Fabrication of Implantable, Enzyme-Immobilized Glutamate Sensors for the Monitoring of Glutamate Concentration Changes in Vitro and in Vivo

Abstract
:1. Introduction
2. Results and Discussion
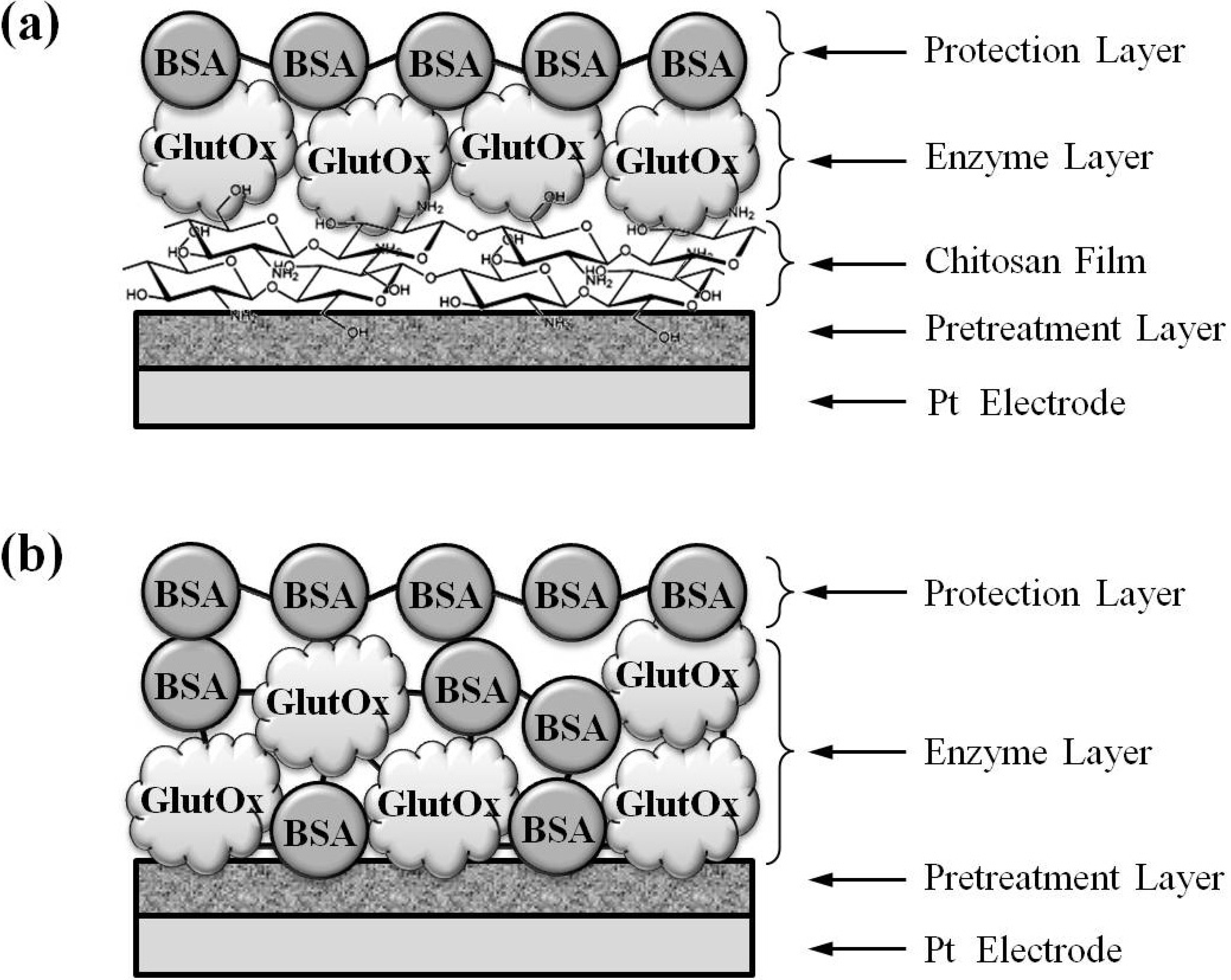
2.1. Evaluation of Glutamate Sensors Prepared by Method 1 and Method 2
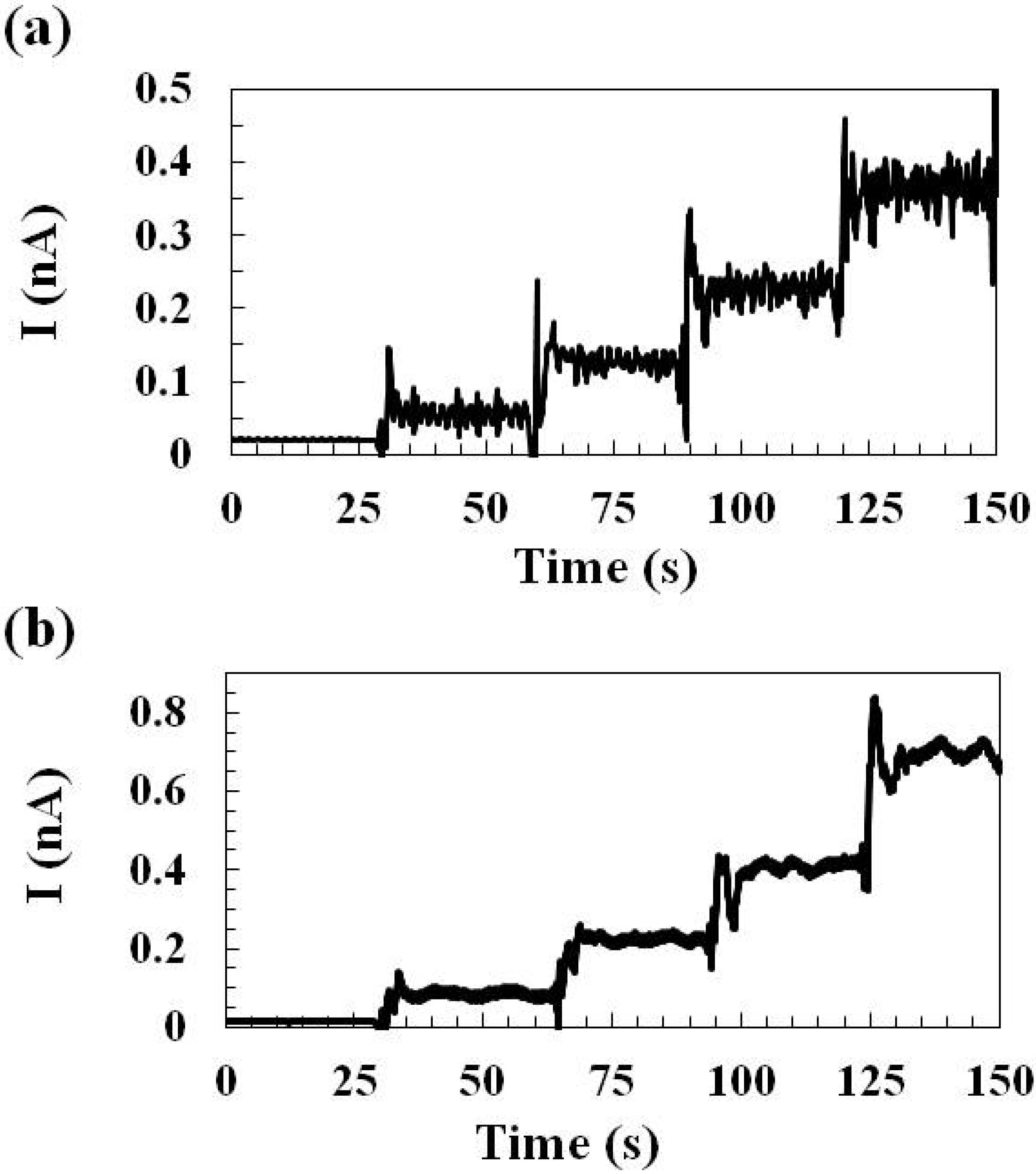
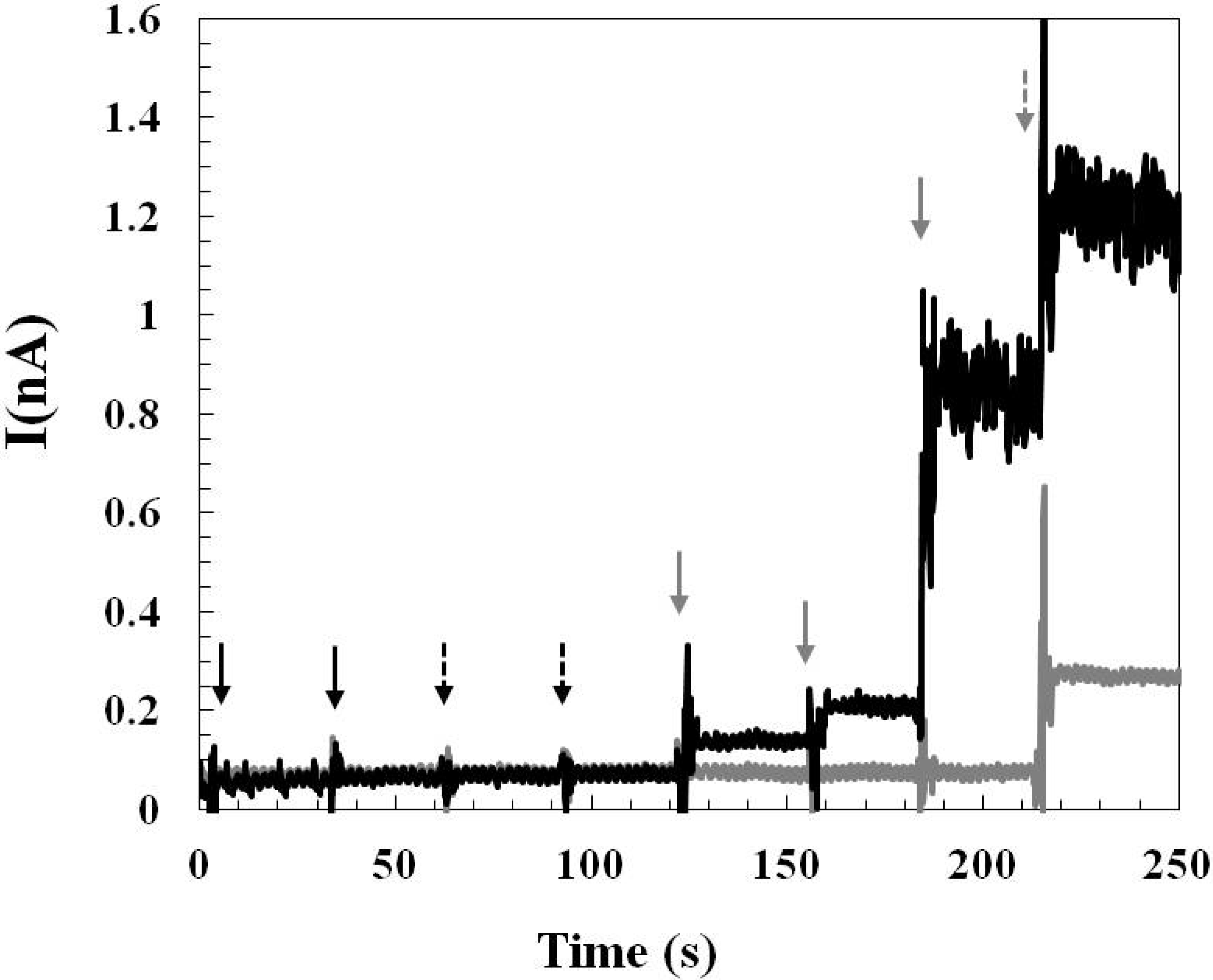
2.1.1. Sensing Current Response

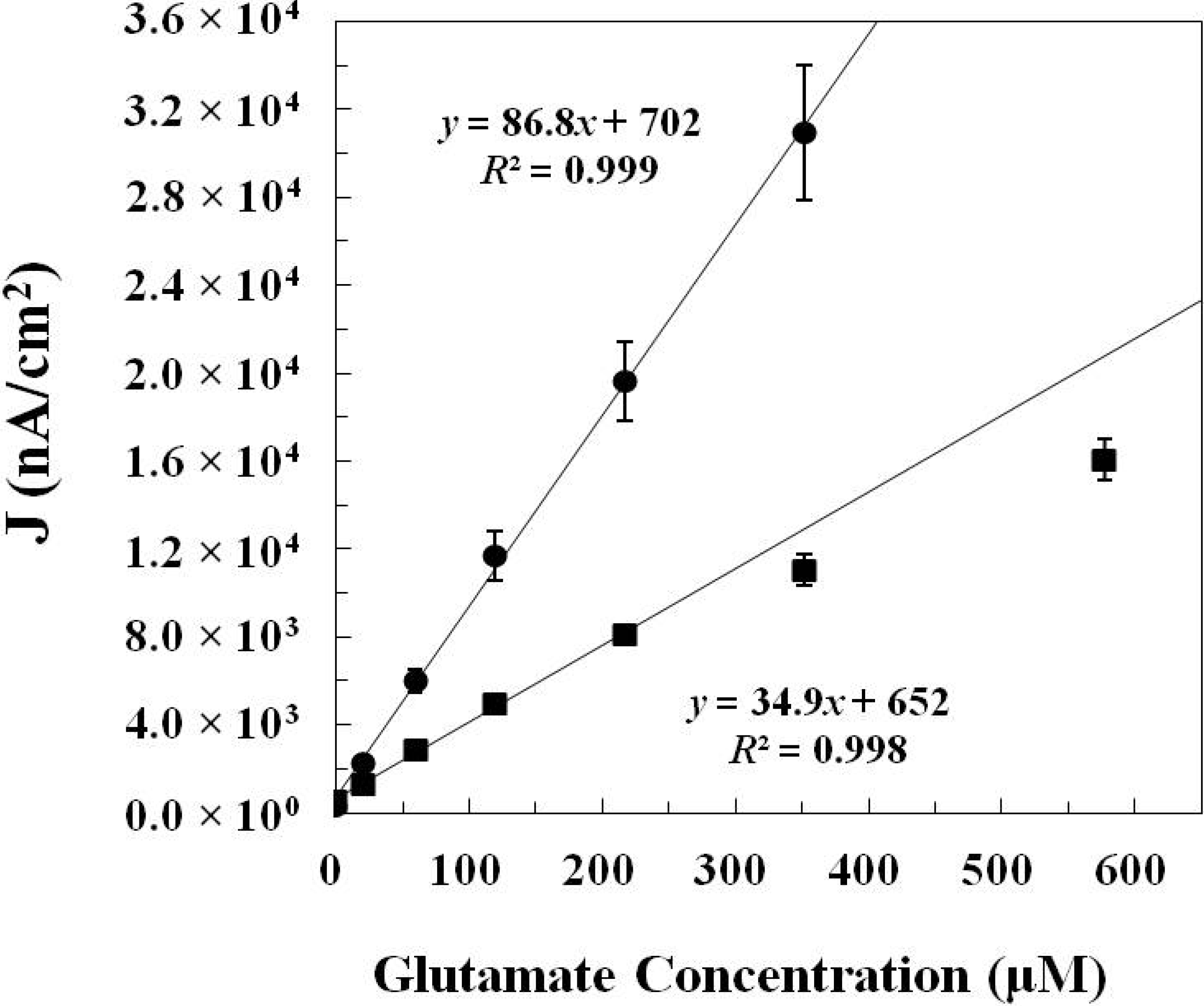
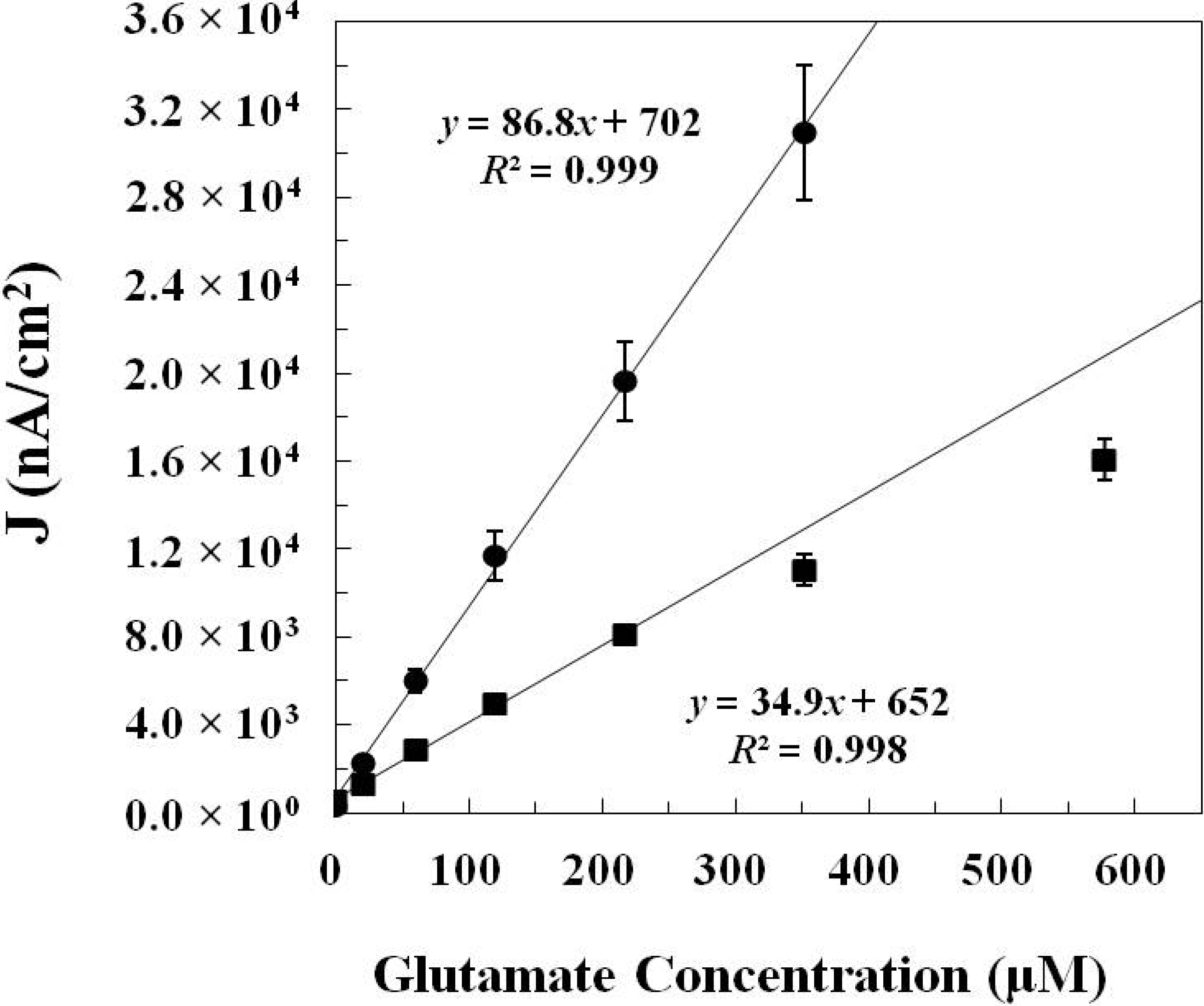
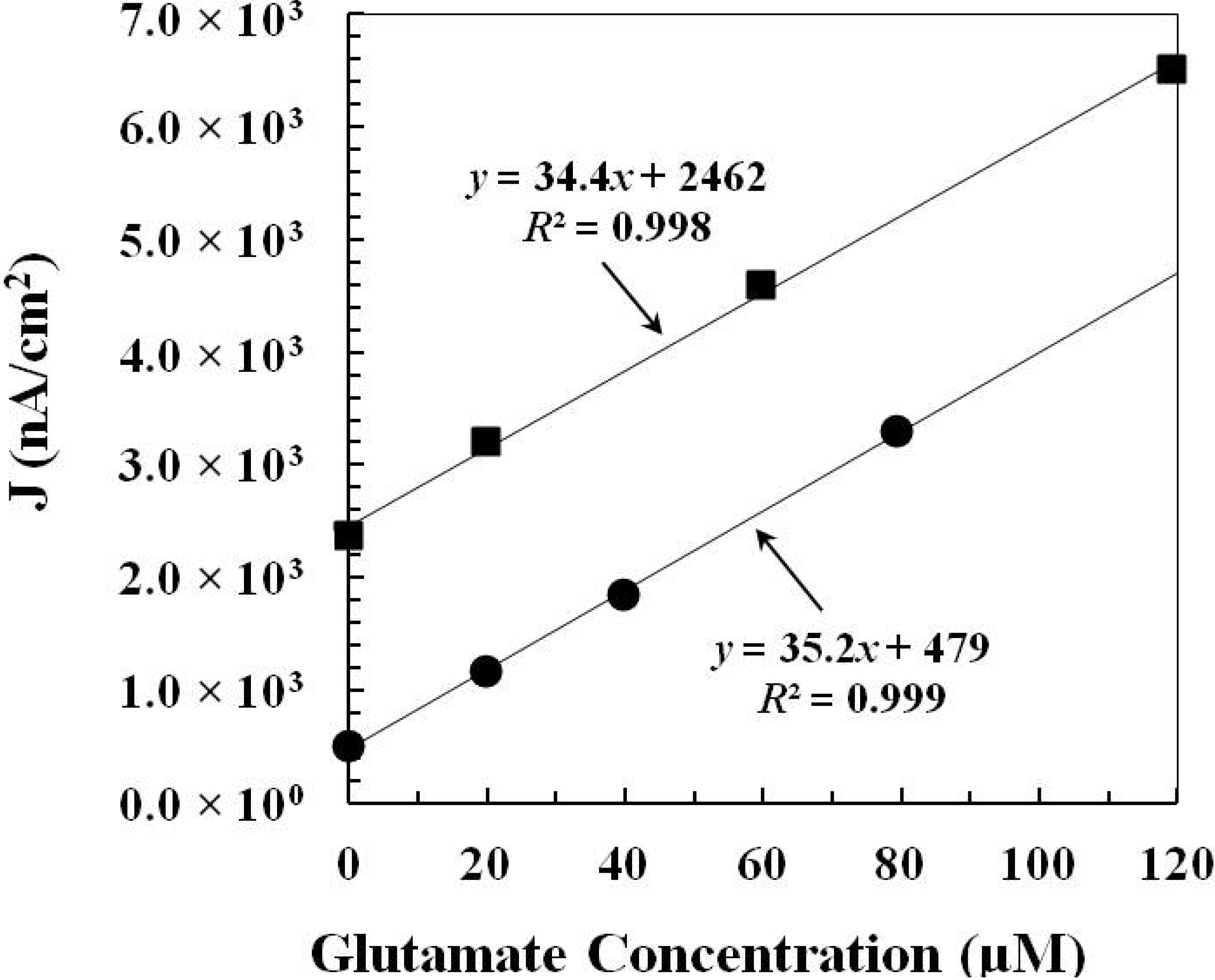
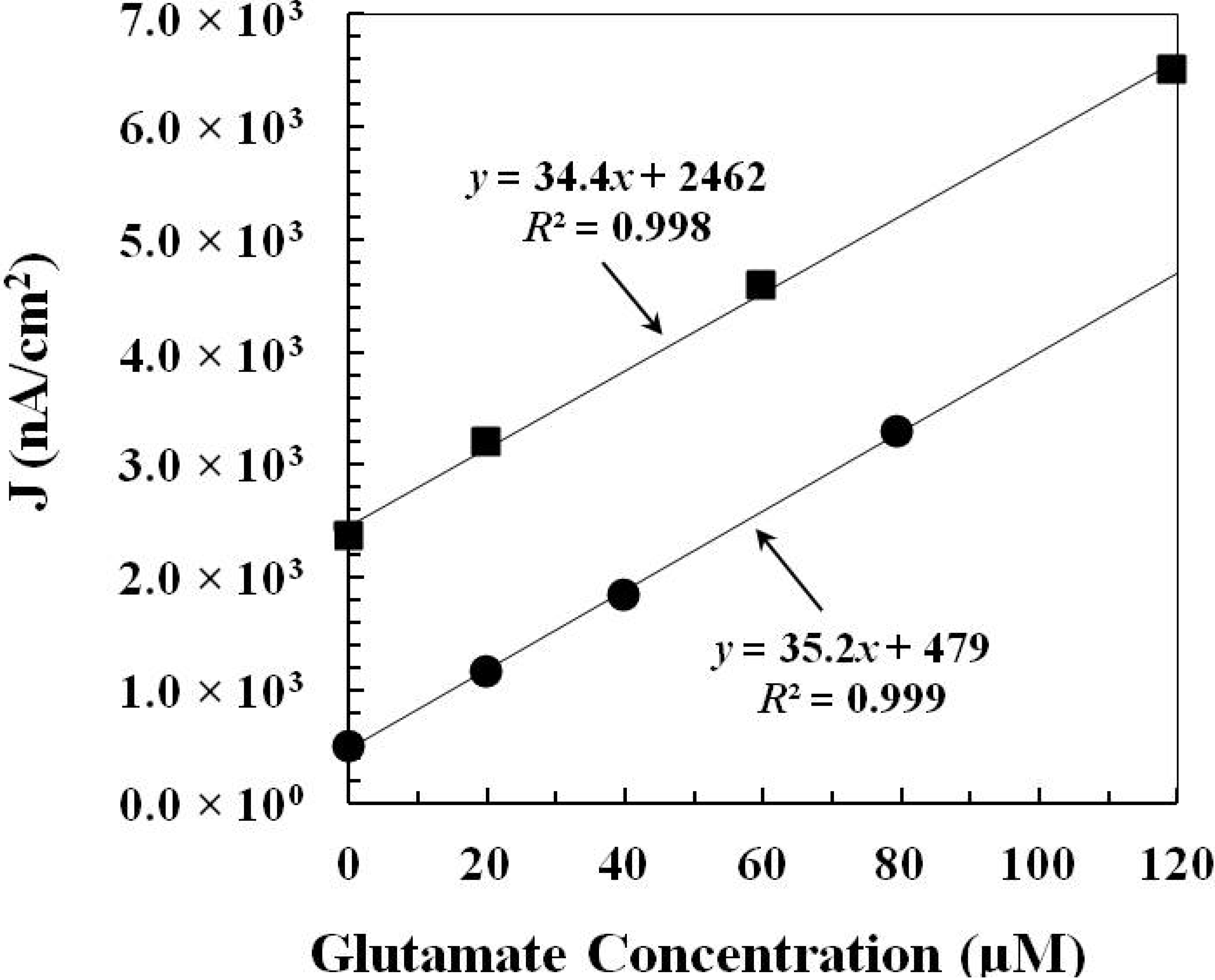
2.1.2. In Vitro Calibration

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GlutOx Immobilization Method | Linear Range (μM) | Sensitivity (nA·μM−1·cm−2) | Limit of Detection (μM) | Response Time (s) |
|---|---|---|---|---|
| Adsorption with electrodeposited chitosan (Method 1) | 20–217 | 34.9 ± 4.8 (N = 8) | 2.5 ± 1.1 | <2 |
| Crosslinking with BSA and glutaraldehyde (Method 2) | 20–352 | 86.8 ± 8.8 (N = 12) | 6.5 ± 1.7 | <5 |
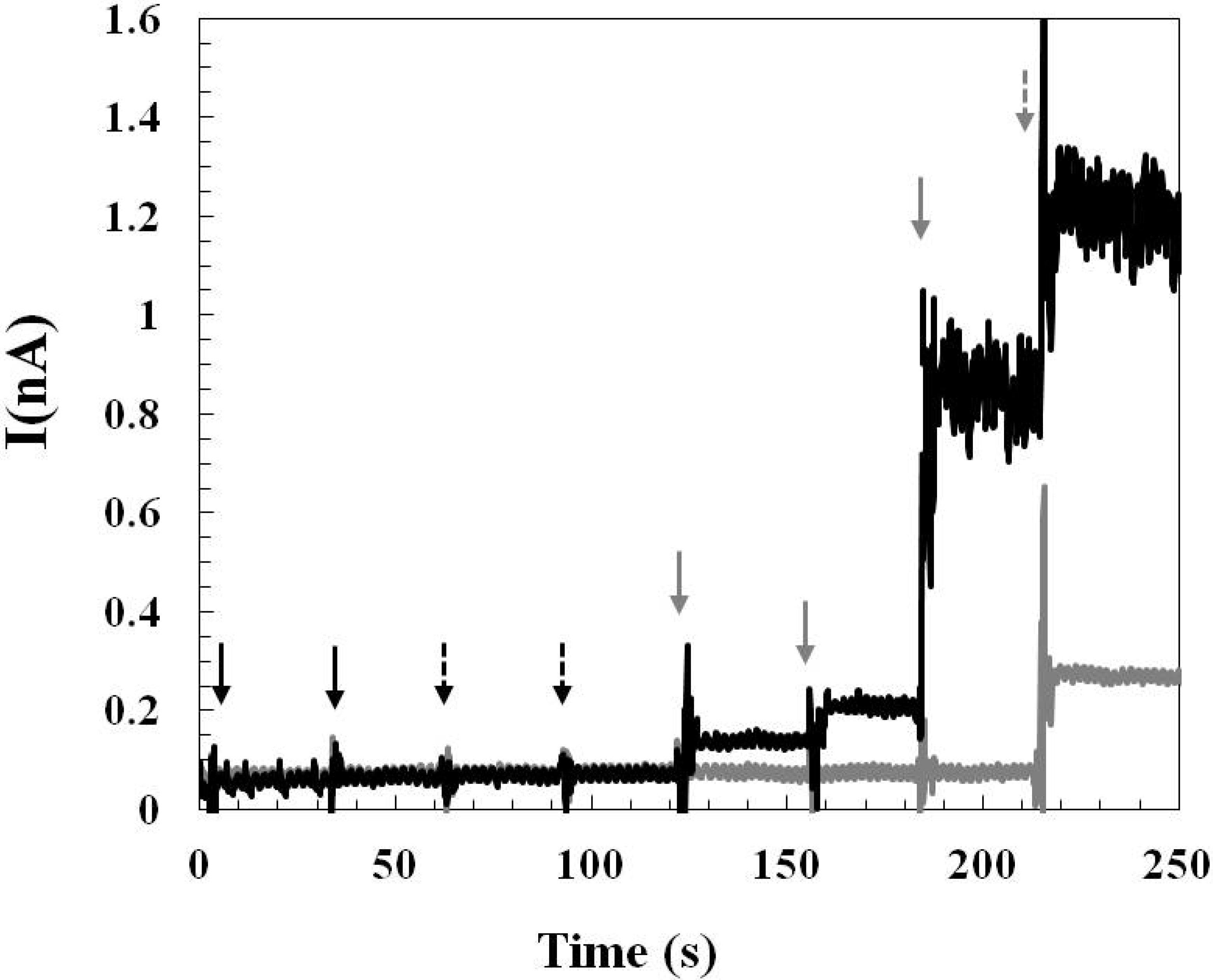
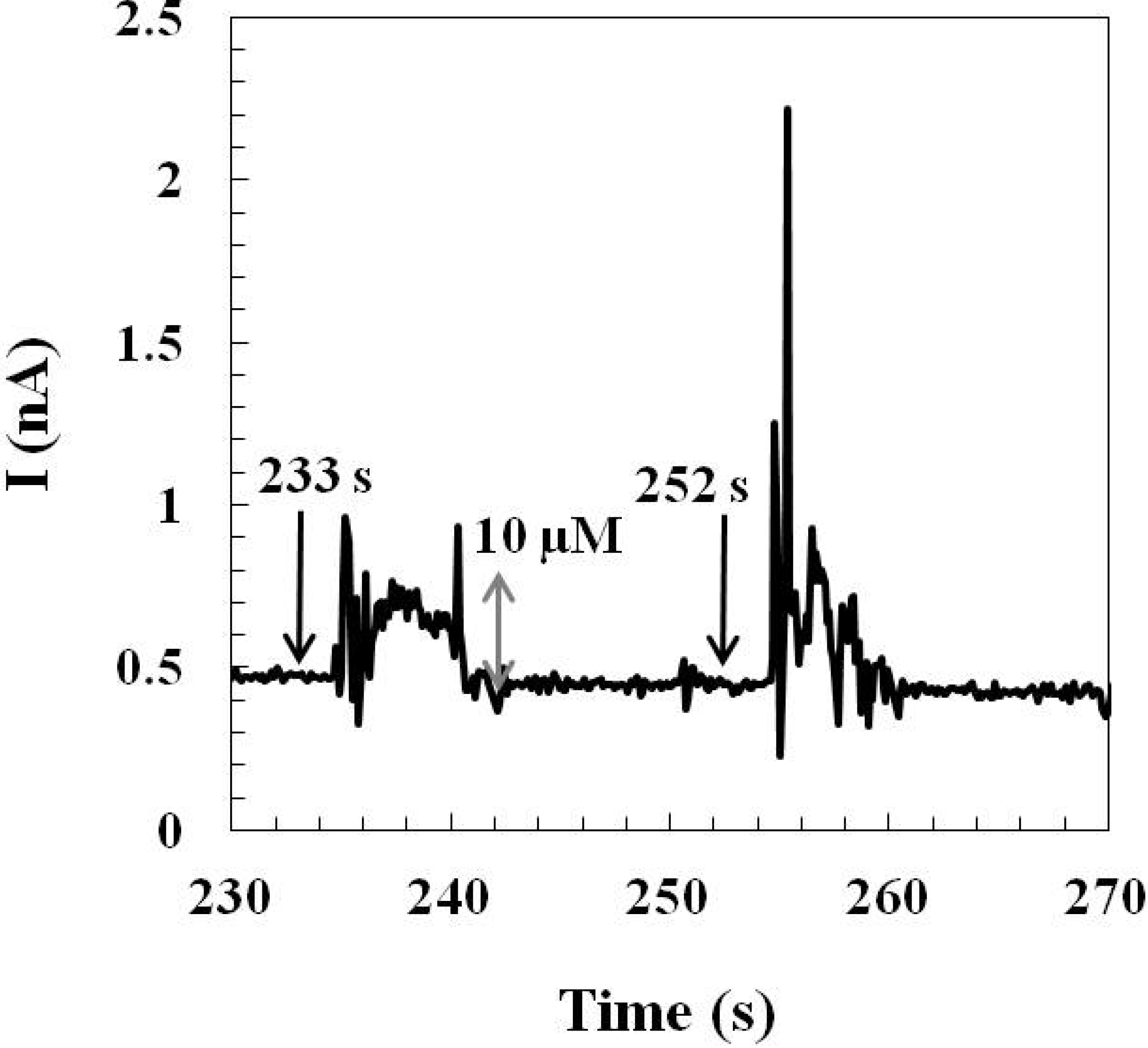
2.1.3. Effect of Interference and Sensor Stability
2.2. The Applicability of the Glutamate Sensors for in Vivo Study
2.2.1. Pre-Calibration and Post-Calibration
2.2.2. Monitoring Glutamate Release in the Rat Hypothalamus

3. Experimental Section
3.1. Materials
3.2. Instrumentation
3.3. Preparation of Micromachined Microelectrodes
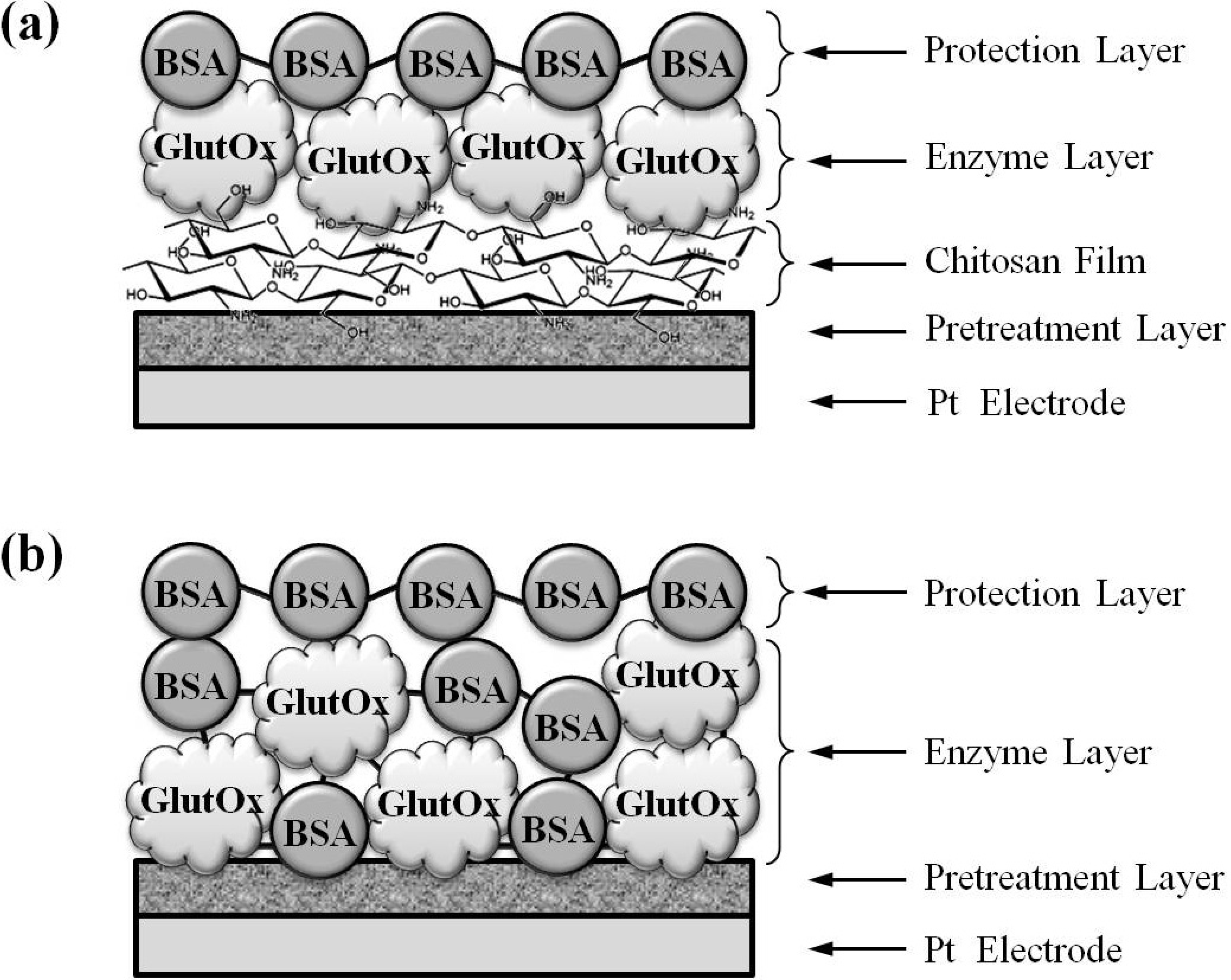
3.4. Fabrication of Glutamate Sensors
3.5. Preparation of Ag/AgCl Wire Reference Electrodes for in Vivo Measurements
3.6. Implantation of Glutamate Sensors into the Rat Brain
3.7. In Vivo Experiments
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Arima, J.; Tamura, T.; Kusakabe, H.; Ashiuchi, M.; Yagi, T.; Tanaka, H.; Inagaki, K. Recombinant expression, biochemical characterization and stabilization through proteolysis of an l-glutamate oxidase from Streptomyces sp. X-119–6. J. Biochem. 2003, 134, 805–812. [Google Scholar]
- Arima, J.; Sasaki, C.; Sakaguchi, C.; Mizuno, H.; Tamura, T.; Kashima, A.; Kusakabe, H.; Sugio, S.; Inagaki, K. Structural characterization of L-glutamate oxidase from Streptomyces sp. X-119–6. FEBS J. 2009, 276, 3894–3903. [Google Scholar]
- Cunningham, A.J. Introduction to Bioanalytical Sensors; Wiley-Interscience: New York, NY, USA, 1998; pp. 167–203. [Google Scholar]
- Kusakabe, H.; Midorikawa, Y.; Fujishima, T.; Kuninaka, A.; Yoshino, H. Purification and properties of a new enzyme, L-glutamate oxidase, from Streptomyces sp. X-119–6 grown on wheat bran. Agric. Biol. Chem. 1983, 47, 1323–1328. [Google Scholar]
- Burmeister, J.J.; Pomerleau, F.; Palmer, M.; Day, B.K.; Huettl, P.; Gerhardt, G.A. Improved ceramic-based multisite microelectrode for rapid measurements of L-glutamate in the CNS. J. Neurosci. Methods 2002, 119, 163–171. [Google Scholar]
- Zhang, M.; Mullens, C.; Gorski, W. Chitosan-glutamate oxidase gels: Synthesis, characterization, and glutamate determination. Electroanalysis 2005, 17, 2114–2120. [Google Scholar]
- Hamdi, N.; Wang, J.; Walker, E.; Maidment, N.T.; Monbouquette, H.G. An electroenzymatic l-glutamate microbiosensor selective against dopamine. J. Electroanal. Chem. 2006, 591, 33–40. [Google Scholar]
- Wassum, K.M.; Tolosa, V.M.; Wang, J.; Walker, E.; Monbouquette, H.G.; Maidment, N.T. Silicon wafer-based platinum microelectrode array biosensor for near real-time measurement of glutamate in vivo. Sensors 2008, 8, 5023–5036. [Google Scholar]
- Tseng, T.T.-C.; Monbouquette, H.G. Implantable microprobe with arrayed microsensors for combined amperometric monitoring of the neurotransmitters, glutamate and dopamine. J. Electroanal. Chem. 2012, 682, 141–146. [Google Scholar]
- Wassum, K.M.; Tolosa, V.M.; Tseng, T.C.; Balleine, B.W.; Monbouquette, H.G.; Maidment, N.T. Transient extracellular glutamate events in the basolateral amygdala track reward-seeking actions. J. Neurosci. 2012, 8, 2734–2746. [Google Scholar]
- Khan, R.; Gorski, W.; Garcia, C.D. Nanomolar detection of glutamate at a biosensor based on screen-printed electrodes modified with carbon nanotubes. Electroanalysis 2011, 23, 2357–2363. [Google Scholar]
- Tseng, T.T.-C.; Yao, J.; Chan, W.-C. Selective enzyme immobilization on arrayed microelectrodes for the application of sensing neurotransmitters. Biochem. Eng. J. 2013, 78, 146–153. [Google Scholar]
- Cosnier, S.; Innocent, C.; Allien, L.; Poitry, S.; Tsacopoulos, M. An electrochemical method for making enzyme microsensors. Application to the detection of dopamine and glutamate. Anal. Chem. 1997, 69, 968–971. [Google Scholar]
- Zhang, M.; Mullens, C.; Gorski, W. Amperometric glutamate biosensor based on chitosan enzyme film. Electrochim. Acta 2006, 51, 4528–4532. [Google Scholar]
- Cooper, J.M.; Pritchard, D.J. Biomolecular sensors for neurotransmitter determination: Electrochemical immobilization of glutamate oxidase at microelectrodes in a poly(o-phenylenediamine) film. J. Mater. Sci. Mater. Electron. 1994, 5, 111–116. [Google Scholar]
- Ryan, M.R.; Lowry, J.P.; O’Neill, R.D. Biosensor for neurotransmitter l-glutamic acid designed for efficient use of l-glutamate oxidase and effective rejection of interference. Analyst 1997, 122, 1419–1424. [Google Scholar]
- Kulagina, N.V.; Shankar, L.; Michael, A.C. Monitoring glutamate and ascorbate in the extracellular space of brain tissue with electrochemical microsensors. Anal. Chem. 1999, 71, 5093–5100. [Google Scholar]
- Migneault, I.; Dartiguenave, C.; Bertrand, M.J.; Waldron, K.C. Glutaraldehyde: Behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking. BioTechniques 2004, 37, 790–802. [Google Scholar]
- Fernandes, R.; Wu, L.Q.; Chen, T.H.; Yi, H.M.; Rubloff, G.W.; Ghodssi, R.; Bentley, W.E.; Payne, G.F. Electrochemically induced deposition of a polysaccharide hydrogel onto a patterned surface. Langmuir 2003, 19, 4058–4062. [Google Scholar]
- Xu, C.; Cai, H.; Xu, Q.; He, P.; Fang, Y. Characterization of single-stranded DNA on chitosan-modified electrode and its application to the sequence-specific DNA detection. Fresenius J. Anal. Chem. 2001, 369, 428–432. [Google Scholar]
- Wu, L.Q.; Chen, T.; Wallace, K.K.; Vazquez-Duhalt, R.; Payne, G.F. Enzymatic coupling of phenol vapors onto chitosan. Biotechnol. Bioeng. 2001, 76, 325–332. [Google Scholar]
- Chen, L.; Gorski, W. Bioinorganic composites for enzyme electrodes. Anal. Chem. 2001, 73, 2862–2868. [Google Scholar]
- Wei, X.; Cruz, J.; Gorski, W. Integration of enzymes and electrodes: Spectroscopic and electrochemical studies of chitosan-enzyme films. Anal. Chem. 2002, 74, 5039–5046. [Google Scholar]
- Zhang, M.; Smith, A.; Gorski, W. Carbon nanotube-chitosan system for electrochemical sensing based on dehydrogenase enzymes. Anal. Chem. 2004, 76, 5045–5050. [Google Scholar]
- Du, D.; Ding, J.; Cai, J.; Zhang, A. One-step electrochemically deposited interface of chitosan–gold nanoparticles for acetylcholinesterase biosensor design. J. Electroanal. Chem. 2007, 605, 53–60. [Google Scholar]
- Zhang, M.; Mullens, C.; Gorski, W. Coimmobilization of dehydrogenases and their cofactors in electrochemical biosensors. Anal. Chem. 2007, 79, 2446–2450. [Google Scholar]
- Karra, S.; Gorski, W. Signal amplification in enzyme-based amperometric biosensors. Anal. Chem. 2013, 85, 10573–10580. [Google Scholar]
- Koev, S.T.; Dykstra, P.H.; Luo, X.; Rubloff, G.W.; Bentley, W.E.; Payne, G.F.; Ghodssi, R. Chitosan: An integrative biomaterial for lab-on-a-chip devices. Lab Chip 2010, 10, 3026–3042. [Google Scholar]
- Qin, S.; van der Zeyden, M.; Oldenziel, W.H.; Cremers, T.I.F.H.; Westerink, B.H.C. Microsensors for in vivo measurement of glutamate in brain tissue. Sensors 2008, 8, 6860–6884. [Google Scholar]
- Putzbach, W.; Ronkainen, N.J. Immobilization techniques in the fabrication of nanomaterial-based electrochemical biosensors: A review. Sensors 2013, 13, 4811–4840. [Google Scholar]
- Oldenziel, W.; Dijkstra, G.; Cremers, T.; Westerink, B. In vivo monitoring of extracellular glutamate in the brain with a microsensor. Brain Res. 2006, 1118, 34–42. [Google Scholar]
- Lowry, J.P.; Ryan, M.R.; O’Neill, R.D. Behaviourally induced changes in extracellular levels of brain glutamate monitored at 1 s resolution with an implanted biosensor. Anal. Commun. 1998, 35, 87–89. [Google Scholar]
- Rutherford, E.C.; Pomerleau, F.; Huettl, P.; Strömberg, I.; Gerhardt, G.A. Chronic second-by-second measures of L-glutamate in the central nervous system of freely moving rats. J. Neurochem. 2007, 102, 712–722. [Google Scholar]
- Pomerleau, F.; Day, B.K.; Huettl, P.; Burmeister, J.J.; Gerhardt, G.A. Real time in vivo measures of l-glutamate in the rat central nervous system using ceramic-based multisite microelectrode arrays. Ann. N. Y. Acad. Sci. 2003, 1003, 454–457. [Google Scholar]
- van den Pol, A.N.; Trombley, P.Q. Glutamate neurons in hypothalamus regulate excitatory transmission. J. Neurosci. 1993, 13, 2829–2836. [Google Scholar]
- John, J.; Ramanathan, L.; Siegel, J.M. Rapid changes in glutamate levels in the posterior hypothalamus across sleep-wake states in freely behaving rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R2041–R2049. [Google Scholar]
- Hamdi, N.; Wang, J.; Monbouquette, H.G. Polymer films as permselective coatings for H2O2-sensing electrodes. J. Electroanal. Chem. 2005, 581, 258–264. [Google Scholar]
- Gerhardt, G.A.; Oke, A.F.; Nagy, G.; Moghaddam, B.; Adams, R.N. Nafion-coated electrodes with high selectivity for CNS electrochemistry. Brain Res. 1984, 290, 390–395. [Google Scholar]
- Paxinos, G.; Watson, C. The Rat Brain: In Stereotaxic Coordinates, 6th ed; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Sample Availability: Not available.
© 2014 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tseng, T.T.-C.; Chang, C.-F.; Chan, W.-C. Fabrication of Implantable, Enzyme-Immobilized Glutamate Sensors for the Monitoring of Glutamate Concentration Changes in Vitro and in Vivo. Molecules 2014, 19, 7341-7355. https://doi.org/10.3390/molecules19067341
Tseng TT-C, Chang C-F, Chan W-C. Fabrication of Implantable, Enzyme-Immobilized Glutamate Sensors for the Monitoring of Glutamate Concentration Changes in Vitro and in Vivo. Molecules. 2014; 19(6):7341-7355. https://doi.org/10.3390/molecules19067341
Chicago/Turabian StyleTseng, Tina T.-C., Cheng-Fu Chang, and Wen-Chin Chan. 2014. "Fabrication of Implantable, Enzyme-Immobilized Glutamate Sensors for the Monitoring of Glutamate Concentration Changes in Vitro and in Vivo" Molecules 19, no. 6: 7341-7355. https://doi.org/10.3390/molecules19067341