: 178.64, 159.97, 144.27, 134.04, 132.22, 131.11, 129.11, 128.65, 128.27, 127.64, 126.75, 126.07, 122.85, 36.06, 34.50, 25.06, 23.36, 14.47. ESI-HRMS [M − Na]−: (m/z) calcd. for C18H17BrN3O2: 386.0504 (79Br), found: 386.0509; calcd. for C18H17BrN3O2: 388.0484 (81Br), found: 388.0498.
Sodium 3-(5-bromo-4-(4-isopropylnaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (1k): White solid; 0.41 g (100%); m.p. 194 °C (dec). 1H-NMR (MeOH-d4, 400 MHz) δ: 8.35 (d, J = 8.4 Hz, 1H), 7.57–7.69 (m, 4H), 7.14 (d, J = 8.4 Hz, 1H), 3.86-3.92 (m, 1H), 2.66-2.78 (m, 2H), 2.53–2.61 (m, 1H), 2.42–2.50 (m, 1H), 1.46 (d, J = 6.8 Hz, 3H), 1.46 (d, J = 6.8 Hz, 3H). 13C-NMR (MeOH-d4, 100 MHz) δ: 178.99, 160.08, 150.11, 133.54, 132.23, 131.13, 129.02, 128.48, 128.30, 127.84, 125.45, 122.95, 122.77, 34.82, 29.98, 23.83, 23.50. ESI-HRMS [M − Na]−: (m/z) calcd. for C18H17BrN3O2: 386.0504 (79Br), found: 386.0508; calcd. for C18H17BrN3O2: 388.0484 (81Br), found: 388.0494.
Sodium 3-(5-bromo-4-(4-methoxynaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (1l): White solid; 0.39 g (98%); m.p. 211 °C (dec). 1H-NMR (MeOH-d4, 400 MHz) δ: 8.36-8.38 (m, 1H), 7.57–7.62 (m, 3H), 7.09 (d, J = 8.4 Hz, 1H), 7.05–7.07 (m, 1H), 4.11 (s, 3H), 2.72–2.77 (m, 2H), 2.53–2.61 (m, 1H), 2.44–2.50 (m, 1H). 13C-NMR (MeOH-d4, 100 MHz) δ: 178.60, 160.20, 158.96, 132.70, 131.71, 129.92, 128.86, 127.60, 127.38, 124.02, 122.59, 122.01, 104.54, 56.63, 34.57, 23.37. ESI-HRMS [M − Na]−: (m/z) calcd. for C16H13BrN3O3: 374.0140 (79Br), found: 374.0137; calcd. for C16H13BrN3O3: 376.0120 (81Br), found: 376.0115.
Sodium 3-(5-bromo-4-(4-ethoxynaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (1m): White solid; 0.40 g (96%); m.p. 194 °C (dec). 1H-NMR (MeOH-d4, 400 MHz) δ: 8.38–8.41 (m, 1H), 7.58–7.61 (m, 3H), 7.05–7.07 (m, 2H), 4.33 (q, J = 7.1 Hz, 2H), 2.67–2.77 (m, 2H), 2.47–2.64 (m, 2H), 1.58 (t, J = 7.0 Hz, 3H). 13C-NMR (MeOH-d4, 100 MHz) δ: 178.02, 160.05, 158.23, 132.75, 131.73, 129.87, 128.84, 127.51, 127.45, 124.08, 122.33, 121.98, 105.17, 65.59, 33.99, 23.13, 14.97. ESI-HRMS [M − Na]−: (m/z) calcd. for C17H15BrN3O3: 388.0297 (79Br), found: 388.0299; calcd. for C17H15BrN3O3: 390.0276 (81Br), found: 390.0283.
Sodium 3-(5-bromo-4-(4-bromonaphth-1-yl)-4H-1,2,4-triazol-3-yl)propionate (1n): White solid; 0.43 g (97%); m.p. 204 °C (dec). 1H-NMR (MeOH-d4, 400 MHz) δ: 8.40 (d, J = 8.8 Hz, 1H), 8.05 (d, J = 7.6 Hz, 1H), 7.76–7.81 (m, 1H), 7.69–7.73 (m, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.22 (d, J = 8.4 Hz, 1H), 2.71–2.76 (m, 2H), 2.55–2.63 (m, 1H), 2.46–2.52 (m, 1H). 13C-NMR (MeOH-d4, 100 MHz) δ: 178.68, 159.89, 134.02, 131.96, 131.84, 131.03, 130.63, 130.38, 130.09, 129.09, 128.72, 127.02, 123.14, 34.54, 23.36. ESI-HRMS [M − Na]−: (m/z) calcd. for C15H10Br2N3O2: 423.9119, found: 423.9115.
Sodium 3-(5-bromo-4-((4-bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)propionate (1o): White solid; 0.46 g (100%); m.p. 152.5 °C (dec). 1H-NMR (MeOH-d4, 400 MHz) δ: 8.32–8.34 (m, 1H), 8.18–8.20 (m, 1H), 7.71–7.75 (m, 3H), 6.43 (d, J = 7.6 Hz, 1H), 5.84 (s, 2H), 2.94 (t, J = 7.4 Hz, 2H), 2.65 (t, J = 7.4 Hz, 2H). 13C-NMR (MeOH-d4, 100 MHz) δ: 178.62, 159.53, 133.22, 132.53, 131.92, 131.65, 130.71, 129.04, 128.94, 128.80, 124.18, 124.01, 123.78, 46.84, 34.65, 22.95. ESI-HRMS [M − Na]−: (m/z) calcd. for C16H12Br2N3O2: 437.9276, found: 437.9272.
Sodium 3-(5-bromo-4-(2-(4-bromonaphth-1-yl)ethyl)-4H-1,2,4-triazol-3-yl)propionate (1p): White solid; 0.47 g (99%); m.p. 167 °C (dec). 1H-NMR (MeOH-d4, 400 MHz) δ: 8.24–8.28 (m, 1H), 8.07–8.11 (m, 1H), 7.68 (d, J = 7.6 Hz, 1H), 7.61–7.66 (m, 2H), 7.07 (d, J = 7.6 Hz, 1H), 4.42 (t, J = 7.0 Hz, 2H), 3.54 (t, J = 6.8 Hz, 2H), 2.69 (t, J = 7.0 Hz, 2H), 2.57 (t, J = 7.2 Hz, 2H). 13C-NMR (MeOH-d4, 100 MHz) δ: 178.88, 158.84, 134.75, 134.50, 133.41, 130.81, 130.53, 129.16, 128.92, 128.60, 128.48, 124.85, 123.29, 46.60, 34.83, 33.24, 22.69. ESI-HRMS [M − Na]−: (m/z) calcd. for C17H14Br2N3O2: 451.9432, found: 451.9426.
Sodium 4-(5-bromo-4-((4-bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)butanoate (1q): White solid; 0.47 g (98%); m.p. 166 °C (dec). 1H-NMR (MeOH-d4, 400 MHz) δ: 8.30–8.33 (m, 1H), 8.16–8.19 (m, 1H), 7.71–7.75 (m, 3H), 6.39 (d, J = 8.0 Hz, 1H), 5.79 (s, 2H), 2.78 (t, J = 7.8 Hz, 2H), 2.21 (t, J = 7.2 Hz, 2H), 1.95–2.02 (m, 2H). 13C-NMR (MeOH-d4, 100 MHz) δ: 180.55, 159.43, 133.19, 132.49, 131.85, 131.78, 130.65, 129.05, 128.97, 128.91, 124.18, 124.04, 123.66, 46.83, 37.21, 25.94, 24.57. ESI-HRMS [M − Na]−: (m/z) calcd. for C17H14Br2N3O2: 451.9432, found: 451.9434.
Sodium 5-(5-bromo-4-((4-bromonaphth-1-yl)methyl)-4H-1,2,4-triazol-3-yl)pentanoate (1r): White solid; 0.48 g (98%); m.p. 146–149 °C. 1H-NMR (MeOH-d4, 400 MHz) δ: 8.32–8.34 (m, 1H), 8.16–8.19 (m, 1H), 7.71–7.76 (m, 3H), 6.42 (d, J = 8.0 Hz, 1H), 5.77 (s, 2H), 2.75 (t, J = 7.4 Hz, 2H), 2.11 (t, J = 7.4 Hz, 2H), 1.67–1.74 (m, 2H), 1.56–1.63 (m, 2H). 13C-NMR (MeOH-d4, 100 MHz) δ: 181.17, 159.48, 133.23, 132.52, 131.86, 131.78, 130.67, 129.09, 129.01, 128.98, 124.17, 124.13, 123.84, 46.83, 37.64, 27.66, 26.65, 26.05. ESI-HRMS [M-Na]−: (m/z) calcd. for C18H16Br2N3O2: 465.9589, found: 465.9588.



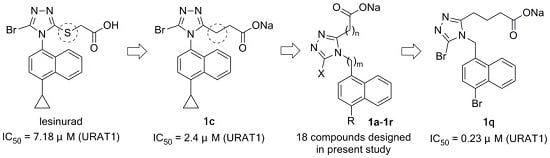
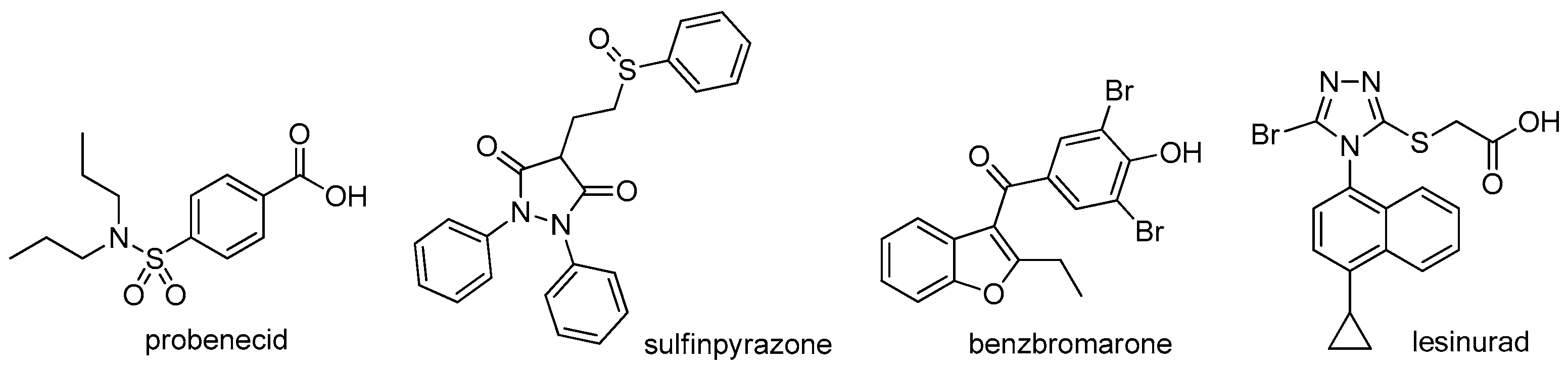

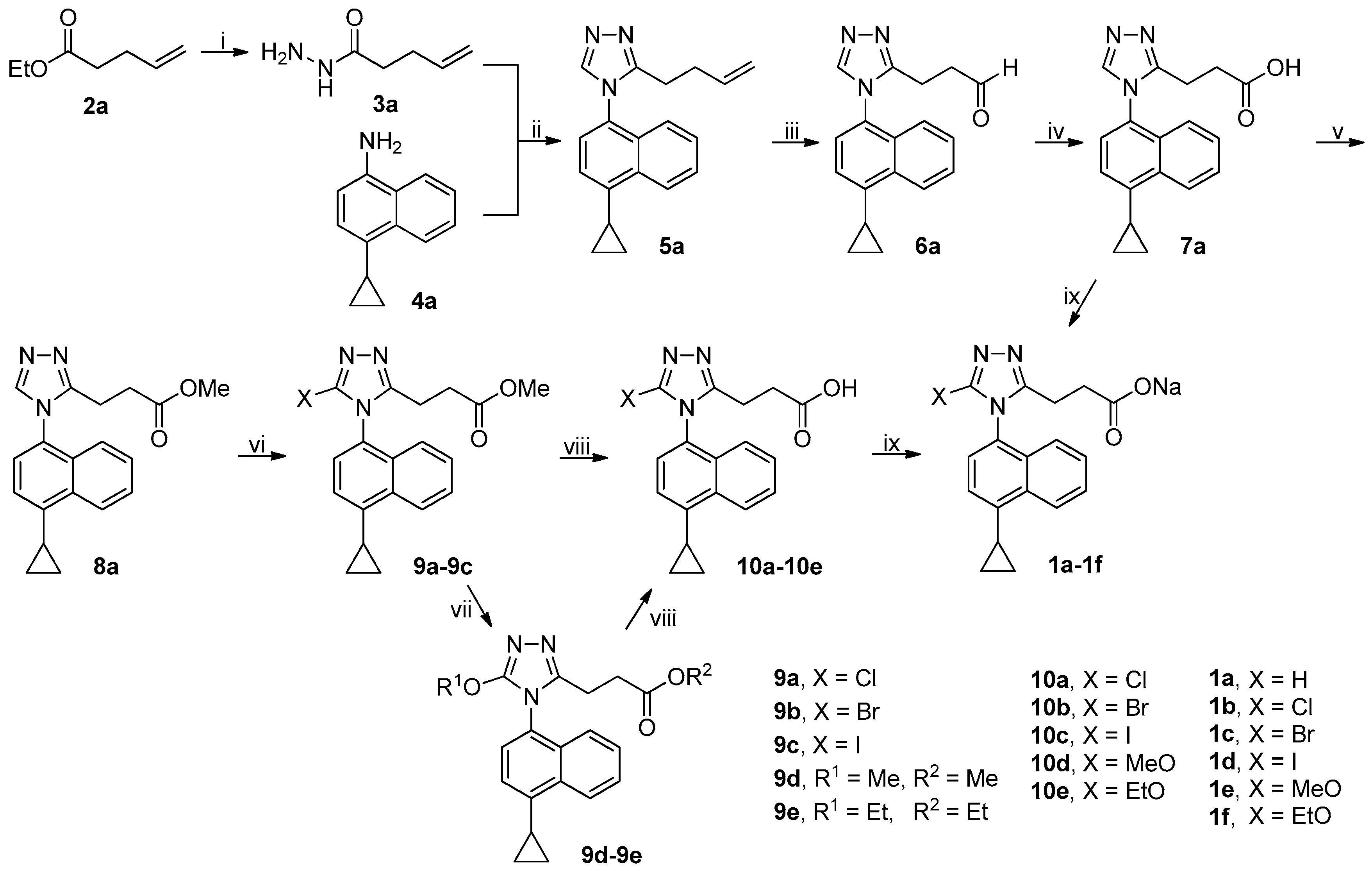
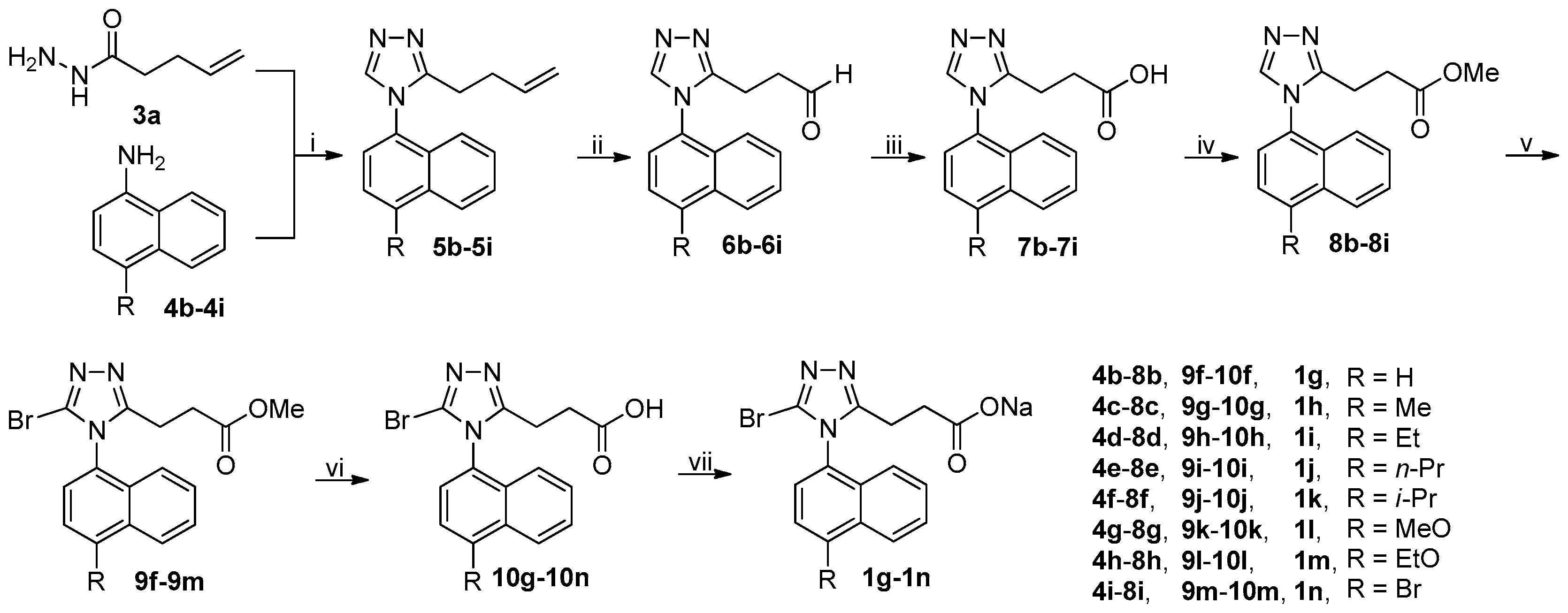
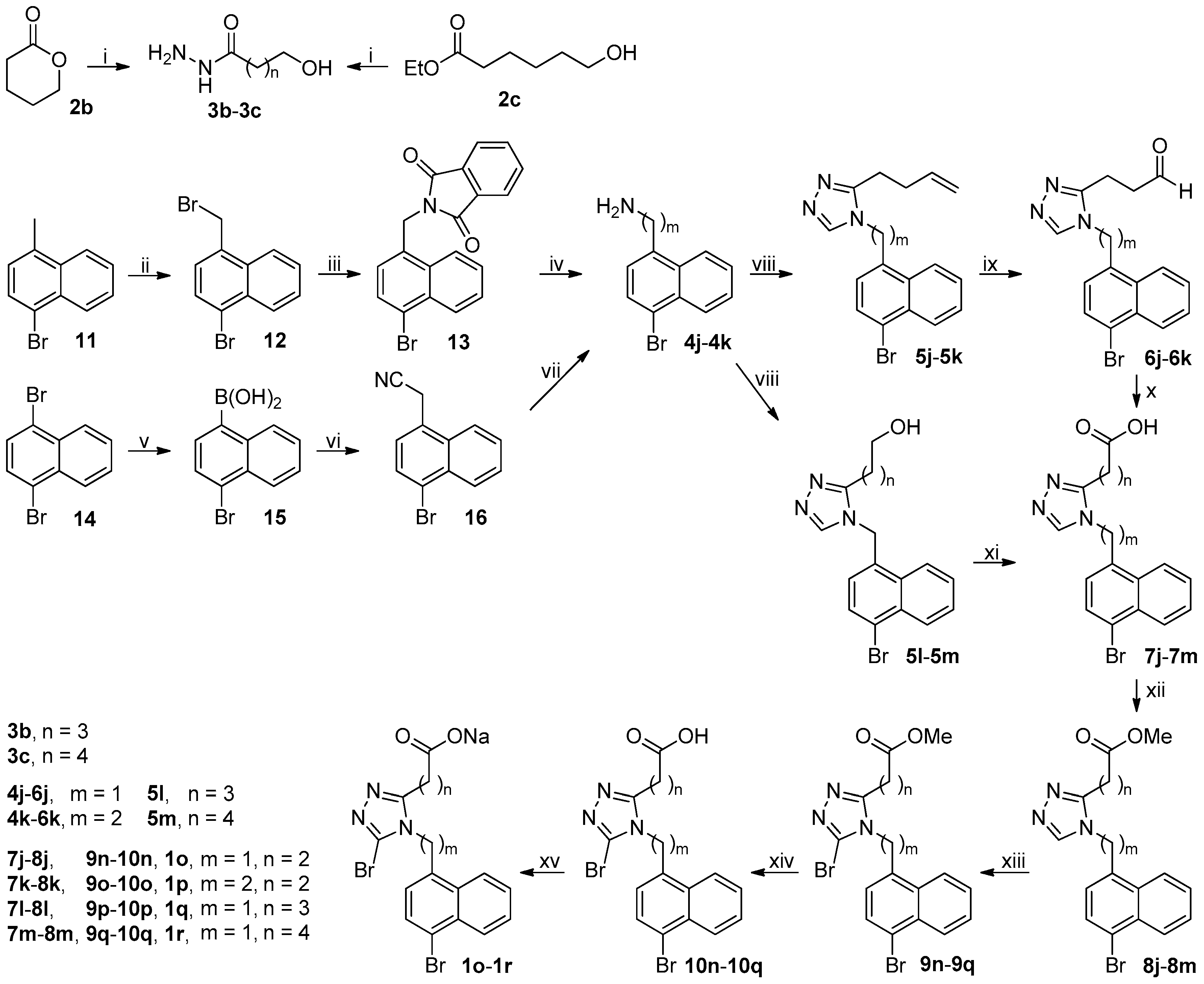

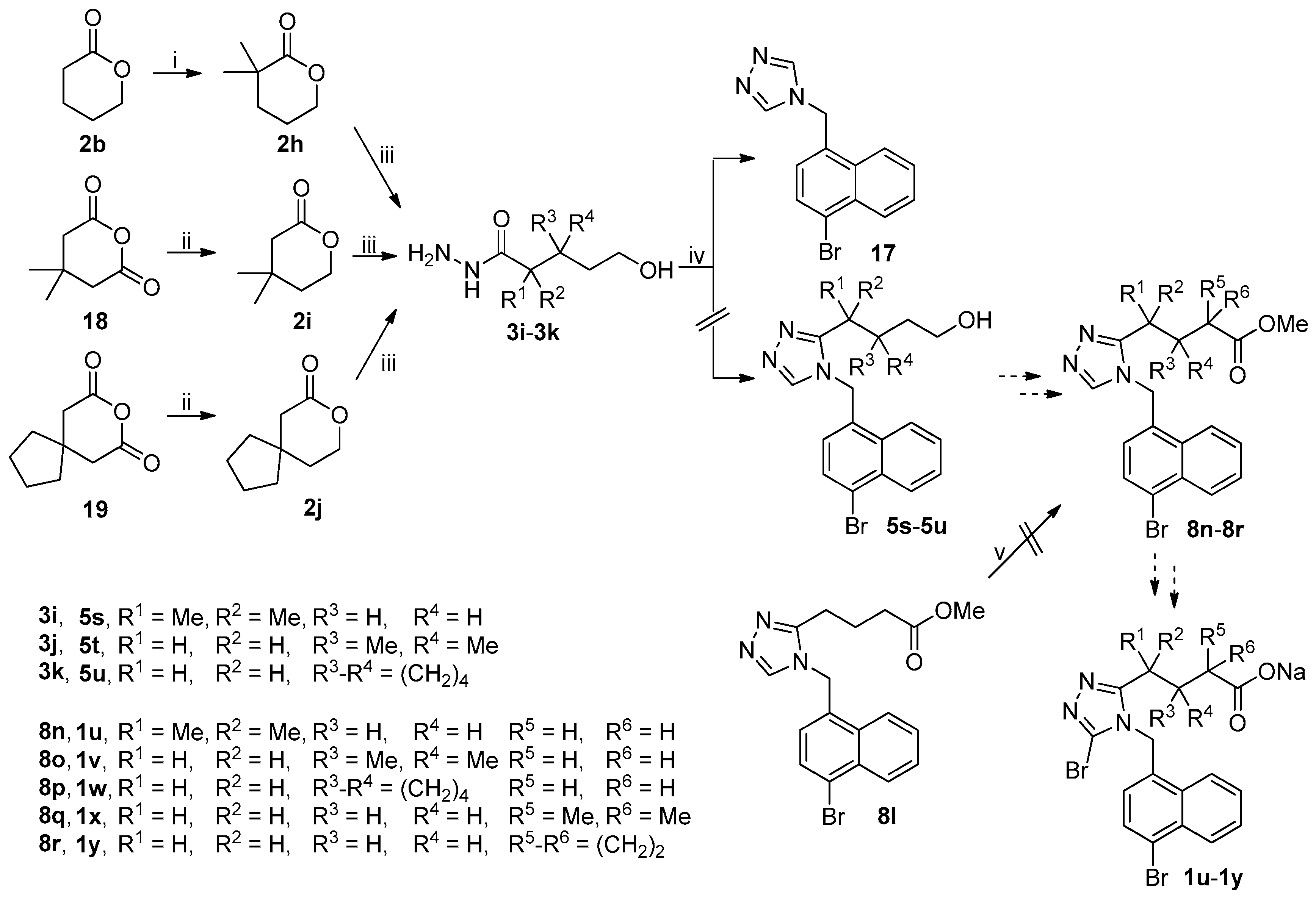

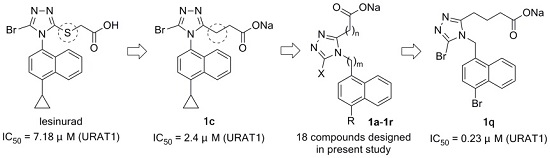{kind=link}
{kind=link}
{kind=link}
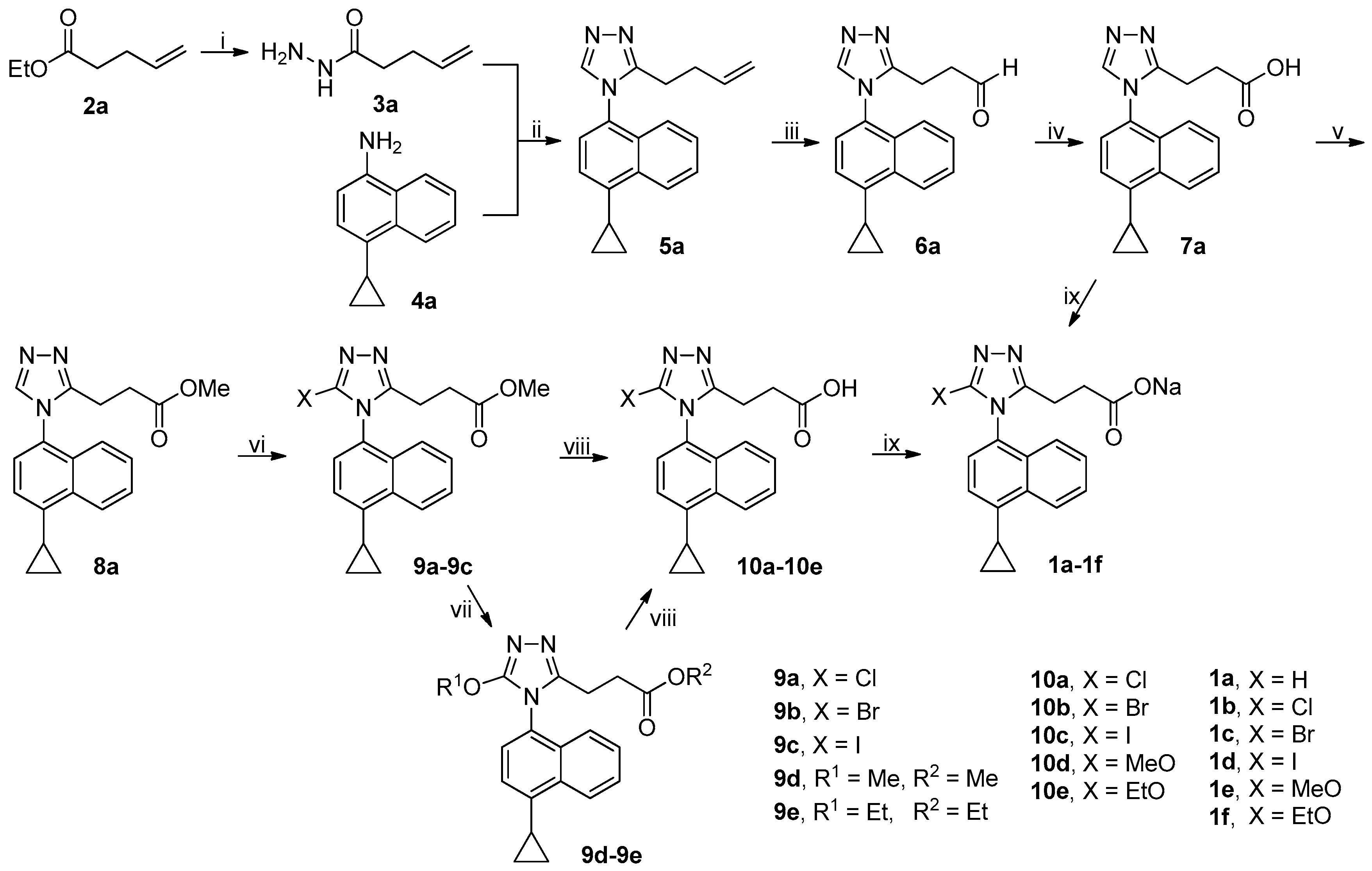{kind=link}
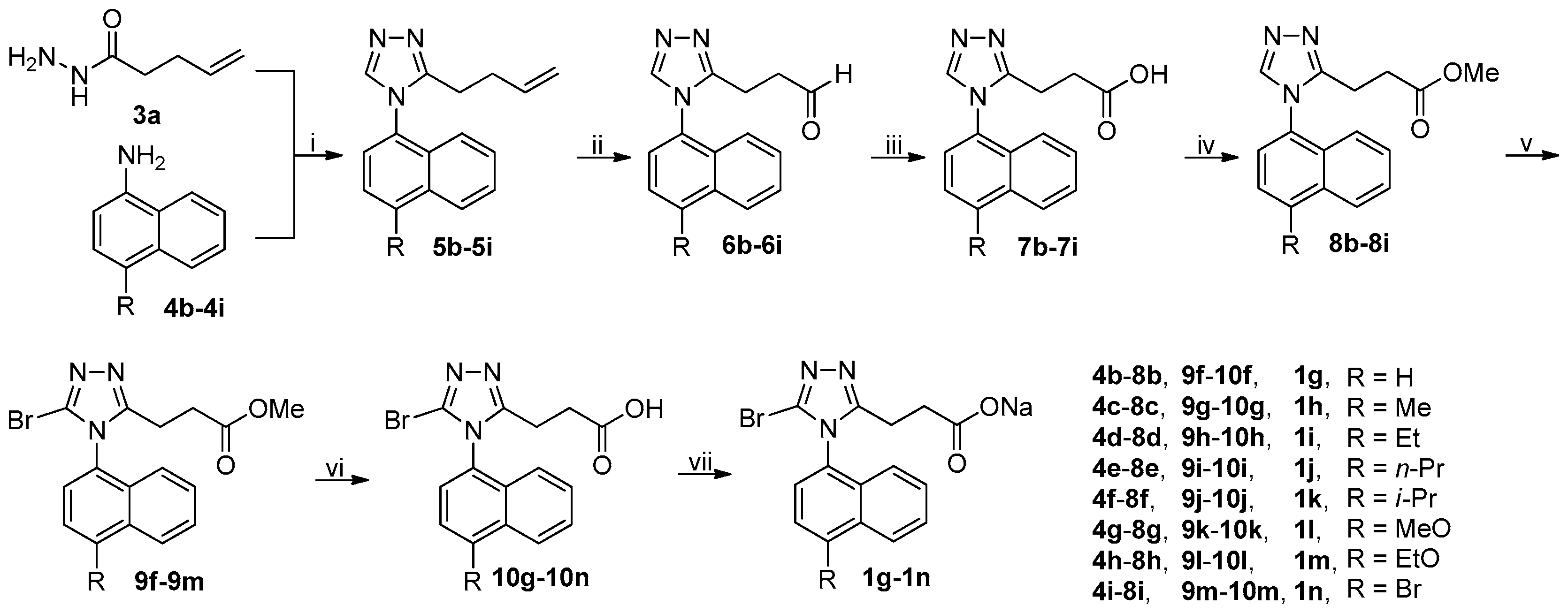{kind=link}
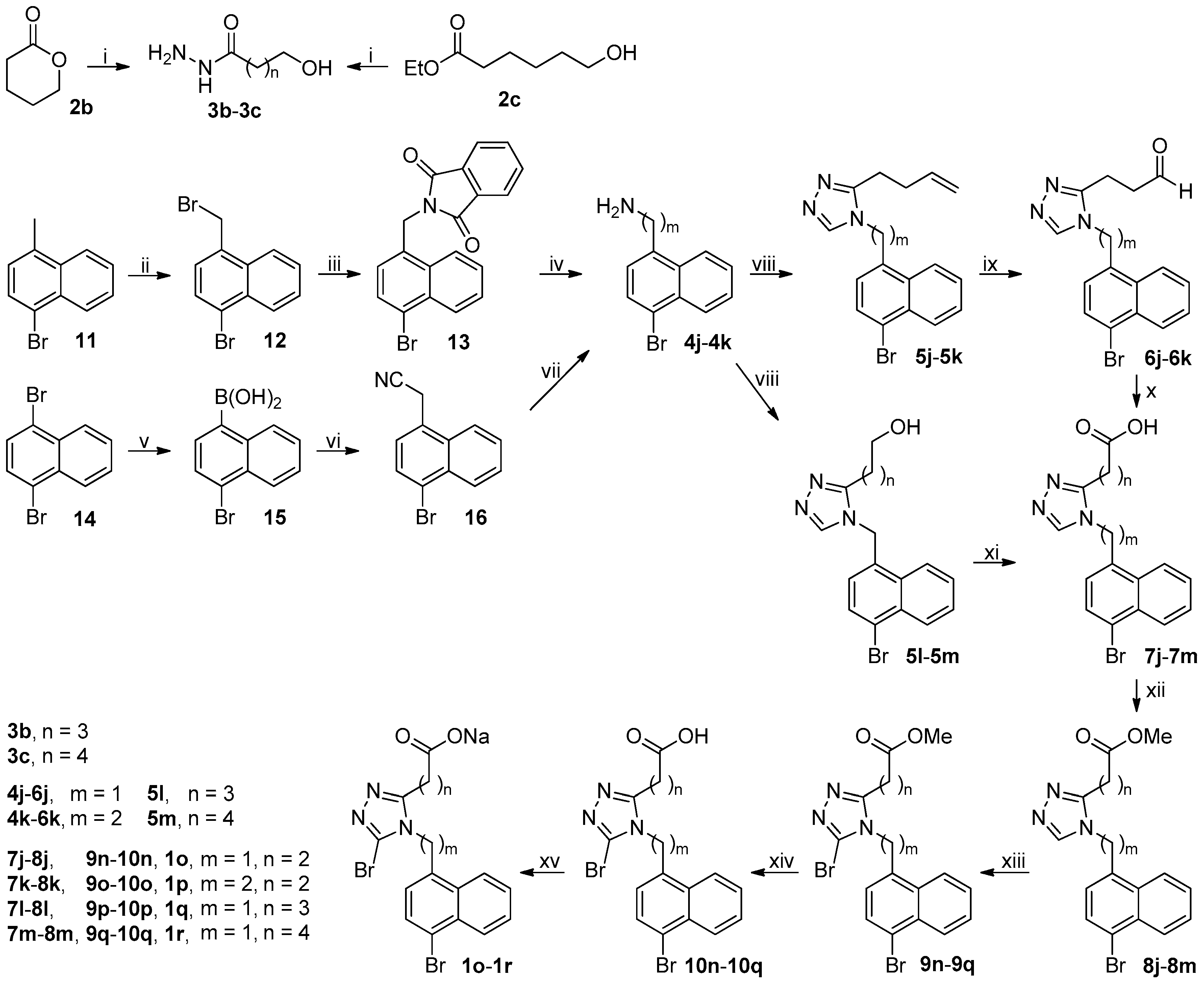{kind=link}
{kind=link}
{kind=link}
