
Halofuginone — The Multifaceted Molecule

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
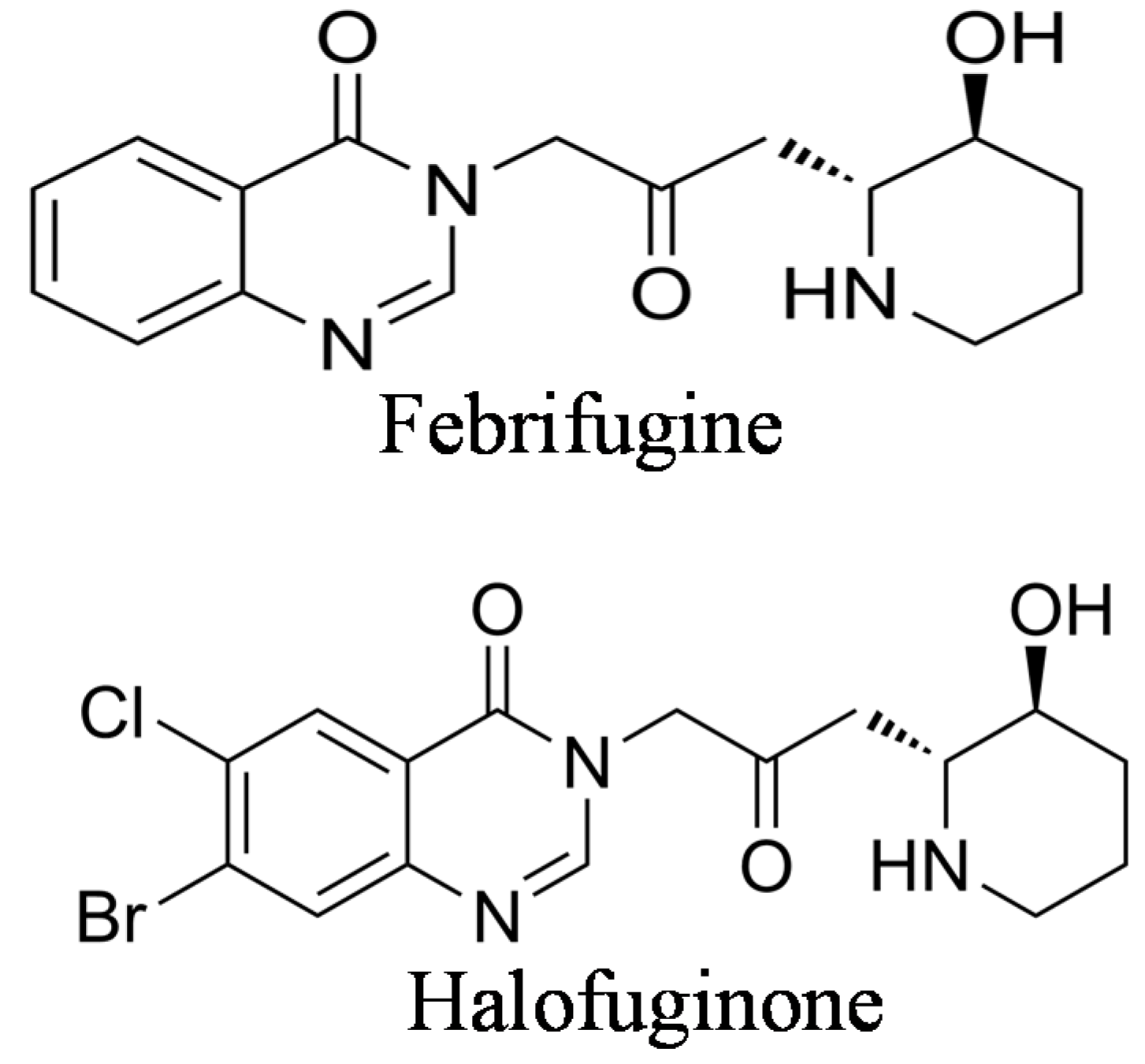
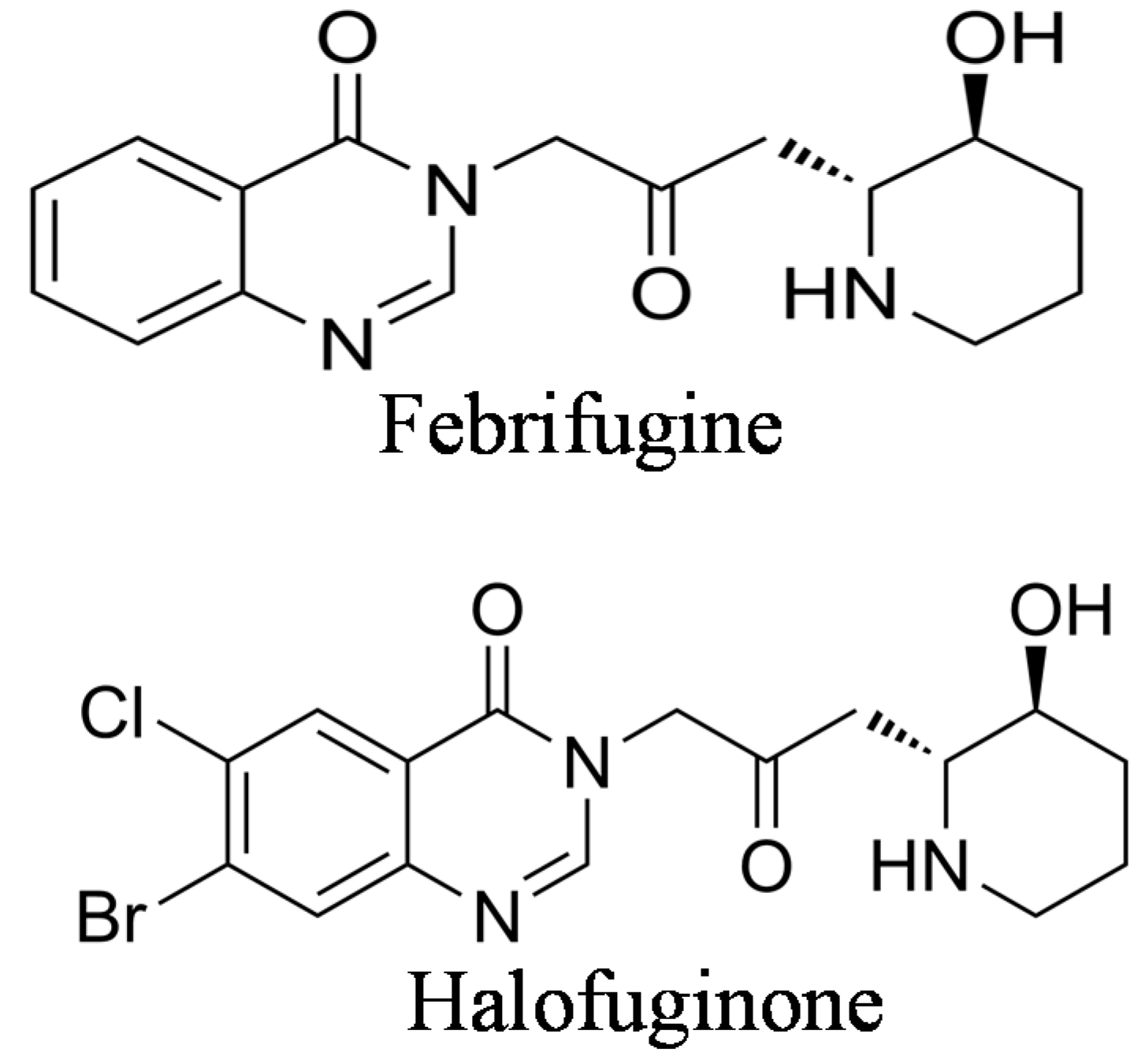
2. The Origin of Halofuginone and Its Synthesis
3. Halofuginone as an Antimalaria Therapy
4. Halofuginone as Antiprotozoan in Poultry and in Ruminants
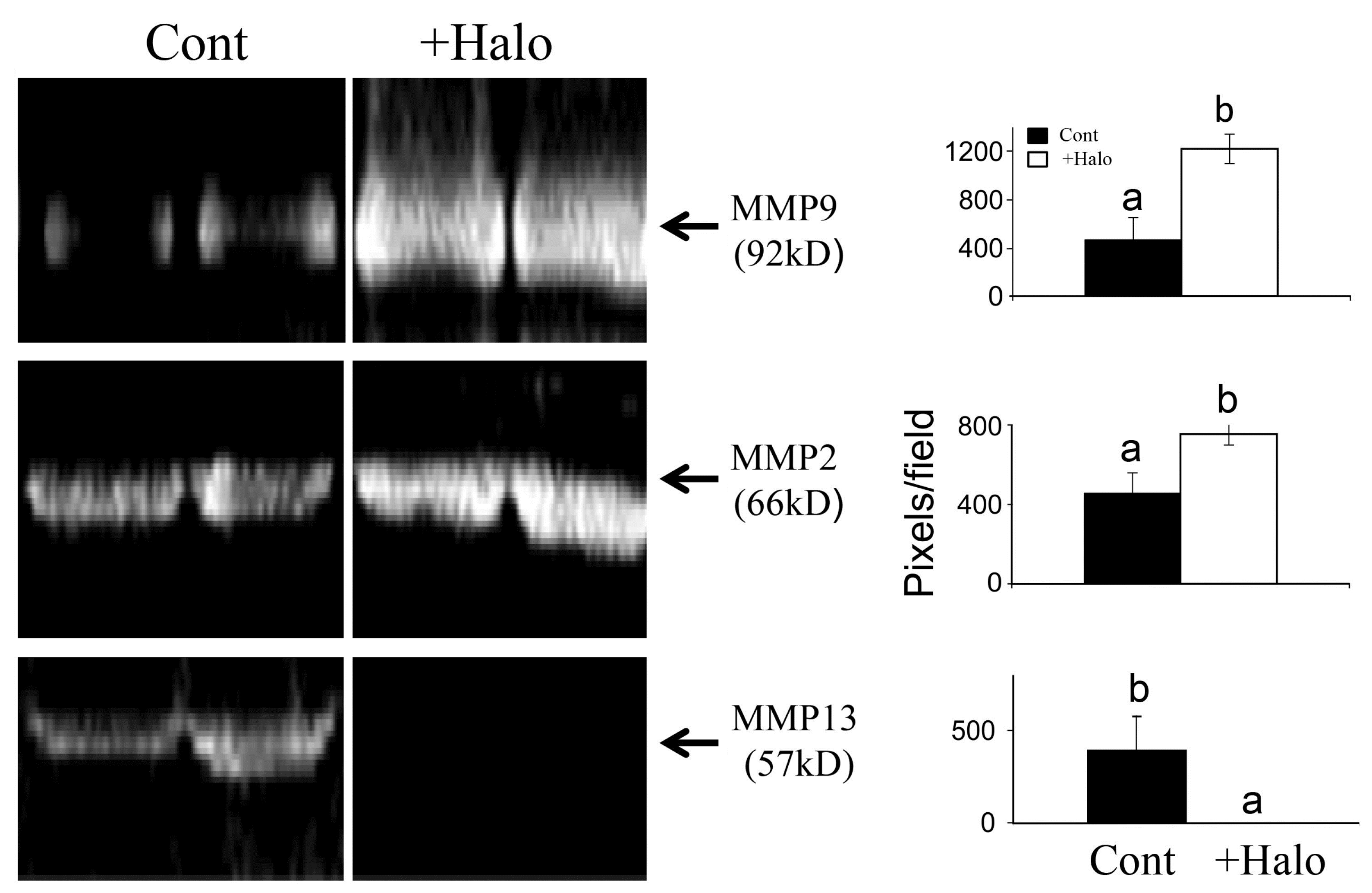
5. Halofuginone as an Antifibrotic Agent

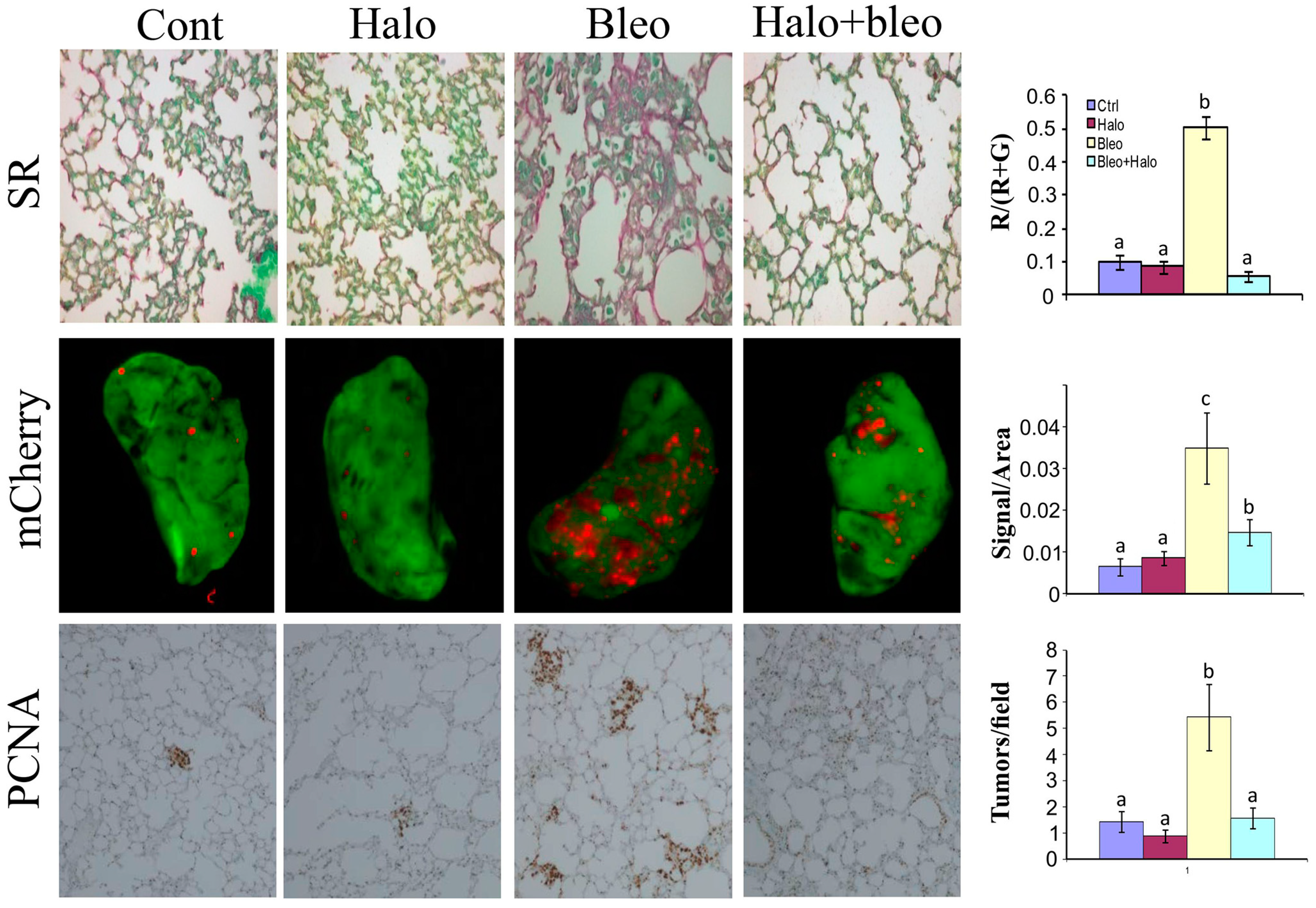
6. Halofuginone in Cancer, Angiogenesis, and Metastasis
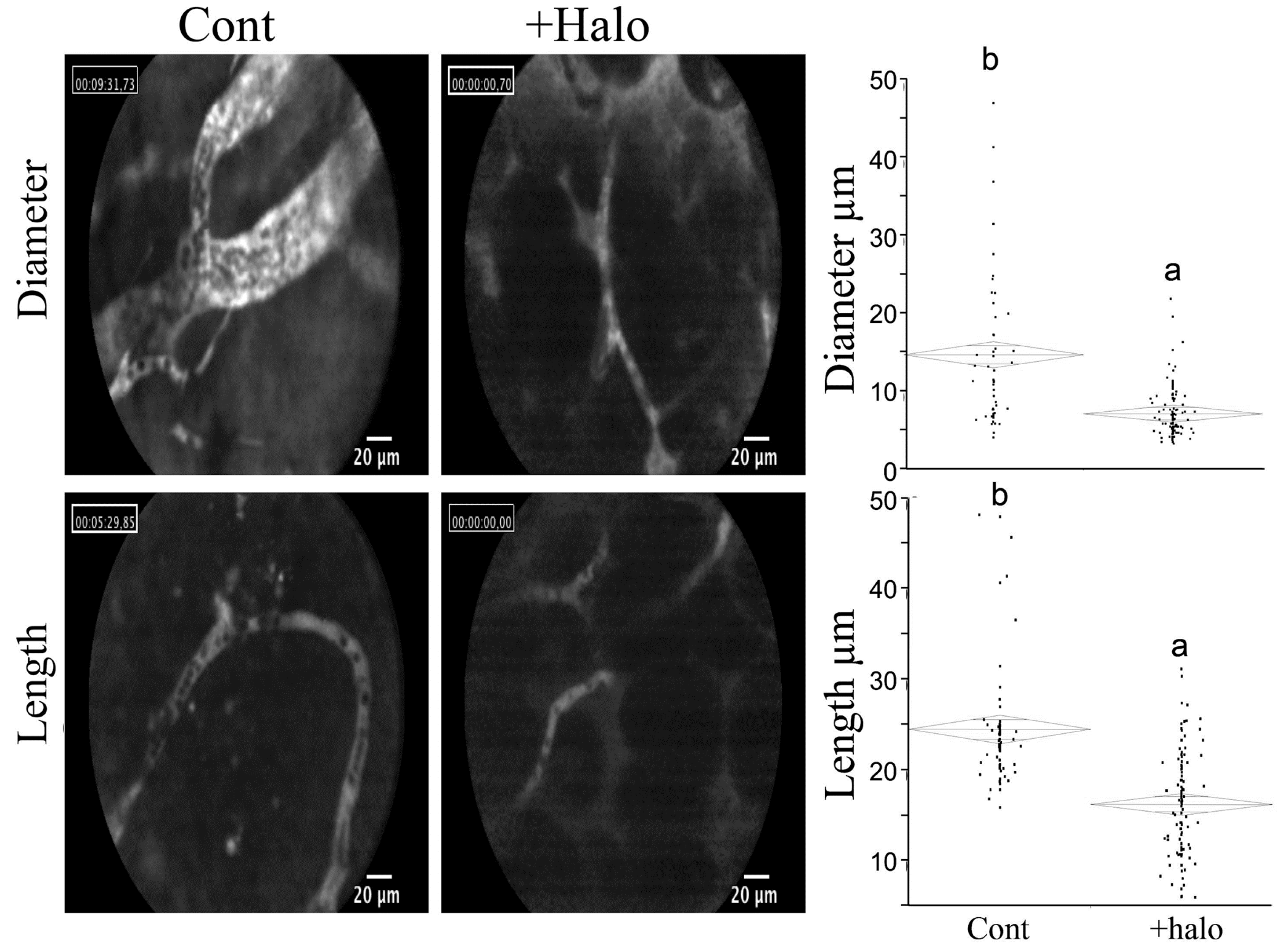
7. Halofuginone and Apoptosis
8. Halofuginone Inflammation and Autoimmunity
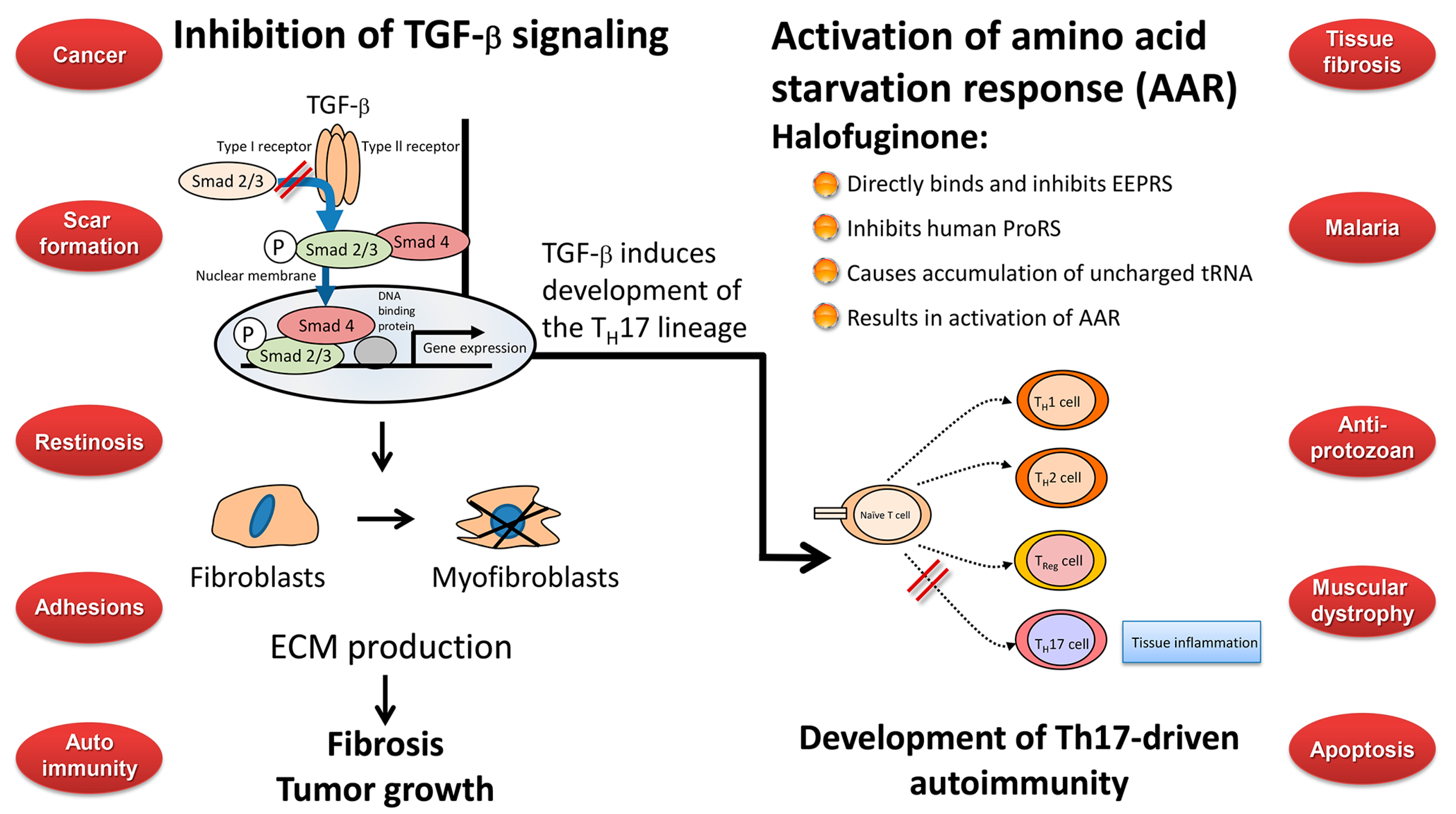
9. Any Links between the Biological Activities of Halofuginone?
10. Human Clinical Efficacy of Halofuginone

11. Concluding Remarks

Acknowledgments
Conflicts of Interest
References
- Ziegler, S.; Pries, V.; Hedberg, C.; Waldmann, H. Target identification for small bioactive molecules: Finding the needle in the haystack. Angew. Chem. Int. Ed. Engl. 2013, 52, 2744–2792. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.B.; Mallikarjuna, P.R.; Fabian, D.A.; Gorajana, A.; Lim, C.L.; Tan, E.L. Bioactive molecules: Current trends in discovery, synthesis, delivery and testing. IeJSME 2013, 7 (Suppl. 1), S32–S46. [Google Scholar]
- Jang, C.S.; Fu, F.Y.; Huang, K.C.; Wang, C.Y. Pharmacology of Ch’ang Shan (Dichroa febrifuga), a Chinese antimalarial herb. Nature 1948, 161, 400–401. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, F.A.; Spencer, C.F.; Folkers, J.A. Alkaloids of Dichroa febrifuga, Lour. J. Am. Chem. Soc. 1947, 70, 2091–2093. [Google Scholar] [CrossRef]
- Jang, C.S.; Fu, F.Y.; Wang, C.Y.; Huang, K.C.; Lu, G.; Chou, T.C. Ch’ang Shan, a Chinese antimalarial herb. Science 1946, 103, 59. [Google Scholar] [CrossRef] [PubMed]
- Koepfli, J.B.; Mead, J.F.; Brockman, J.A., Jr. An alkaloid with high antimalarial activity from Dichroa febrifuga. J. Am. Chem. Soc. 1947, 69. [Google Scholar] [CrossRef]
- Koepfli, J.B.; Mead, J.F.; Brockman, J.A., Jr. Alkaloids of Dichroa febrifuga. I. Isolation and degradative studies. J. Am. Chem. Soc. 1949, 71, 1048–1054. [Google Scholar]
- Coatney, G.R.; Cooper, W.C.; Culwell, W.B.; White, W.C.; Imboden, C.A., Jr. Studies in human malaria. XXV. Trial of febrifugine, an alkaloid obtained from Dichroa febrifuga Lour. against the Chesson strain of Plasmodium vivax. J. Natl. Malar. Soc. 1950, 9, 183–186. [Google Scholar] [PubMed]
- Tang, W.; Eisenbrand, G. Chinese Drugs of Plant Origin; Springer-Verlag: Berlin, Germany, 1992; pp. 455–457. [Google Scholar]
- Fishman, M.; Cruickshank, P.A. Febrifugine antimalarial agents. I. Pyridine analogs of febrifugine. J. Med. Chem. 1970, 13, 155–156. [Google Scholar]
- Chien, P.L.; Cheng, C.C. Structural modification of febrifugine. Some methylenedioxy analogs. J. Med. Chem. 1970, 13, 867–870. [Google Scholar] [CrossRef] [PubMed]
- De Smet, P.A.G.M. The role of plant-derived drugs and herbal medicines. Drugs 1997, 54, 801–840. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.R.; Schaub, R.E.; McEvoy, F.J.; Williams, J.H. An antimalarial alkaloid from Hydrangea. J. Org. Chem. 1952, 17, 133–137. [Google Scholar]
- Jiang, S.; Zeng, Q.; Gettayacamin, M.; Tungtaeng, A.; Wannaying, S.; Lim, A.; Hansukjariya, P.; Okunji, C.O.; Zhu, S.; Fang, D. Antimalarial activities and therapeutic properties of febrifugine analogs. Antimicrob. Agents Chemother. 2005, 49, 1169–7116. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Banerjee, A.; Ghosh, A.K.; Chatterjee, T.K. Synthesis and antimalarial evaluation of some 4-quinazolinone derivatives based on febrifugine. J. Adv. Pharm. Technol. Res. 2010, 1, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, H.; Yamamoto, K.; Horoiwa, S.; Hirai, S.; Kasahara, R.; Hariguchi, N.; Matsumoto, M.; Oshima, Y. Exploration of a new type of antimalarial compounds based on febrifugine. J. Med. Chem. 2006, 49, 4698–4706. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, H.; Horoiwa, S.; Kasahara, R.; Hariguchi, N.; Matsumoto, M.; Oshima, Y. Synthesis of febrifugine derivatives and development of an effective and safe tetrahydroquinazoline-type antimalarial. Eur. J. Med. Chem. 2014, 76, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Chatterjee, T.K. Pharmacophore modeling and 3D quantitative structure-activity relationship analysis of febrifugine analogues as potent antimalarial agent. J. Adv. Pharm. Technol. Res. 2013, 4, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Takaya, Y.; Tasaka, H.; Chiba, T.; Uwai, K.; Tanitsu, M.; Kim, H.S. New type of febrifugine analogues, bearing a quinolizidine moiety, show potent antimalarial activity against plasmodium malaria parasite. J. Med. Chem. 1999, 42, 3163–3166. [Google Scholar] [CrossRef] [PubMed]
- Pinion, J.L.; Bilgili, S.F.; Eckman, M.K.; Hess, J.B. The effects of halofuginone and salinomycin, alone and in combination, on live performance and skin characteristics of broilers. Poult. Sci. 1995, 74, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.F.; Sun, B.B.; Yue, Y.Y.; Yu, H.J.; Zhang, H.L.; Zhou, Q.J.; Du, A.F. Anticoccidial effect of halofuginone hydrobromide against Eimeria tenella with associated histology. Parasitol. Res. 2012, 111, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Daugschies, A.; Gässlein, U.; Rommel, M. Comparative efficacy of anticoccidials under the conditions of commercial broiler production and in battery trials. Vet. Parasitol. 1998, 76, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Peeters, J.E.; Villacorta, I.; Naciri, M.; Vanopdenbosch, E. Specific serum and local antibody responses against Cryptosporidium parvum during medication of calves with halofuginone lactate. Infect. Immun. 1993, 61, 4440–4445. [Google Scholar] [PubMed]
- Kamberov, Y.G.; Kim, J.; Mazitschek, R.; Kuo, W.P.; Whitman, M. Microarray profiling reveals the integrated stress response is activated by halofuginone in mammary epithelial cells. BMC Res. Notes 2011, 4, 381. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Ueno, M.; Suzuki, R.; Ishitani, H.; Kim, H.S.; Wataya, Y. Catalytic asymmetric synthesis of antimalarial alkaloids febrifugine and isofibrifugine and their biological activity. J. Org. Chem. 1999, 64, 6833–6841. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Ogasawara, K. A diastereocontrolled synthesis of (+)-febrifugine: A potent antimalarial piperidine alkaloid. Org. Lett. 2000, 2, 3193–3195. [Google Scholar] [CrossRef] [PubMed]
- Samant, B.S.; Sukhthankar, M.G. Synthesis and comparison of antimalarial activity of febrifugine derivatives including halofuginone. Med. Chem. 2009, 5, 293–300. [Google Scholar] [CrossRef]
- McLaughlin, N.P.; Evans, P. Dihydroxylation of vinyl sulfones: Stereoselective synthesis of (+)- and (−)-febrifugine and halofuginone. J. Org. Chem. 2010, 75, 518–521. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Azuma, K.; Abe, H.; Sasaki, K.; Harayama, T. Re-revision of the stereo structure of piperidine lactone, an intermediate in the synthesis of febrifugine. Chem. Pharm. Bull. (Tokyo) 2002, 50, 1011–1012. [Google Scholar] [CrossRef]
- McLaughlin, N.P.; Evans, P.; Pines, M. The chemistry and biology of febrifugine and halofuginone. Bioorg. Med. Chem. 2014, 22, 1993–2004. [Google Scholar] [CrossRef] [PubMed]
- Geary, T.G.; Divo, A.A.; Jensen, J.B. Stage specific actions of antimalarial drugs on Plasmodium falciparum in culture. Am. J. Trop. Med. Hyg. 1989, 40, 240–244. [Google Scholar] [PubMed]
- Derbyshire, E.R.; Mazitschek, R.; Clardy, J. Characterization of Plasmodium liver stage inhibition by halofuginone. ChemMedChem 2012, 7, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Jain, V.; Kikuchi, H.; Oshima, Y.; Sharma, A.; Yogavel, M. Structural and functional analysis of the antimalarial drug target prolyl-tRNA synthetase. J. Struct. Funct. Genomics 2014, 15, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Sun, L.; Yang, X.L.; Schimmel, P. ATP-directed capture of bioactive herbal-based medicine on human tRNA synthetase. Nature 2013, 494, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Chapman, H.D. Milestones in avian coccidiosis research: A review. Poult. Sci. 2014, 93, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Daugschies, A.; Najdrowski, M. Eimeriosis in cattle: Current understanding. J. Vet. Med. B Infect. Dis. Vet. Public Health 2005, 52, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Silverlås, C.; Björkman, C.; Egenvall, A. Systematic review and meta-analyses of the effects of halofuginone against calf cryptosporidiosis. Prev. Vet. Med. 2009, 91, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Folz, S.D.; Lee, B.L.; Nowakowski, L.H.; Conder, G.A. Anticoccidial evaluation of halofuginone, lasalocid, maduramicin, monensin and salinomycin. Vet. Parasitol. 1988, 28, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.Q.; Fukata, T.; Gilbert, J.M.; McDougald, L.R. Evaluation of anticoccidial drugs in chicken embryos. Parasitol. Res. 1991, 77, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Fitz-Coy, S.H.; Edgar, S.A. Pathogenicity and control of Eimeria mitis infections in broiler chickens. Avian Dis. 1992, 36, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Constable, P.D. Treatment of calf diarrhea: Antimicrobial and ancillary treatments. Vet. Clin. North Am. Food Anim. Pract. 2009, 25, 101–120. [Google Scholar] [CrossRef] [PubMed]
- De Waele, V.; Speybroeck, N.; Berkvens, D.; Mulcahy, G.; Murphy, T.M. Control of cryptosporidiosis in neonatal calves: Use of halofuginone lactate in two different calf rearing systems. Prev. Vet. Med. 2010, 96, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Klein, P. Preventive and therapeutic efficacy of halofuginone-lactate against Cryptosporidium parvum in spontaneously infected calves: A centralised, randomised, double-blind, placebo-controlled study. Vet. J. 2008, 177, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Giadinis, N.D.; Papadopoulos, E.; Panousis, N.; Papazahariadou, M.; Lafi, S.Q.; Karatzias, H. Effect of halofuginone lactate on treatment and prevention of lamb cryptosporidiosis: An extensive field trial. J. Vet. Pharmacol. Ther. 2007, 30, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Shahiduzzaman, M.; Dyachenko, V.; Obwaller, A.; Unglaube, S.; Daugschies, A. Combination of cell culture and quantitative PCR for screening of drugs against Cryptosporidium parvum. Vet. Parasitol. 2009, 162, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.R.; Heckeroth, A.R.; Najdrowski, M.; Daugschies, A.; Schollmeyer, D.; Miculka, C. (2R,3S)-(+)- and (2S,3R)-(−)-halofuginone lactate: Synthesis, absolute configuration, and activity against Cryptosporidium parvum. Bioorg. Med. Chem. Lett. 2007, 17, 4140–4143. [Google Scholar] [CrossRef] [PubMed]
- Pines, M.; Nagler, A. Halofuginone—A novel anti-fibrotic therapy. Drug Future 1996, 21, 569–599. [Google Scholar]
- Pines, M.; Vlodavsky, I.; Nagler, A. Halofuginone—A novel anti-fibrotic therapy. Gen. Pharmacol. 1997, 30, 445–450. [Google Scholar] [CrossRef]
- Pines, M.; Vlodavsky, I.; Nagler, A. Halofuginone: From veterinary use to human therapy. Drug Dev. Res. 2000, 50, 371–378. [Google Scholar] [CrossRef]
- Pines, M. Targeting TGFβ signaling to inhibit fibroblasts activation as a therapy for fibrosis and cancer. Expt. Opin. Drug Dis. 2008, 3, 11–20. [Google Scholar] [CrossRef]
- Pines, M. Halofuginone for fibrosis, regeneration and cancer in the gastrointestinal tract. World J. Gastroenterol. 2014, 20, 14778–14786. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.F.; Huang, C.W.; Ewel, J.M.; Chang, A.A.; Yuan, C. Halofuginone down-regulates Smad3 expression and inhibits the TGFβ-induced expression of fibrotic markers in human corneal fibroblasts. Mol. Vis. 2012, 18, 479–487. [Google Scholar] [PubMed]
- Halevy, O.; Nagler, A.; Levi-Schaffer, F.; Genina, O.; Pines, M. Inhibition of collagen type I synthesis by skin fibroblasts of graft versus host disease and scleroderma patients: Effect of halofuginone. Biochem. Pharmacol. 1996, 52, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Levi-Schaffer, F.; Nagler, A.; Slavin, S.; Knopov, V.; Pines, M. Inhibition of collagen synthesis and changes in skin morphology in murine graft versus host disease and tight skin mice: Effect of halofuginone. J. Investig. Dermatol. 1996, 106, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Pines, M.; Domb, A.; Ohana, M.; Inbar, J.; Genina, O.; Alexiev, R.; Nagler, A. Reduction in dermal fibrosis in the tight-skin (Tsk) mouse after local application of halofuginone. Biochem. Pharmacol. 2001, 62, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Nagler, A.; Firman, N.; Feferman, R.; Cotev, S.; Pines, M.; Shoshan, S. Reduction in pulmonary fibrosis in vivo by halofuginone. Am. J. Respir. Crit. Care Med. 1996, 154, 1082–1086. [Google Scholar] [CrossRef] [PubMed]
- Nagler, A.; Rivkind, A.I.; Raphael, J.; Levi-Schaffer, F.; Genina, O.; Lavelin, I.; Pines, M. Halofuginone—An inhibitor of collagen type I synthesis—Prevents postoperation abdominal adhesions formation. Ann. Surg. 1998, 227, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Nagler, A.; Genina, O.; Lavelin, I.; Ohana, M.; Pines, M. Halofuginone, an inhibitor of collagen type I synthesis, prevents formation of postoperative adhesions formation in the rat uterine horn model. Am. J. Obstet. Gynecol. 1999, 180, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Nagler, A.; Gofrit, O.; Ohana, M.; Pode, D.; Genina, O.; Pines, M. The effect of halofuginone, an inhibitor of collagen type I synthesis, on urethral stricture formation: In vivo and in vitro study in a rat model. J. Urol. 2000, 164, 1776–1780. [Google Scholar] [CrossRef] [PubMed]
- Pines, M.; Knopov, V.; Genina, O.; Lavelin, I.; Nagler, A. Halofuginone, a specific inhibitor of collagen type I synthesis, prevents dimethylnitrosamine-induced liver cirrhosis. J. Hepatol. 1997, 26, 391–398. [Google Scholar] [CrossRef]
- Gnainsky, Y.; Spira, G.; Paizi, M.; Bruck, R.; Nagler, A.; Naffer Abu-Amar, S.; Geiger, B.; Genina, O.; Monsonego-Ornan, E.; Pines, M. Halofuginone—An inhibitor of collagen synthesis by rat stellate cells—Stimulates insulin-like growth factor binding protein 1 synthesis by hepatocytes. J. Hepatol. 2004, 40, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Zion, O.; Genin, O.; Kawada, N.; Yoshizato, K.; Roffe, S.; Nagler, A.; Iovanna, J.L.; Halevy, O.; Pines, M. Inhibition of transforming growth factor beta signaling by halofuginone as a modality for pancreas fibrosis prevention. Pancreas 2009, 38, 427–435. [Google Scholar] [CrossRef] [PubMed]
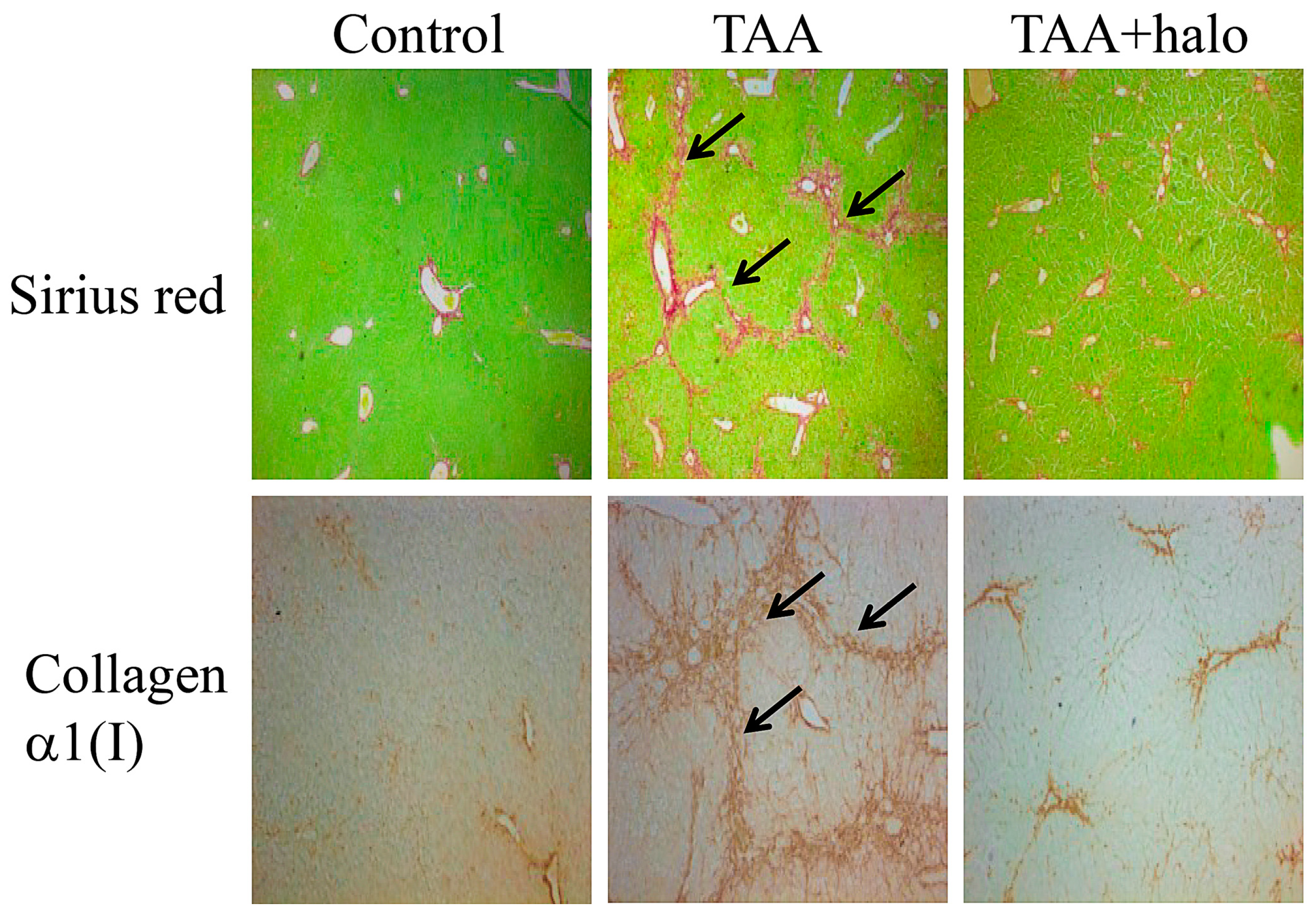
- Bruck, R.; Genina, O.; Aeed, H.; Alexiev, R.; Nagler, A.; Pines, M. Halofuginone to prevent and treat thioacetamide-induced liver fibrosis in rats. Hepatology 2001, 33, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Huebner, K.D.; Jassal, D.S.; Halevy, O.; Pines, M.; Anderson, J.E. Functional resolution of fibrosis in mdx mouse dystrophic heart and skeletal muscle by halofuginone. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1550–H1561. [Google Scholar] [CrossRef] [PubMed]
- Spira, G.; Mawasi, N.; Paizi, M.; Anbinder, N.; Genina, O.; Alexiev, R.; Pines, M. Halofuginone, a collagen type I inhibitor improves liver regeneration in cirrhotic rats. J. Hepatol. 2002, 37, 331–339. [Google Scholar] [CrossRef] [PubMed]
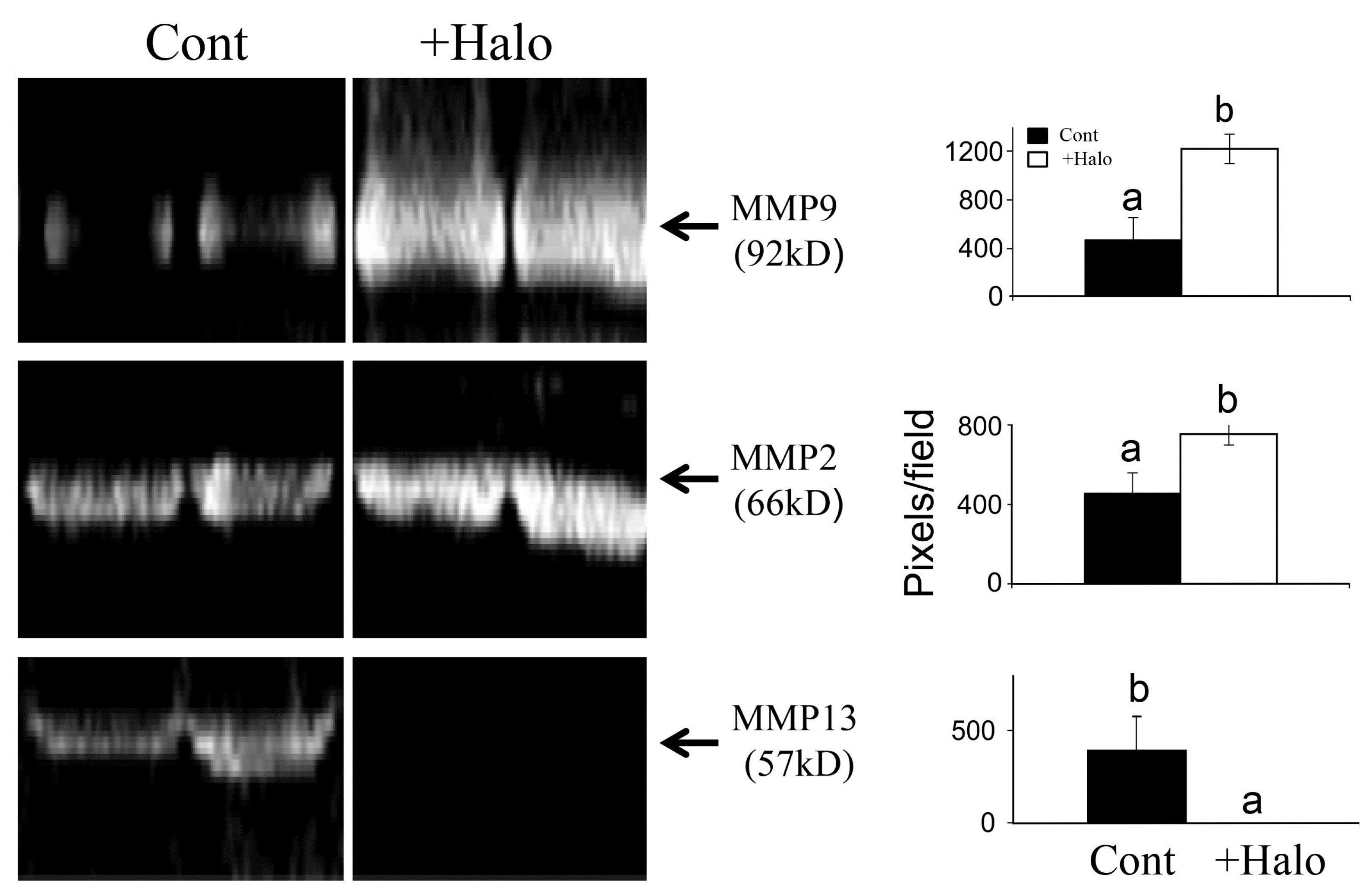
- Popov, Y.; Patsenker, E.; Bauer, M.; Niedobitek, E.; Schulze-Krebs, A.; Schuppan, D. Halofuginone induces matrix metalloproteinases in rat hepatic stellate cells via activation of p38 and NFkappaB. J. Biol. Chem. 2006, 281, 15090–15098. [Google Scholar] [CrossRef] [PubMed]
- Zacharia, E.; Atzmon, R.; Nagler, A.; Shimoni, A.; Peretz, T.; Vlodavsky, I.; Nagler, A. Inhibition of matrix metalloproteinase-2 by halofuginone is mediated by the Egr1 transcription factor. Anticancer Drugs 2012, 23, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Verrecchia, F.; Mauviel, A. Transforming growth factor-beta and fibrosis. World J. Gastroenterol. 2007, 13, 3056–3062. [Google Scholar] [PubMed]
- Kamato, D.; Burch, M.L.; Piva, T.J.; Rezaei, H.B.; Rostam, M.A.; Xu, S.; Zheng, W.; Little, P.J.; Osman, N. Transforming growth factor-β signalling: Role and consequences of Smad linker region phosphorylation. Cell. Signal. 2013, 25, 2017–2024. [Google Scholar] [CrossRef] [PubMed]
- McGaha, T.L.; Phelps, R.G.; Spiera, H.; Bona, C. Halofuginone, an inhibitor of type-I collagen synthesis and skin sclerosis, blocks transforming-growth-factor-beta-mediated Smad3 activation in fibroblasts. J. Investig. Dermatol. 2002, 118, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Turgeman, T.; Hagai, Y.; Huebner, K.; Jassal, D.S.; Anderson, J.E.; Genin, O.; Nagler, A.; Halevy, O.; Pines, M. Prevention of muscle fibrosis and improvement in muscle performance in the mdx mouse by halofuginone. Neuromuscul. Disord. 2008, 18, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Gnainsky, Y.; Kushnirsky, Z.; Bilu, G.; Hagai, Y.; Genina, O.; Volpin, H.; Bruck, R.; Spira, G.; Nagler, A.; Kawada, N.; et al. Gene expression during chemically induced liver fibrosis: Effect of halofuginone on TGF-β signaling. Cell Tissue Res. 2007, 328, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Roffe, S.; Hagai, Y.; Pines, M.; Halevy, O. Halofuginone inhibits Smad3 phosphorylation via the PI3K/Akt and MAPK/ERK pathways in muscle cells: Effect on myotube fusion. Exp. Cell Res. 2010, 316, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Spector, I.; Zilberstein, Y.; Lavy, A.; Nagler, A.; Genin, O.; Pines, M. Involvement of host stroma cells and tissue fibrosis in pancreatic tumor development in transgenic mice. PLoS One 2012, 7, e41833. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.W.; Wang, B.; Goel, S.A.; Little, C.; Takayama, T.; Shi, X.D.; Roenneburg, D.; DiRenzo, D.; Kent, K.C. Halofuginone stimulates adaptive remodeling and preserves re-endothelialization in balloon-injured rat carotid arteries. Circ. Cardiovasc. Interv. 2014, 7, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Nevo, Y.; Halevy, O.; Genin, O.; Turgeman, T.; Harel, M.; Biton, E.; Reif, S.; Pines, M. Inhibition of fibrosis in laminin-α2-deficient congenital muscular dystrophy mice: Effect of halofuginone. Muscle Nerve 2010, 4, 218–229. [Google Scholar] [CrossRef]
- Halevy, O.; Genin, O.; Barzilai-Tutsch, H.; Pima, Y.; Levi, O.; Moshe, I.; Pines, M. Inhibition of muscle fibrosis and improvement of muscle histopathology in dysferlin knock-out mice treated with halofuginone. Histol. Histopathol. 2013, 28, 211–226. [Google Scholar] [PubMed]
- Fromes, Y.; Bouyon, S.; Nagi, S.; Roussel, V.; Genin, O.; Levi, O.; Halevy, O.; Pines, M. Inhibition of fibrosis and improving function of the myopathic hamster cardiac muscle by halofuginone. J. Exp. Clin. Cardiol. 2014, 20, 2351–2383. [Google Scholar]
- Genin, O.; Rechavi, G.; Nagler, A.; Ben-Itzhak, O.; Nazemi, K.; Pines, M. Myofibroblasts in pulmonary and brain metastases of alveolar soft-part sarcoma: A novel target for treatment? Neoplasia 2008, 10, 940–948. [Google Scholar] [PubMed]
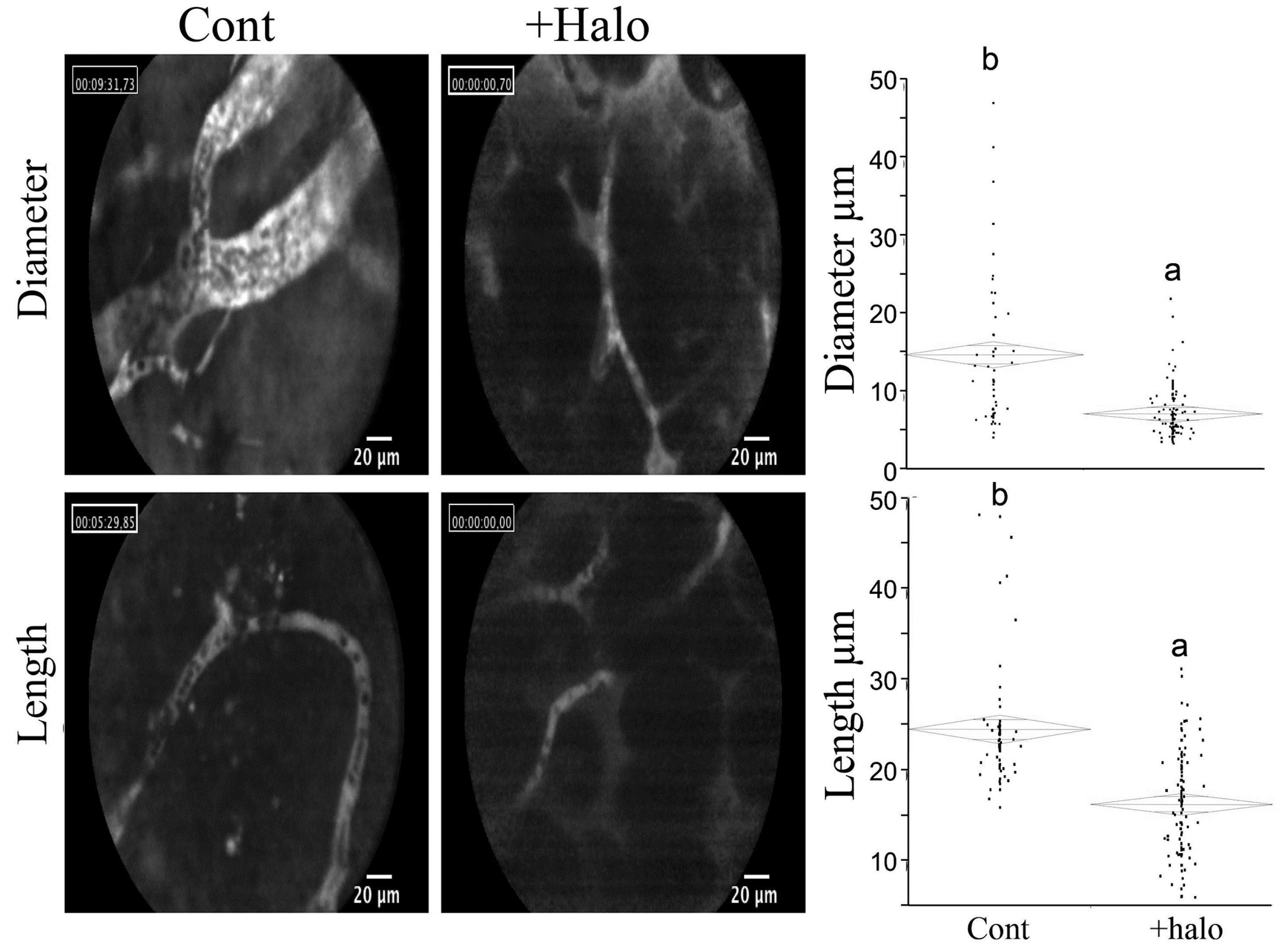
- Elkin, M.; Miao, H.Q.; Nagler, A.; Aingorn, E.; Reich, R.; Hemo, I.; Dou, H.L.; Pines, M.; Vlodavsky, I. Halofuginone: A potent inhibitor of critical steps in angiogenesis progression. FASEB J. 2000, 14, 2477–2485. [Google Scholar] [CrossRef] [PubMed]
- Abramovitch, R.; Dafni, H.; Neeman, M.; Nagler, A.; Pines, M. Inhibition of neovascularization and tumor growth, and facilitation of wound repair, by halofuginone, an inhibitor of collagen type I synthesis. Neoplasia 1999, 1, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Elkin, M.; Ariel, I.; Miao, H.Q.; Nagler, A.; Pines, M.; de-Groot, N.; Hochberg, A.; Vlodavsky, I. Inhibition of bladder carcinoma angiogenesis, stromal support, and tumor growth by halofuginone. Cancer Res. 1999, 59, 4111–4118. [Google Scholar] [PubMed]
- Pinthus, J.H.; Sheffer, Y.; Nagler, A.; Fridman, E.; Mor, Y.; Genina, O.; Pines, M. Inhibition of Wilms tumor xenograft progression by halofuginone is accompanied by activation of WT-1 gene expression. J. Urol. 2005, 174, 1527–1531. [Google Scholar] [CrossRef] [PubMed]
- Van Kempen, L.C.; Rijntjes, J.; Mamor-Cornelissen, I.; Vincent-Naulleau, S.; Gerritsen, M.J.; Ruiter, D.J.; van Dijk, M.C.; Geffrotin, C.; van Muijen, G.N. Type I collagen expression contributes to angiogenesis and the development of deeply invasive cutaneous melanoma. Int. J. Cancer 2008, 122, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Spector, I.; Honig, H.; Kawada, N.; Nagler, A.; Genin, O.; Pines, M. Inhibition of pancreatic stellate cell activation by halofuginone prevents pancreatic xenograft tumor development. Pancreas 2010, 39, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Gavish, Z.; Pinthus, J.H.; Barak, V.; Ramon, J.; Nagler, A.; Eshhar, Z.; Pines, M. Growth inhibition of prostate cancer xenografts by halofuginone. Prostate 2002, 51, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Taras, D.; Blanc, J.F.; Rullier, A.; Dugot-Senant, N.; Laurendeau, I.; Bièche, I.; Pines, M.; Rosenbaum, J. Halofuginone suppresses the lung metastasis of chemically induced hepatocellular carcinoma in rats through MMP inhibition. Neoplasia 2006, 8, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Juárez, P.; Mohammad, K.S.; Yin, J.J.; Fournier, P.G.; McKenna, R.C.; Davis, H.W.; Peng, X.H.; Niewolna, M.; Javelaud, D.; Chirgwin, J.M.; et al. Halofuginone inhibits the establishment and progression of melanoma bone metastases. Cancer Res. 2012, 72, 6247–6256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheffer, Y.; Leon, O.; Pinthus, J.H.; Nagler, A.; Mor, Y.; Genin, O.; Iluz, M.; Kawada, N.; Yoshizato, K.; Pines, M. Inhibition of fibroblast to myofibroblast transition by halofuginone contributes to the chemotherapy-mediated antitumoral effect. Mol. Cancer Ther. 2007, 6, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Salvesen, G. Regulated cell death: Signaling and mechanisms. Ann. Rev. Cell Dev. Biol. 2014, 30, 337–356. [Google Scholar] [CrossRef]
- Grudzien, M.M.; Low, P.S.; Manning, P.C.; Arredondo, M.; Belton, R.J., Jr.; Nowak, R.A. The antifibrotic drug halofuginone inhibits proliferation and collagen production by human leiomyoma and myometrial smooth muscle cells. Fertil. Steril. 2010, 93, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- De Figueiredo-Pontes, L.L.; Assis, P.A.; Santana-Lemos, B.A.; Jácomo, R.H.; Lima, A.S.; Garcia, A.B.; Thomé, C.H.; Araújo, A.G.; Panepucci, R.A.; Zago, M.A.; et al. Halofuginone has anti-proliferative effects in acute promyelocytic leukemia by modulating the transforming growth factor beta signaling pathway. PLoS One 2011, 6, e26713. [Google Scholar] [CrossRef] [PubMed]
- Leiba, M.; Jakubikova, J.; Klippel, S.; Mitsiades, C.S.; Hideshima, T.; Tai, Y.T.; Leiba, A.; Pines, M.; Richardson, P.G.; Nagler, A.; et al. Halofuginone inhibits multiple myeloma growth in vitro and in vivo and enhances cytotoxicity of conventional and novel agents. Br. J. Haematol. 2012, 157, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.L.; Guan, Q.; Nguan, C.Y.; Du, C. Halofuginone suppresses T cell proliferation by blocking proline uptake and inducing cell apoptosis. Int. Immunopharmacol. 2013, 16, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.L.; Park, S.Y.; Kim, Y.H.; Park, G.; Lee, S.J. Halofuginone induces the apoptosis of breast cancer cells and inhibits migration via downregulation of matrix metalloproteinase-9. Int. J. Oncol. 2014, 44, 309–318. [Google Scholar] [PubMed]
- Bodanovsky, A.; Guttman, N.; Barzilai-Tutsch, H.; Genin, O.; Levy, O.; Pines, M.; Halevy, O. Halofuginone improves muscle-cell survival in muscular dystrophies. Biochim. Biophys. Acta 2014, 1843, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Hasan, S.; Sharma, S.; Nagra, S.; Yamaguchi, D.T.; Wong, D.; Bh, H.; Hossain, A. Th17 cells in inflammation and autoimmunity. Autoimmun. Rev. 2014, 13, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Shabgah, A.G.; Fattahi, E.; Shahneh, F.Z. Interleukin-17 in human inflammatory diseases. Postepy Dermatol. Alergol. 2014, 31, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Sundrud, M.S.; Koralov, S.B.; Feuerer, M.; Calado, D.P.; Kozhaya, A.E.; Rhule-Smith, A.; Lefebvre, R.E.; Unutmaz, D.; Mazitschek, R.; Waldner, H.; et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science 2009, 324, 1334–1338. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Robertson, L.; Gallinetti, J.; Mejia, P.; Vose, S.; Charlip, A.; Chu, T.; Mitchell, J.R. Surgical stress resistance induced by single amino acid deprivation requires Gcn2 in mice. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef]
- Pietrella, D.; Rachini, A.; Pines, M.; Pandey, N.; Mosci, P.; Bistoni, F.; d’Enfert, C.; Vecchiarelli, A. Th17 cells and IL-17 in protective immunity to vaginal candidiasis. PLoS One 2011, 6, e22770. [Google Scholar] [CrossRef] [PubMed]
- Deselm, C.J.; Zou, W.; Teitelbaum, S.L. Halofuginone prevents estrogen-deficient osteoporosis in mice. J. Cell. Biochem. 2012, 113, 3086–3092. [Google Scholar] [CrossRef] [PubMed]
- Park, M.K.; Park, J.S.; Park, E.M.; Lim, M.A.; Kim, S.M.; Lee, D.G.; Baek, S.Y.; Yang, E.J.; Woo, J.W.; Lee, J.; et al. Halofuginone ameliorates autoimmune arthritis in mice by regulating the balance between Th17 and Treg cells and inhibiting osteoclastogenesis. Arthritis Rheumatol. 2014, 66, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Tian, J.; Zeng, L.; Pan, B.; Li, Z.; Song, G.; Chen, W.; Xu, K. Halofugine prevents cutaneous graft versus host disease by suppression of Th17 differentiation. Hematology 2012, 17, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Keller, T.L.; Zocco, D.; Sundrud, M.S.; Hendrick, M.; Edenius, M.; Yum, J.; Kim, Y.J.; Lee, H.K.; Cortese, J.F.; Wirth, D.F.; et al. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat. Chem. Biol. 2012, 8, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lee, E.H.; Park, M.; Kim, J.H.; Kim, J.; Kim, S.; Jeon, Y.H.; Hwang, K.Y. Conformational changes in human prolyl-tRNA synthetase upon binding of the substrates proline and ATP and the inhibitor halofuginone. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 2136–2145. [Google Scholar] [CrossRef] [PubMed]
- Hernández, A.S. Helper (TH1, TH2, TH17) and regulatory cells (Treg, TH3, NKT) in rheumatoid arthritis. Reumatol. Clin. 2009, 5 (Suppl. S1), 1–5. [Google Scholar]
- Ho, A.W.; Gaffen, S.L. IL-17RC: A partner in IL-17 signaling and beyond. Semin. Immunopathol. 2010, 32, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.P.; Fujii, H.; Zhou, A.X.; Creemers, J.; Unutmaz, D.; Shevach, E.M. Regulation of the expression of GARP/latent TGF-β1 complexes on mouse T cells and their role in regulatory T cell and Th17 differentiation. J. Immunol. 2013, 190, 5506–5515. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Lopes, J.E.; Chong, M.M.; Ivanov, I.I.; Min, R.; Victora, G.D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-β-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORct function. Nature 2008, 453, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, N.; Kang, J. SMAD regulatory networks construct a balanced immune system. Immunology 2013, 139, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pinchuk, I.V.; Morris, K.T.; Nofchissey, R.A.; Earley, R.B.; Wu, J.Y.; Ma, T.Y.; Beswick, E.J. Stromal cells induce Th17 during Helicobacter pylori infection and in the gastric tumor microenvironment. PLoS One 2013, 8, e53798. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhang, B.; Shen, R.W.; Liu, J.B.; Gao, M.H.; Geng, X.; Li, Y.; Li, Y.Y.; Zhang, W. The effect of antifibrotic drug halofugine on Th17 cells in concanavalin A-induced liver fibrosis. Scand. J. Immunol. 2014, 79, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Ghoreschi, K.; Laurence, A.; Yang, X.P.; Tato, C.M.; McGeachy, M.J.; Konkel, J.E.; Ramos, H.L.; Wei, L.; Davidson, T.S.; Bouladoux, N.; et al. Generation of pathogenic TH17 cells in the absence of TGF-β signalling. Nature 2010, 467, 967–971. [Google Scholar]
- Coombes, J.L.; Siddiqui, K.R.R.; Arancibia-Cárcamo, C.V.; Hall, J.; Sun, C.M.; Belkaid, Y.; Powrie, F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β- and retinoic acid-dependent mechanism. J. Exp. Med. 2007, 204, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Meis, J.F.; Ponnudurai, T.; Mons, B.; van Belkum, A.; van Eerd, P.M.; Druilhe, P.; Schellekens, H. Plasmodium falciparum: Studies on mature exoerythrocytic forms in the liver of the chimpanzee, Pan troglodytes. Exp. Parasitol. 1990, 70, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vanderberg, J.P.; Khan, Z.M.; Stewart, M.J. Induction of hepatic inflammatory response by Plasmodium berghei sporozoites protects BALB/c mice against challenge with Plasmodium yoelii sporozoites. J. Parasitol. 1993, 79, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.M.; Vanderberg, J.P. Specific inflammatory cell infiltration of hepatic schizonts in BALB/c mice immunized with attenuated Plasmodium yoelii sporozoites. Int. Immunol. 1992, 4, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.L. Prediction of fibrosis progression in chronic viral hepatitis. Clin. Mol. Hepatol. 2014, 20, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Schacker, T.W.; Nguyen, P.L.; Beilman, G.J.; Wolinsky, S.; Larson, M.; Reilly, C.; Haase, A.T. Collagen deposition in HIV-1 infected lymphatic tissues and T cell homeostasis. J. Clin. Investig. 2002, 110, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Nagler, A.; Pines, M. Topical treatment of cutaneous chronic graft versus host disease (cGvHD) with halofuginone: A novel inhibitor of collagen type I synthesis. Transplantation 1999, 68, 1806–1809. [Google Scholar] [CrossRef] [PubMed]
- Koon, H.B.; Fingleton, B.; Lee, J.Y.; Geyer, J.T.; Cesarman, E.; Parise, R.A.; Egorin, M.J.; Dezube, B.J.; Aboulafia, D.; Krown, S.E. Phase II AIDS Malignancy Consortium trial of topical halofuginone in AIDS-related Kaposi sarcoma. J. Acquir. Immune Defic. Syndr. 2011, 56, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Pines, M.; Snyder, D.; Yarkoni, S.; Nagler, A. Halofuginone to treat fibrosis in chronic graft versus host disease and scleroderma. Biol. Blood Marrow Transplant. 2003, 9, 417–425. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, M.J.; Dumez, H.; Verweij, J.; Yarkoni, S.; Snyder, D.; Lacombe, D.; Marréaud, S.; Yamaguchi, T.; Punt, C.J.; van Oosterom, A. EORTC New Drug Development Group (NDDG). Phase I and pharmacokinetic study of halofuginone, an oral quinazolinone derivative in patients with advanced solid tumours. Eur. J. Cancer 2006, 42, 1768–1774. [Google Scholar] [CrossRef] [PubMed]
- Leiba, M.; Cahalon, L.; Shimoni, A.; Lider, O.; Zanin-Zhorov, A.; Hecht, I.; Sela, U.; Vlodavsky, I.; Nagler, A. Halofuginone inhibits NF-kappaB and p38 MAPK in activated T cells. J. Leukoc. Biol. 2006, 80, 399–406. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pines, M.; Spector, I. Halofuginone — The Multifaceted Molecule. Molecules 2015, 20, 573-594. https://doi.org/10.3390/molecules20010573
Pines M, Spector I. Halofuginone — The Multifaceted Molecule. Molecules. 2015; 20(1):573-594. https://doi.org/10.3390/molecules20010573
Chicago/Turabian StylePines, Mark, and Itai Spector. 2015. "Halofuginone — The Multifaceted Molecule" Molecules 20, no. 1: 573-594. https://doi.org/10.3390/molecules20010573