
Characterisation of the Broadly-Specific O-Methyl-transferase JerF from the Late Stages of Jerangolid Biosynthesis

Abstract
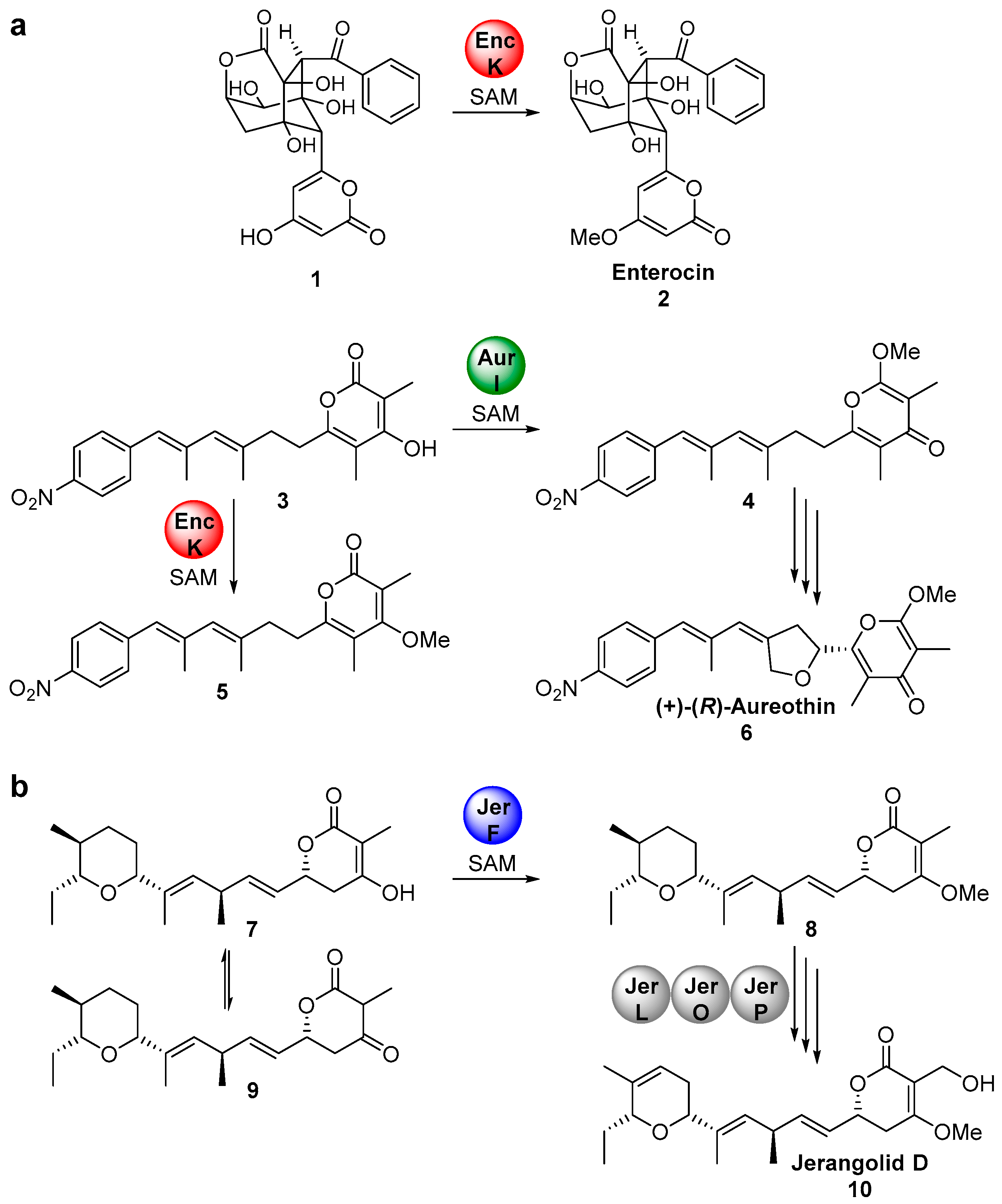
:1. Introduction
2. Results
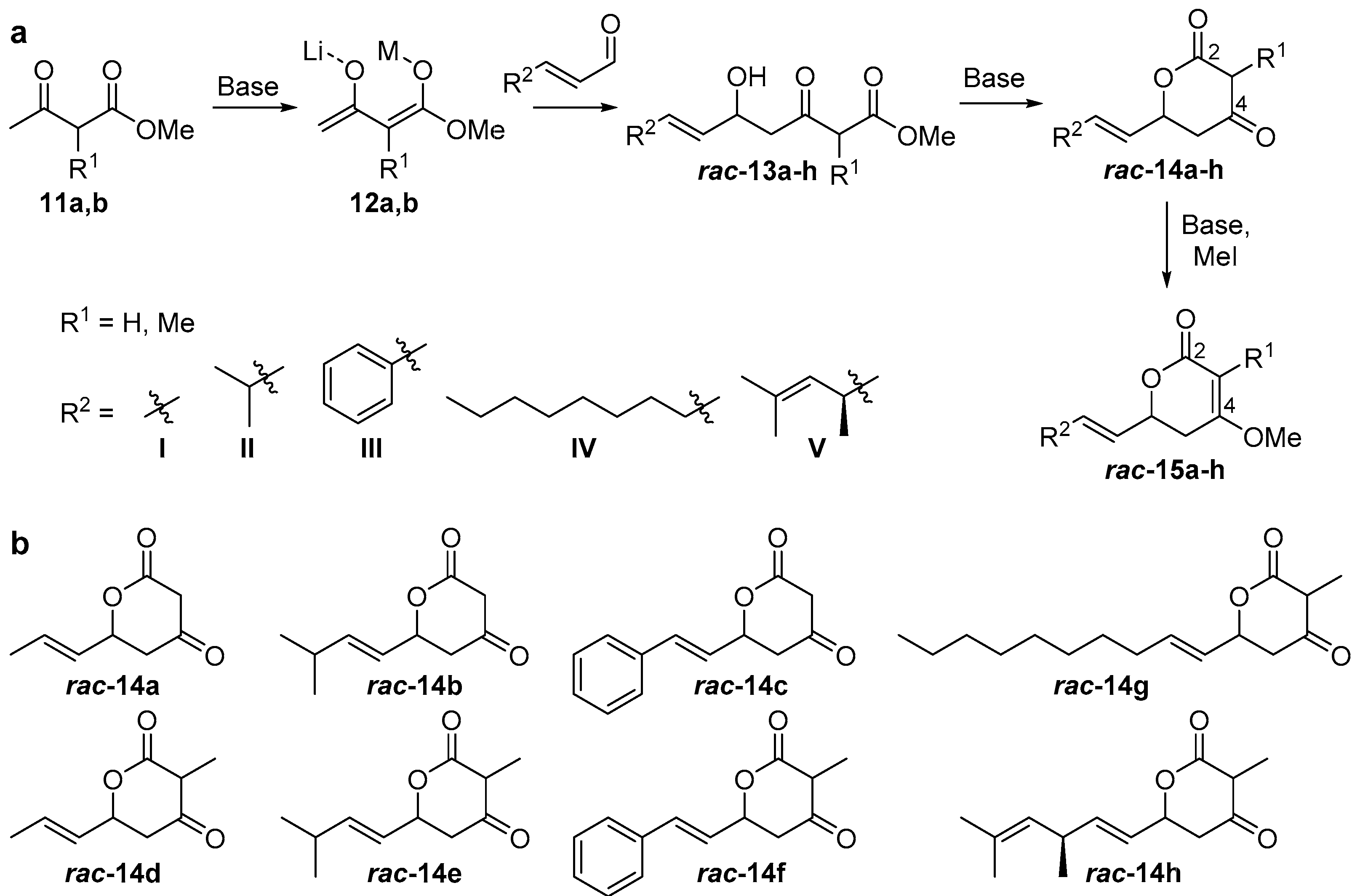
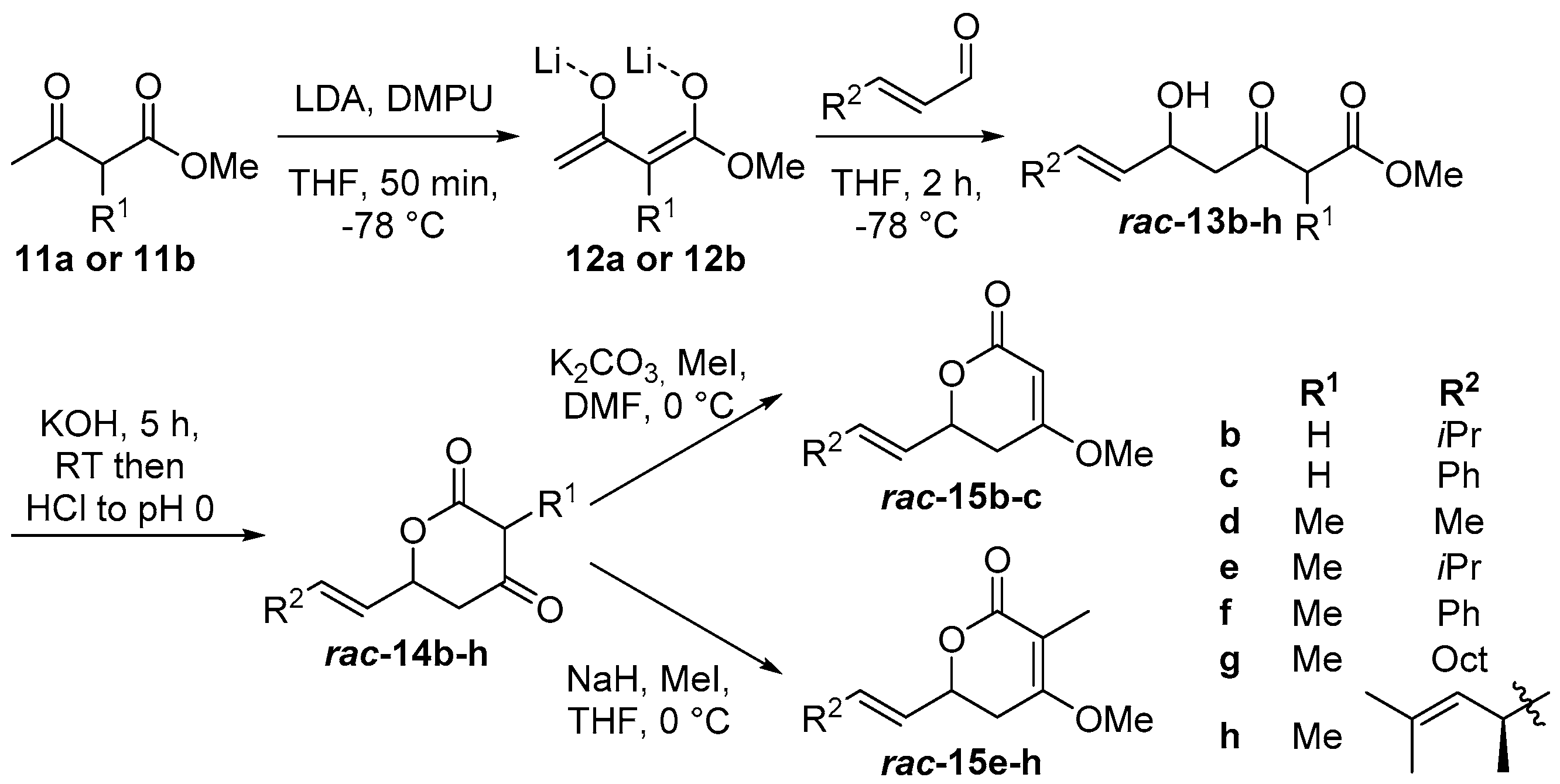
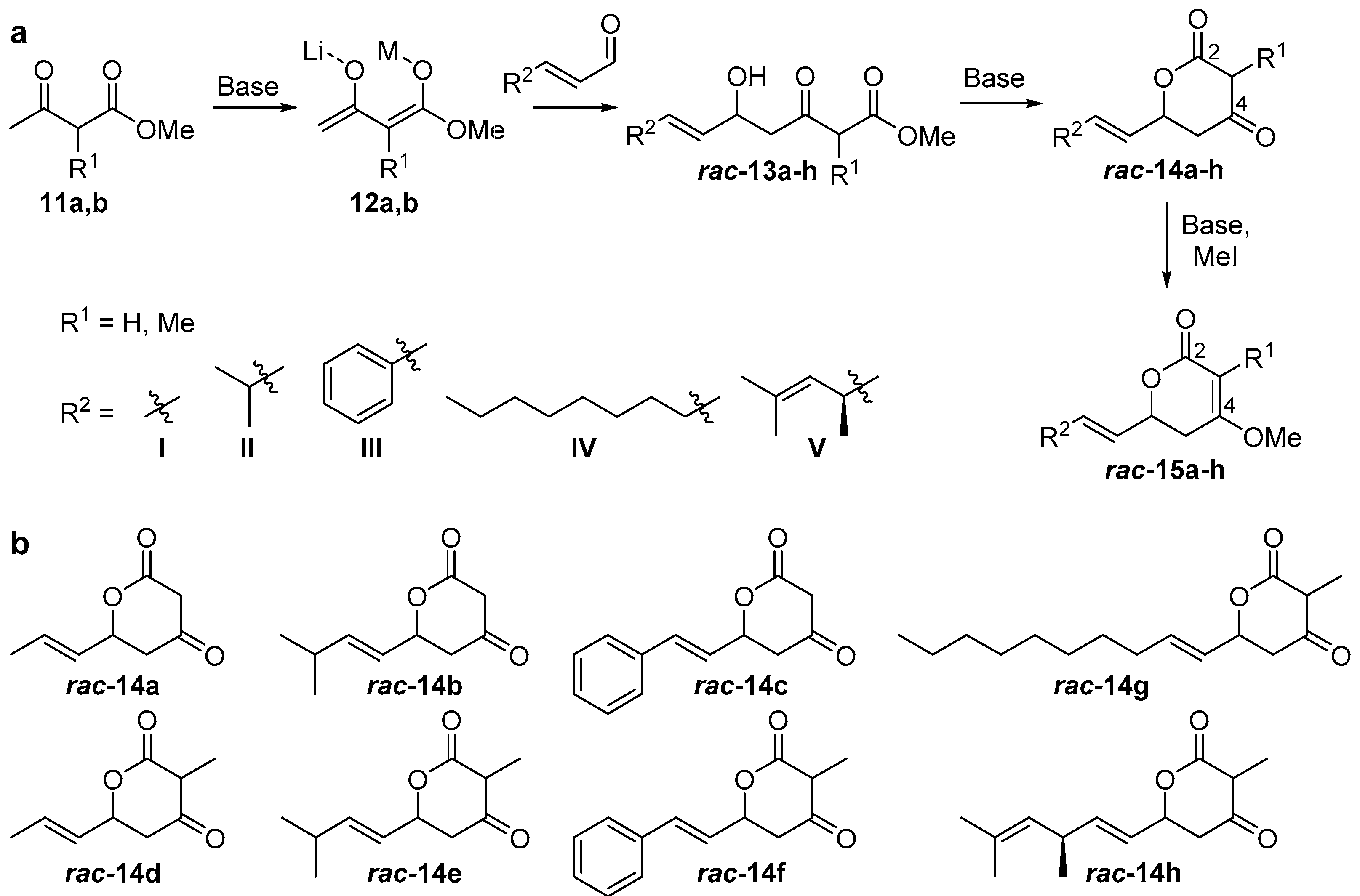
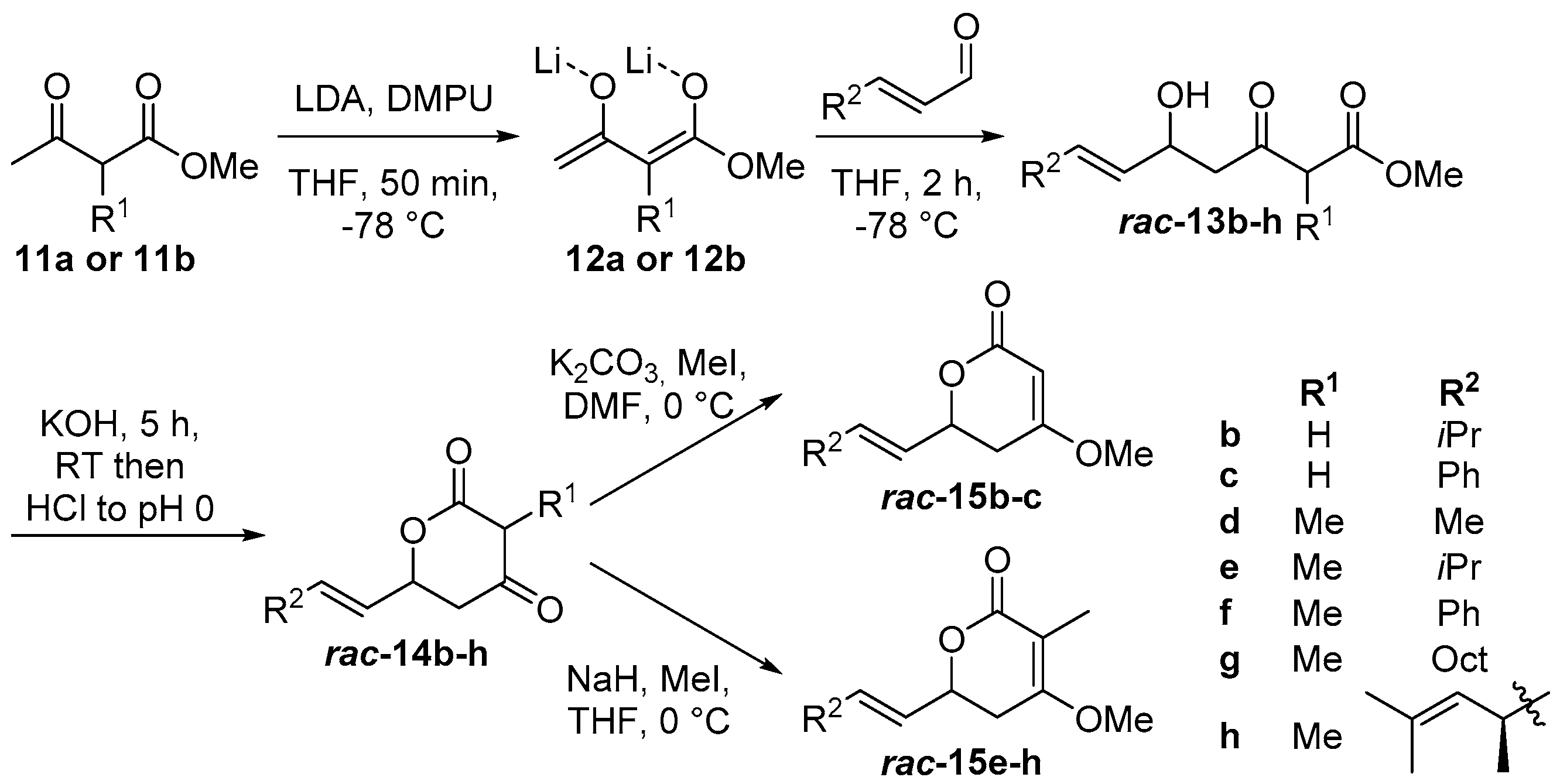
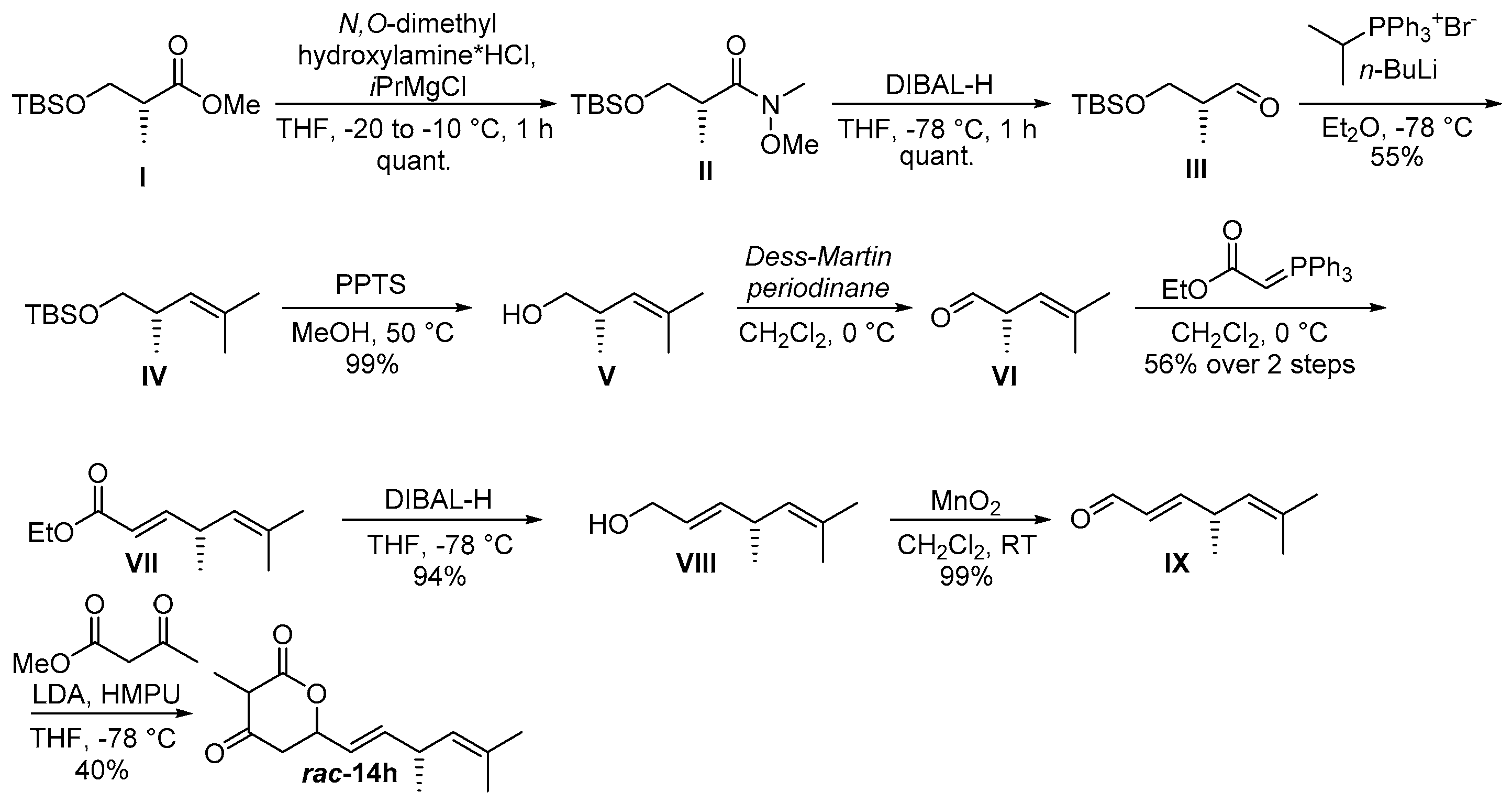
2.1. Synthesis of Substrate Surrogates
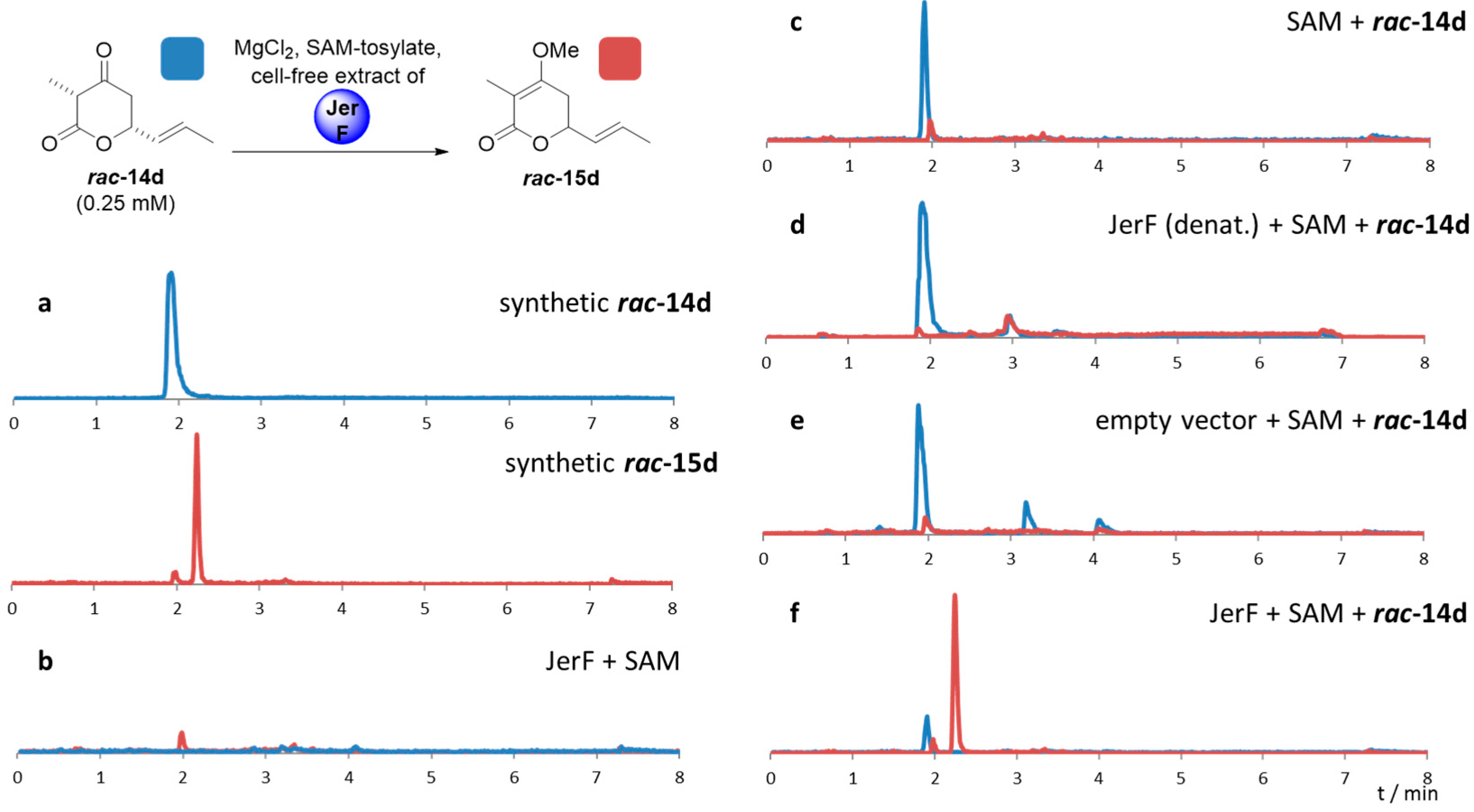
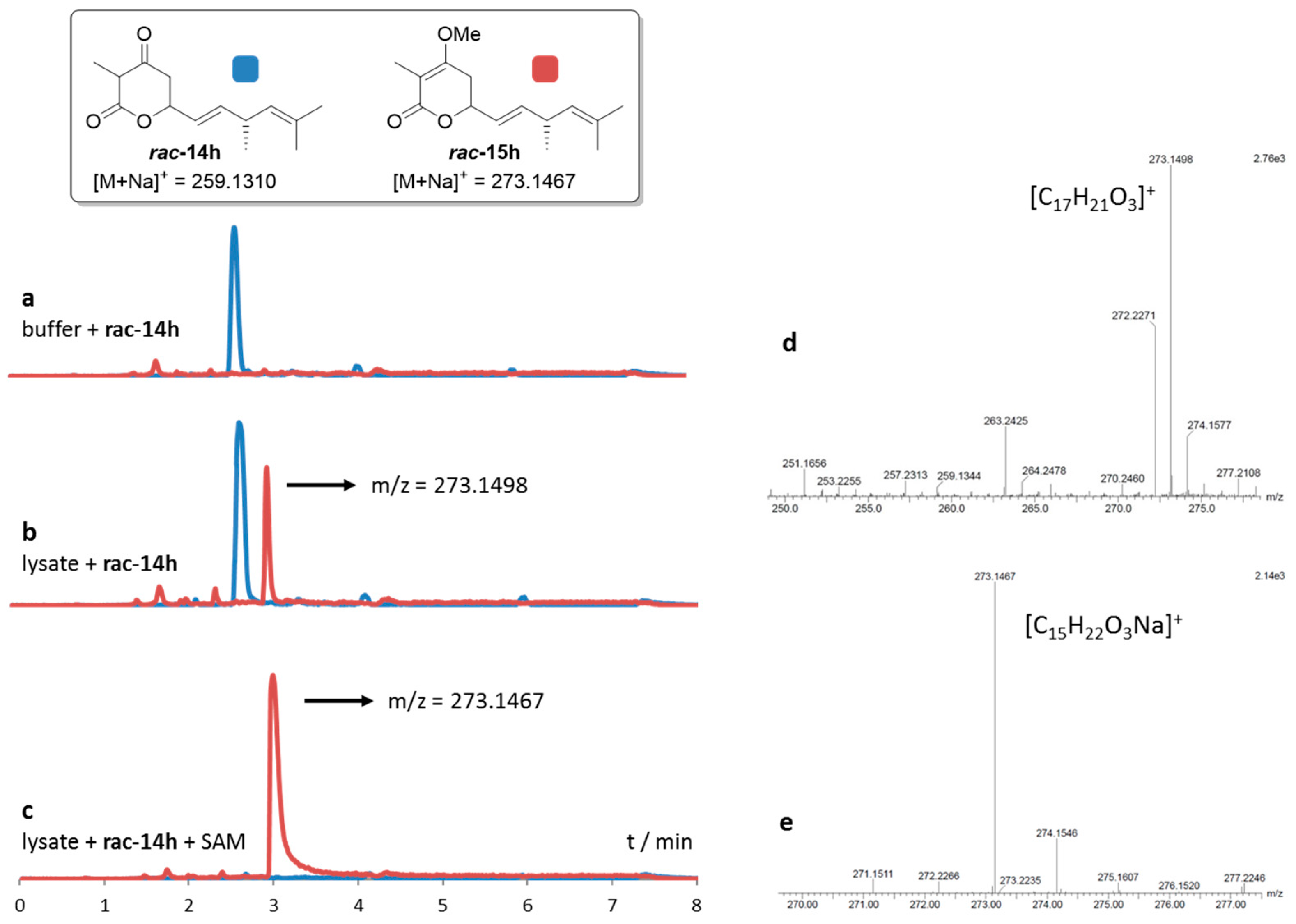
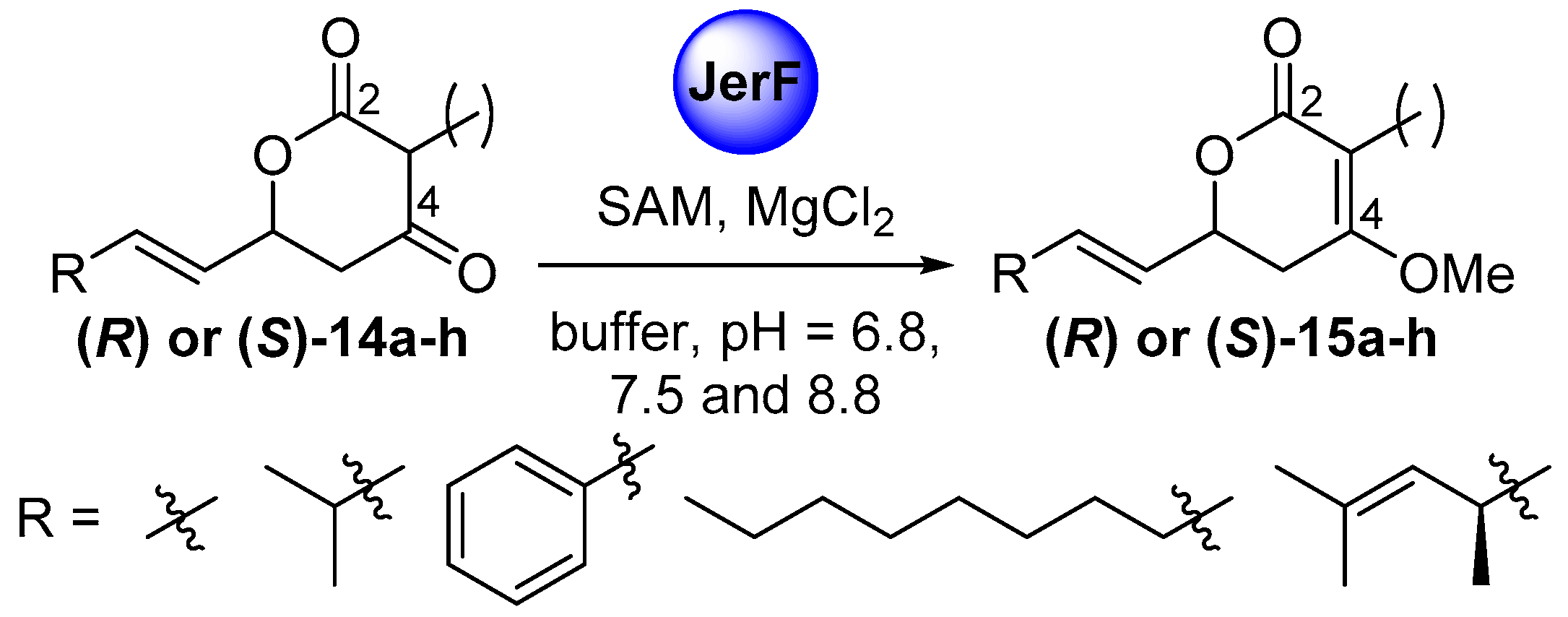
2.2. Cloning, Expression and Establishment of Assay Conditions
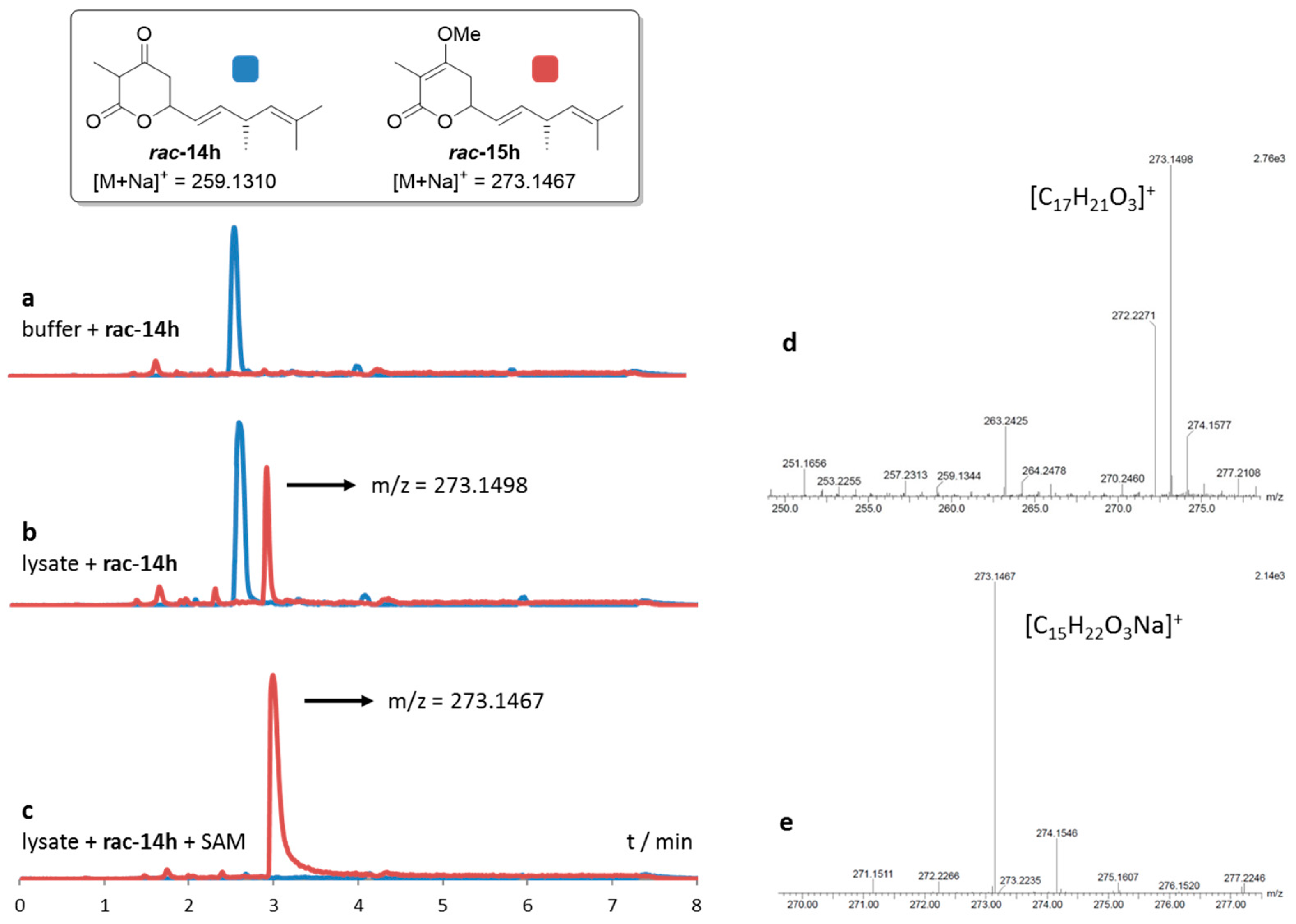
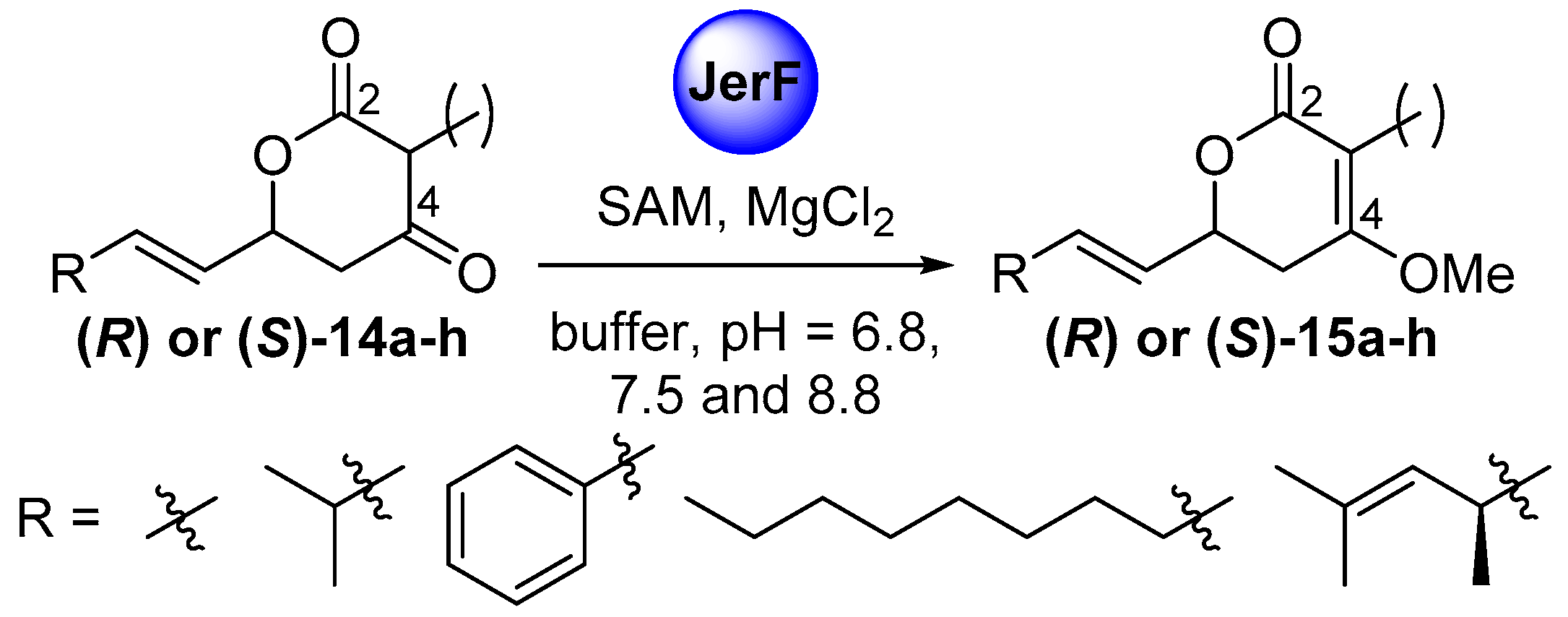
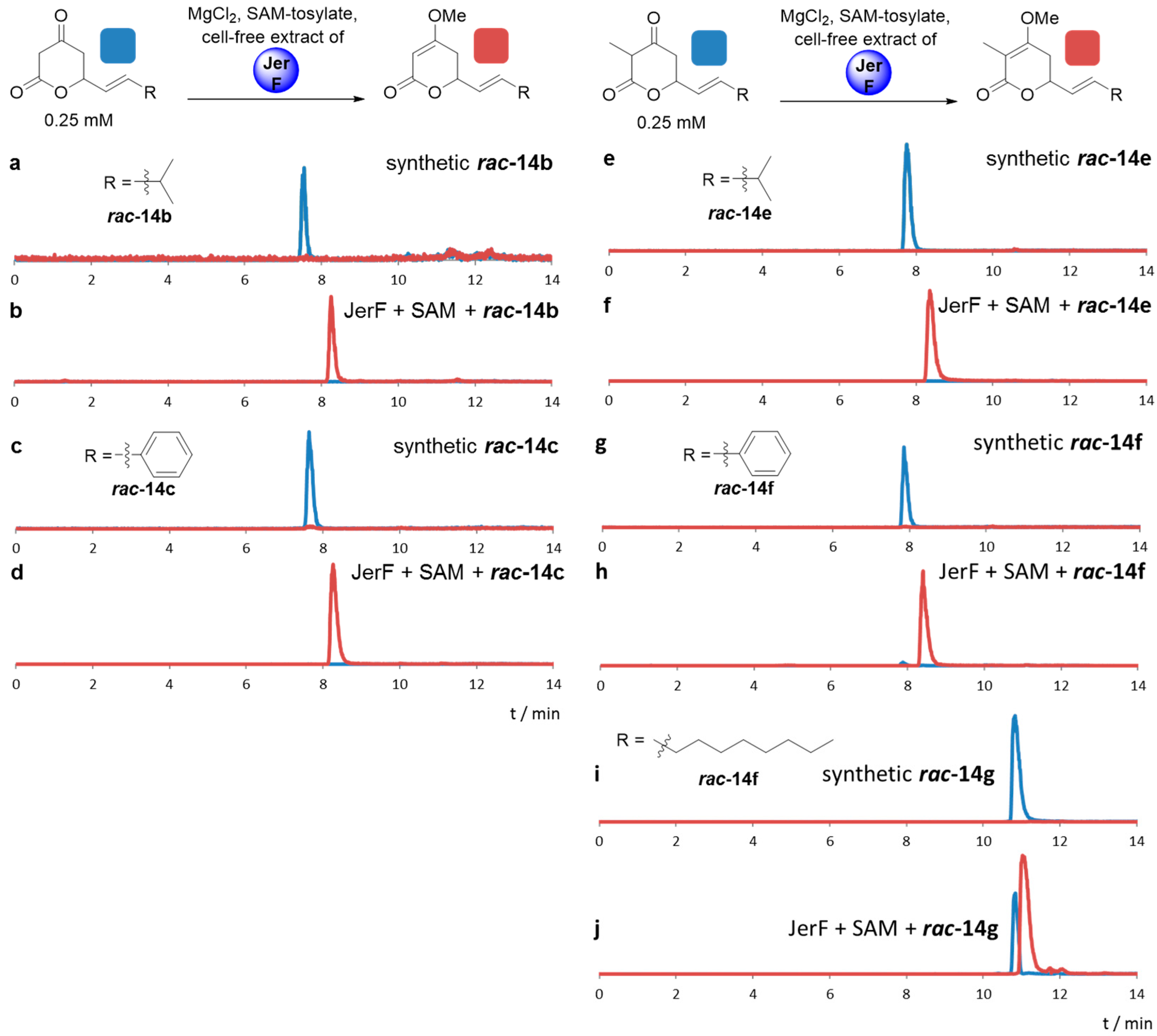
2.3. Comparative Assaying of Synthetic Substrates and Assay Upscaling
3. Discussion
4. Materials and Methods
4.1. General Information
4.1.1. Chemistry Methods and Materials
4.1.2. Biochemistry Methods and Materials
4.2. Synthesis of Substrate Surrogates
4.2.1. General Procedures
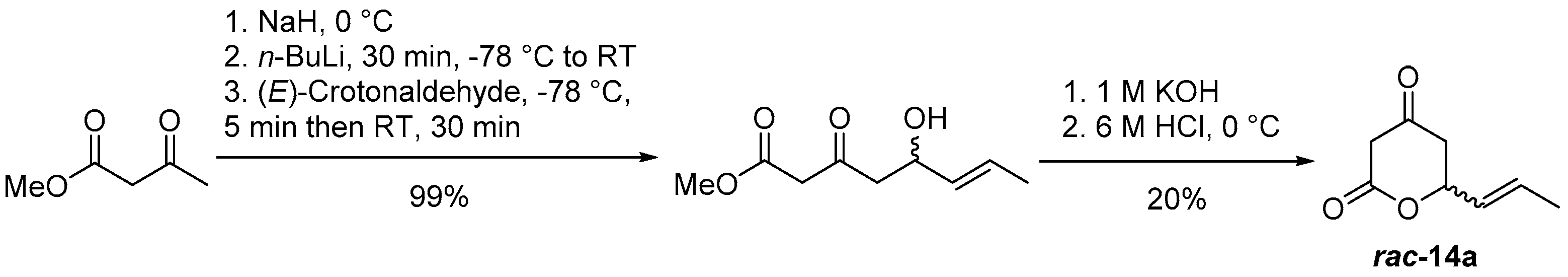
Aldol Reaction
Lactonization
O-Methylation of the Dihydropyran-2,4-diones
O-Methylation of the 3-Methyldihydropyran-2,4-diones
4.2.2. Substrate Synthesis
(E)-6-(Prop-1-en-1-yl)dihydro-2H-pyran-2,4(3H)-dione (rac-14a)
Methyl-(E)-5-hydroxy-2-methyl-3-oxooct-6-enoate (rac-13d)
(E)-3-Methyl-6-(prop-1-en-1-yl)dihydro-2H-pyran-2,4(3H)-dione (rac-14d)
(E)-4-Methoxy-3-methyl-6-(prop-1-en-1-yl)-5,6-dihydro-2H-pyran-2-one (rac-15d)
(E)-6-(3-Methylbut-1-en-1-yl)dihydro-2H-pyran-2,4(3H)-dione (rac-14b)
(E)-4-Methoxy-6-(3-methylbut-1-en-1-yl)-5,6-dihydro-2H-pyran-2-one (rac-15b)
(E)-3-Methyl-6-(3-methylbut-1-en-1-yl)dihydro-2H-pyran-2,4(3H)-dione (rac-14e)
(E)-4-Methoxy-3-methyl-6-(3-methylbut-1-en-1-yl)-5,6-dihydro-2H-pyran- 2-one (rac-15e)
Methyl-(E)-5-hydroxy-3-oxo-7-phenylhept-6-enoate (rac-13c)
(E)-6-Styryldihydro-2H-pyran-2,4(3H)-dione (rac-14c)
(E)-4-Methoxy-6-styryl-5,6-dihydro-2H-pyran-2-one (rac-15c)
(E)-3-Methyl-6-styryldihydro-2H-pyran-2,4-(3H)-dione (rac-14f)
(E)-4-Methoxy-3-methyl-6-styryl-5,6-dihydro-2H-pyran-2-one (rac-15f)
(E)-6-(Dec-1-en-1-yl)-3-methyldihydro-2H-pyran-2,4(3H)-dione (rac-14g)
(E)-6-(Dec-1-en-1-yl)-4-methoxy-3-methyl-5,6-dihydro-2H-pyran-2-on (rac-15g)
Methyl-(R)-3-((tert-Butyldimethylsilyl)oxy)-2-methylpropanoate (I)
(R)-3-((tert-butyldimethylsilyl)oxy)-N-methoxy-N,2-dimethylpropanamide (II)
(R)-3-((tert-Butyldimethylsilyl)oxy)-2-methylpropanal (III)
(S)-tert-Butyl-((2,4-dimethylpent-3-en-1-yl)oxy)dimethylsilane (IV)
(S)-2,4-Dimethylpent-3-en-1-ol (V)
(R,E)-Ethyl-4,6-dimethylhepta-2,5-dienoate (VII)
(R,E)-4,6-Dimethylhepta-2,5-dien-1-ol (VIII)
(R,E)-4,6-Dimethylhepta-2,5-dienal (IX)
((R,E)-3,5-Dimethylhexa-1,4-dien-1-yl)-3-methyldihydro-2H-pyran-2,4(3H)-dione (rac-14h)
4.2.3. Analytical Data
























4.3. Cloning and Enzyme Expression
4.3.1. Molecular Cloning
4.3.2. Protein Sequence
>JerF
4.3.3. Enzyme Expression
4.4. Enzyme Assays
4.4.1. Establishment of Assay Conditions
4.4.2. Comparative Assaying of Synthetic Substrate Surrogates
4.4.3. Semi-preparative Scale Conversions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hoult, J.R.S.; Payá, M. Pharmacological and biochemical actions of simple coumarins: Natural products with therapeutic potential. Gen. Pharmacol. Vasc. Syst. 1996, 27, 713–722. [Google Scholar] [CrossRef]
- Brachmann, A.O.; Brameyer, S.; Kresovic, D.; Hitkova, I.; Kopp, Y.; Manske, C.; Schubert, K.; Bode, H.B.; Heermann, R. Pyrones as bacterial signaling molecules. Nat. Chem. Biol. 2013, 9, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Struck, A.W.; Thompson, M.L.; Wong, L.S.; Micklefield, J. S-Adenosyl-Methionine-Dependent methyl-transferases: Highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. ChemBioChem 2012, 13, 2642–2655. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; He, J.; Hertweck, C. Dissection of the late steps in aureothin biosynthesis. ChemBioChem 2006, 7, 37–39. [Google Scholar] [CrossRef] [PubMed]
- Werneburg, M.; Busch, B.; He, J.; Richter, M.E.A.; Xiang, L.; Moore, B.S.; Roth, M.; Dahse, H.M.; Hertweck, C. Exploiting enzymatic promiscuity to engineer a focused library of highly selective antifungal and antiproliferative aureothin analogues. J. Am. Chem. Soc. 2010, 132, 10407–10413. [Google Scholar] [CrossRef] [PubMed]
- Julien, B.; Tian, Z.-Q.; Reid, R.; Reeves, C.D. Analysis of the ambruticin and jerangolid gene clusters of Sorangium cellulosum reveals unusual mechanisms of polyketide biosynthesis. Chem. Biol. 2006, 13, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Gerth, K.; Washausen, P.; Hofle, G.; Irschik, H.; Reichenbach, H. The jerangolids: A family of new antifungal compounds from Sorangium cellulosum (Myxobacteria). Production, physico-chemical and biological properties of jerangolid A. J. Antibiot. 1996, 49, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Watanabe, M.; Nagai, K.; Suzumura, K.; Suzuki, K.; Tanaka, A. Gamma-pyrone compounds with selective and potent anti-Helicobacter pylori activity. J. Antibiot. 2000, 53, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Miyairi, N.; Sakai, H.; Konomi, T.; Imanaka, H. Enterocin, a new antibiotic taxonomy, isolation and characterisation. J. Antibiot. 1976, 29, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Piel, J.; Hertweck, C.; Shipley, P.R.; Hunt, D.M.; Newman, M.S.; Moore, B.S. Cloning, sequencing and analysis of the enterocin biosynthesis gene cluster from the marine isolate “Streptomyces maritimus”: Evidence for the derailment of an aromatic polyketide synthase. Chem. Biol. 2000, 7, 943–955. [Google Scholar] [CrossRef]
- Cheng, Q.; Xiang, L.; Izumikawa, M.; Meluzzi, D.; Moore, B.S. Enzymatic total synthesis of enterocin polyketides. Nat. Chem. Biol. 2007, 3, 557–558. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Hertweck, C. Iteration as programmed event during polyketide assembly; molecular analysis of the aureothin biosynthetic gene cluster. Chem. Biol. 2003, 10, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, S.; Hahn, F. Opportunities for enzyme catalysis in natural product chemistry. Tetrahedron 2015, 71, 1473–1508. [Google Scholar] [CrossRef]
- Singh, S.; Zhang, J.; Huber, T.D.; Sunkara, M.; Hurley, K.; Goff, R.D.; Wang, G.; Zhang, W.; Liu, C.; Rohr, J.; et al. Facile chemoenzymatic strategies for the synthesis and utilisation of S-Adenosyl-l-Methionine analogues. Angew. Chem. Int. Ed. 2014, 126, 4046–4050. [Google Scholar] [CrossRef]
- Zhang, C.; Weller, R.L.; Thorson, J.S.; Rajski, S.R. Natural product diversification using a non-natural cofactor analogue of S-adenosyl-l-methionine. J. Am. Chem. Soc. 2006, 128, 2760–2761. [Google Scholar] [CrossRef] [PubMed]
- Stecher, H.; Tengg, M.; Ueberbacher, B.J.; RemLer, P.; Schwab, H.; Griengl, H.; Gruber-Khadjawi, M. Biocatalytic Friedel–Crafts alkylation using non-natural cofactors. Angew. Chem. Int. Ed. 2009, 121, 9710–9712. [Google Scholar] [CrossRef]
- Winter, J.M.; Sato, M.; Sugimoto, S.; Chiou, G.; Garg, N.K.; Tang, Y.; Watanabe, K. Identification and characterisation of the chaetoviridin and chaetomugilin gene cluster in Chaetomium globosum reveal dual functions of an iterative highly-reducing polyketide synthase. J. Am. Chem. Soc. 2012, 134, 17900–17903. [Google Scholar] [CrossRef] [PubMed]
- Peters, W.; Willnow, S.; Duisken, M.; Kleine, H.; Macherey, T.; Duncan, K.E.; Litchfield, D.W.; Lüscher, B.; Weinhold, E. Enzymatic site-specific functionalization of protein methyltransferase substrates with alkynes for click labeling. Angew. Chem. Int. Ed. 2010, 122, 5296–5299. [Google Scholar] [CrossRef]
- Schulz, D.; Holstein, J.M.; Rentmeister, A. Ein chemo-enzymatischer Ansatz zur regiospezifischen Modifizierung der RNA-Kappe. Angew. Chem. Int. Ed. 2013, 125, 8028–8032. [Google Scholar] [CrossRef]
- Bothwell, I.R.; Islam, K.; Chen, Y.; Zheng, W.; Blum, G.; Deng, H.; Luo, M. Se-adenosyl-l-selenomethionine cofactor analogue as a reporter of protein methylation. J. Am. Chem. Soc. 2012, 134, 14905–14912. [Google Scholar] [CrossRef] [PubMed]
- Islam, K.; Bothwell, I.; Chen, Y.; Sengelaub, C.; Wang, R.; Deng, H.; Luo, M. Bioorthogonal profiling of protein methylation using azido derivative of S-adenosyl-l-methionine. J. Am. Chem. Soc. 2012, 134, 5909–5915. [Google Scholar] [CrossRef] [PubMed]
- Huckin, S.N.; Weiler, L. Aldol reactions of the dianion of β-keto esters. Tetrahedron Lett. 1971, 12, 4835–4838. [Google Scholar] [CrossRef]
- Andersh, B.; Nguyen, E.T.; van Hoveln, R.J.; Kemmerer, D.K.; Baudo, D.A.; Graves, J.A.; Roark, M.E.; Bosma, W.B. Investigation of the mechanism for the preparation of 6-phenyl-2,4-dioxotetrahydropyrans by the potassium carbonate promoted condensation between acetoacetate esters and benzaldehyde. J. Org. Chem. 2013, 78, 4563–4567. [Google Scholar] [CrossRef] [PubMed]
- Prantz, K.; Mulzer, J. Decarboxylative Grob-Type fragmentations in the synthesis of trisubstituted Z olefins: Application to Peloruside A, Discodermolide, and Epothilone D. Angew. Chem. Int. Ed. 2009, 48, 5030–5033. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Sakaki, J.; Sugita, Y.; Yasuda, S.; Sakoda, H.; Kaneko, C. Two lactone formation reactions from 1,3-dioxin-4-ones having hydroxyalkyl group at the 6-position: Difference in ring opening and closure. Tetrahedron 1991, 47, 5689–5708. [Google Scholar] [CrossRef]
- Singh, S.; Nandurkar, N.S.; Thorson, J.S. Characterisation of the Calicheamicin Orsellinate C2-O-Methyltransferase CalO6. ChemBioChem 2014, 15, 1418–1421. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Anzai, Y.; Kinoshita, K.; Kato, F.; Sherman, D.H. Functional analysis of MycE and MycF, two O-Methyltransferases involved in the biosynthesis of mycinamicin macrolide antibiotics. ChemBioChem 2009, 10, 1297–1301. [Google Scholar] [CrossRef] [PubMed]
- Prantz, K.; Mulzer, J. Synthesis of (Z)-Trisubstituted olefins by decarboxylative Grob-type fragmentations: Epothilone D, Discodermolide, and Peloruside A. Chem. Eur. J. 2010, 16, 485–506. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.A.; Martin, W.H.C.; Hargreaves, J.M.; Wilson, C.; Blake, A.J. The one-pot, multi-component construction of highly substituted tetrahydropyran-4-ones using the Maitland–Japp reaction. Org. Biomol. Chem. 2005, 3, 3551–3563. [Google Scholar] [CrossRef] [PubMed]
- Henkel, B.; Kunath, A.; Schick, H. Enzymes in Organic Synthesis, 11. Enantioselective Lactonization of Methyl 3,5-Dihydroxyalkanoates—An Access to (3R,5S,6E)-3-Hydroxy-7-phenyl-6-hepten-5-olide by Enzyme-Catalyzed Kinetic Resolution in Organic Solvents. Liebigs Ann. Chem. 1992, 1992, 809–811. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds rac-14b–f as well as rac-15b–f are available from the authors.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Conversion a | Crude yield | e.e. b | Scale |
|---|---|---|---|---|
| rac-14c | 42% | n.d. | 84% | 4.6 mg |
| rac-14e | 27% | 38%c | 42% | 4.6 mg |
| rac-14f | 35% | 13%c | <10% | 4.3 mg |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friedrich, S.; Hemmerling, F.; Lindner, F.; Warnke, A.; Wunderlich, J.; Berkhan, G.; Hahn, F. Characterisation of the Broadly-Specific O-Methyl-transferase JerF from the Late Stages of Jerangolid Biosynthesis. Molecules 2016, 21, 1443. https://doi.org/10.3390/molecules21111443
Friedrich S, Hemmerling F, Lindner F, Warnke A, Wunderlich J, Berkhan G, Hahn F. Characterisation of the Broadly-Specific O-Methyl-transferase JerF from the Late Stages of Jerangolid Biosynthesis. Molecules. 2016; 21(11):1443. https://doi.org/10.3390/molecules21111443
Chicago/Turabian StyleFriedrich, Steffen, Franziska Hemmerling, Frederick Lindner, Anna Warnke, Johannes Wunderlich, Gesche Berkhan, and Frank Hahn. 2016. "Characterisation of the Broadly-Specific O-Methyl-transferase JerF from the Late Stages of Jerangolid Biosynthesis" Molecules 21, no. 11: 1443. https://doi.org/10.3390/molecules21111443