[15]aneN4S: Synthesis, Thermodynamic Studies and Potential Applications in Chelation Therapy


 ,
, 
Abstract
:
1. Introduction
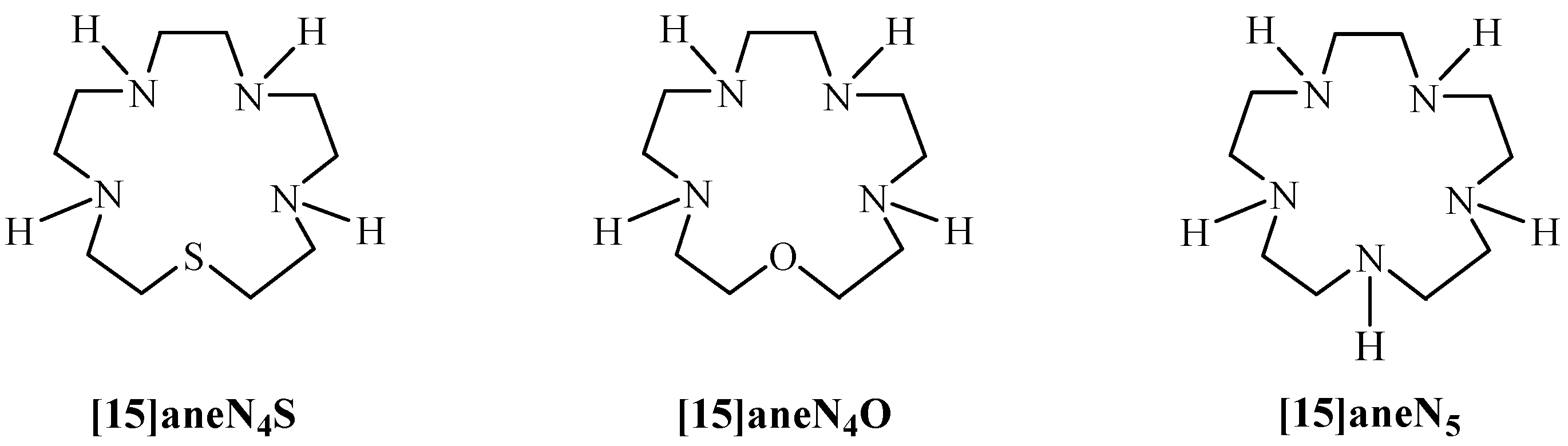
2. Results and Discussion
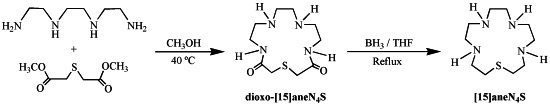
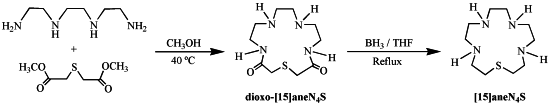
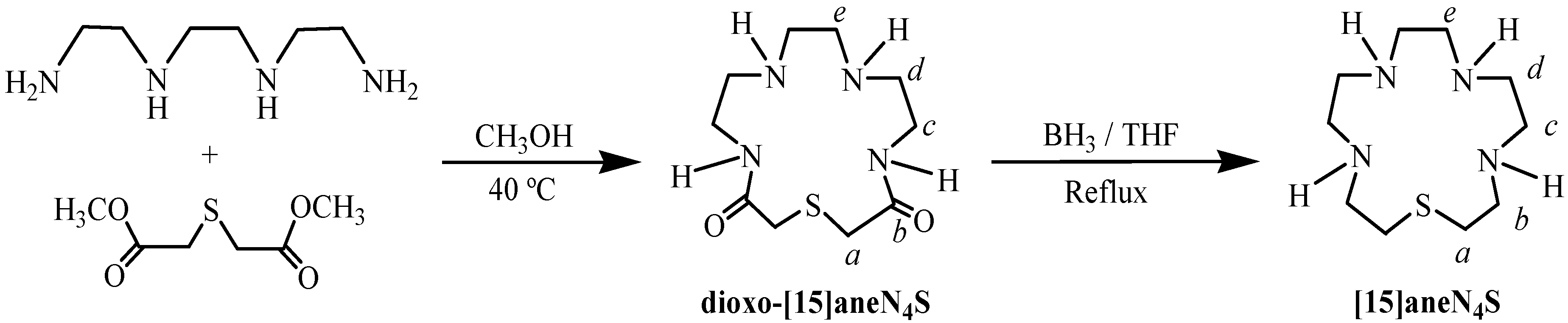
2.1. Synthesis and Characterization
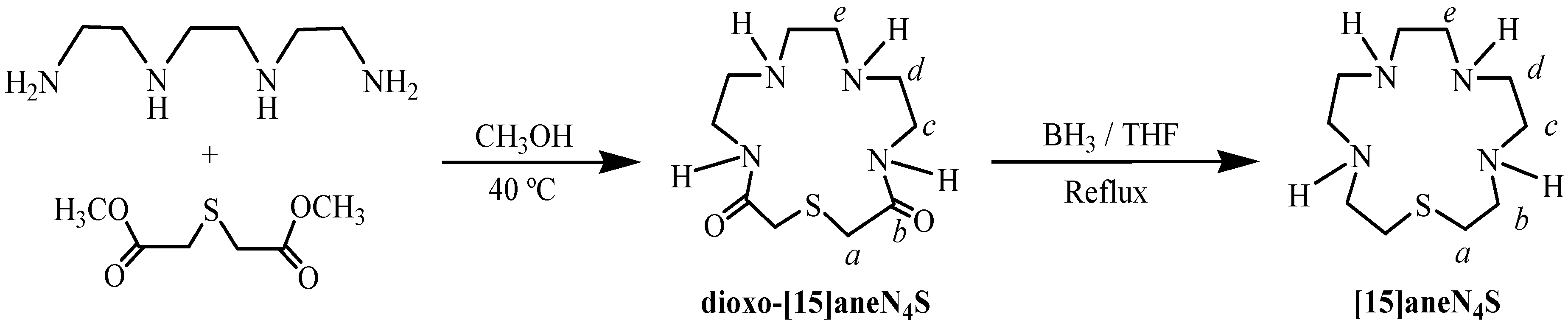
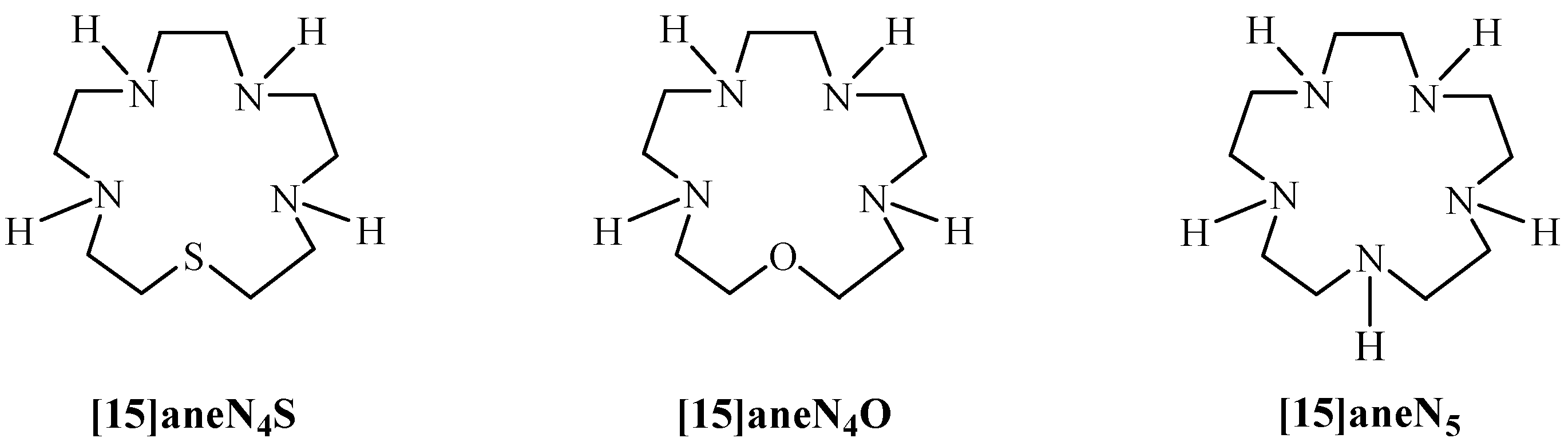
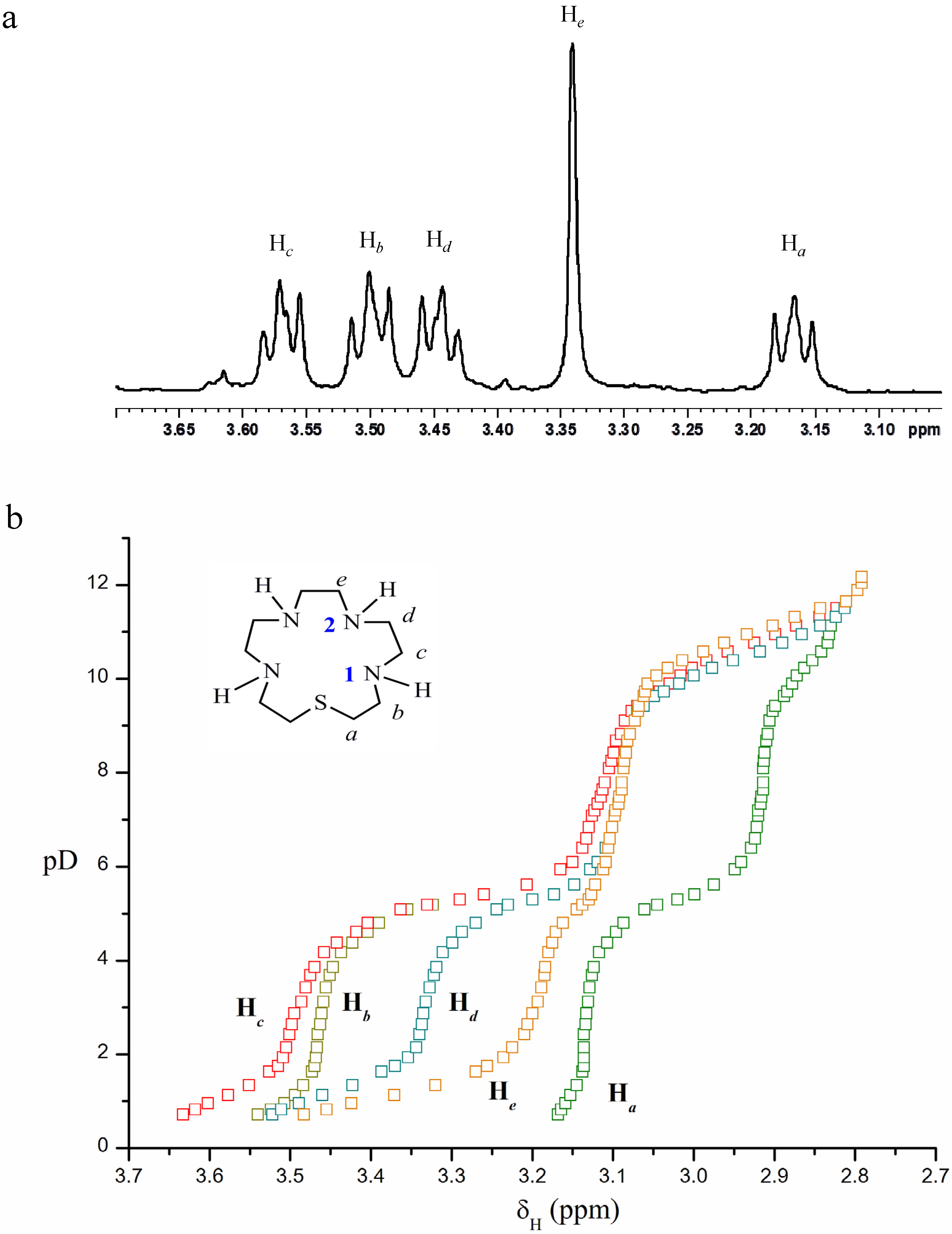
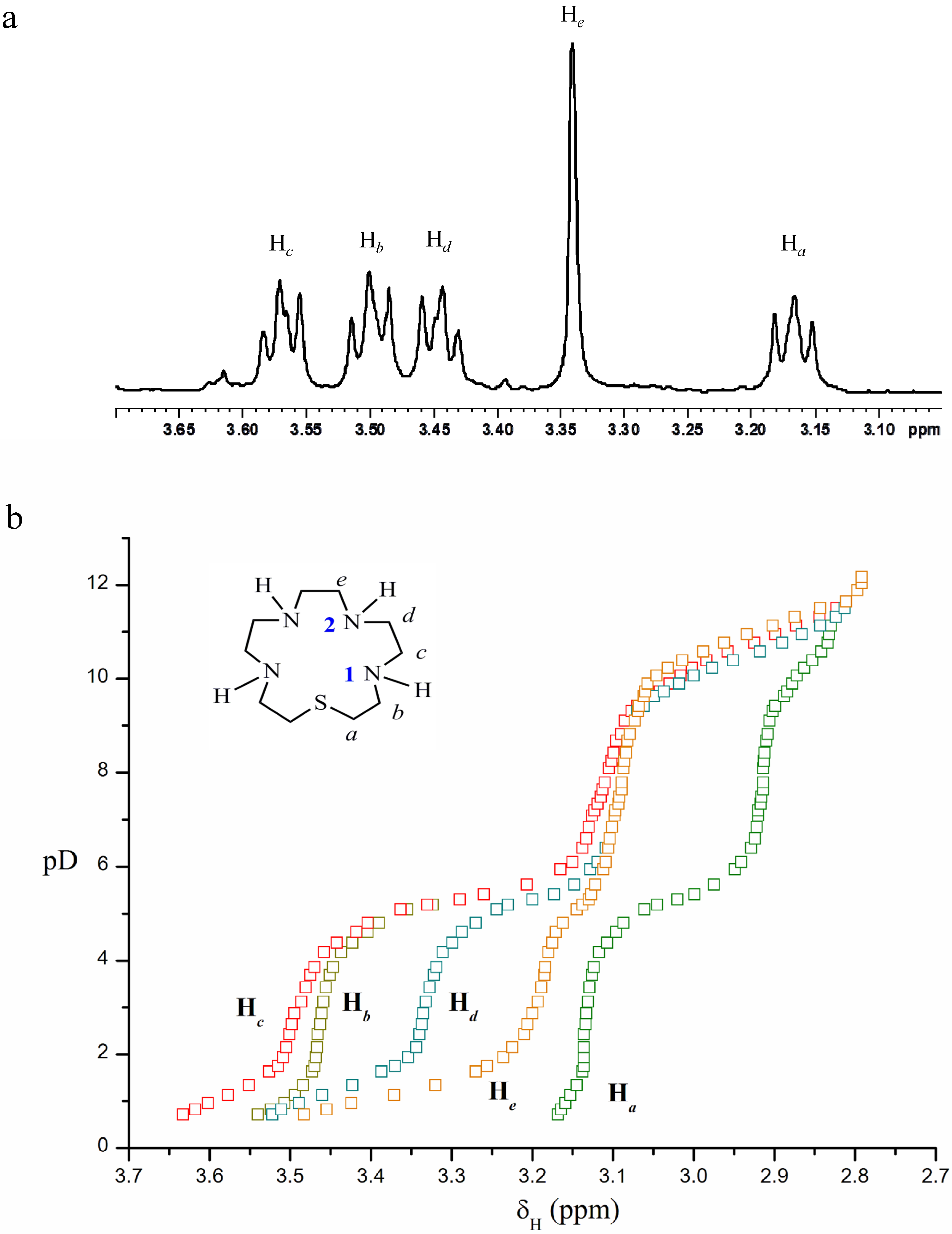
2.2. Acid-Base Behaviour

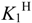
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction equilibrium | [15]aneN4S a,b | [15]aneN4O c | [15]aneN5 d |
|---|---|---|---|
L + H+  HL+ HL+ | 9.51(1) | 9.66 | 10.01 |
| HL+ + H+ H2L2+ | 8.56(2) | 8.77 | 9.28 |
| H2L2+ + H+ H3L3+ | 4.47(4) | 5.30 | 5.87 |
| H3L3+ + H+ H4L4+ | 0.8(2) e | 1.2 | 1.84 |
| L + 4 H+ H4L4+ | 23.34 | 24.93 | 27.00 |
2.3. Thermodynamic Stability of Metal Complexes
| Reaction equilibrium | [15]aneN4S a,b | [15]aneN4O c | [15]aneN5 d |
|---|---|---|---|
| Mn2+ + L MnL2+ | 6.65(2) | 8.53 c | 10.85 e |
| MnL2+ + H+ MnHL3+ | - | - | 5.04 e |
| MnL(OH)+ + H+ MnL2+ | 9.68(4) | - | 11.22 e |
| Fe2+ + L FeL2+ | 10.08(1) | 10.34 c | - |
| FeL2+ + H+ FeHL3+ | 4.83(6) | - | - |
| FeL(OH)+ + H+ FeL2+ | 8.25(7) | pp. | - |
| Co2+ + L CoL2+ | 13.62(6) | 12.72 c | 16.76 f |
| CoL2+ + H+ CoHL3+ | 5.20(7) | - | - |
| Ni2+ + L NiL2+ | 17.95(5) | 14.76 c | 18.1 g |
| NiL2+ + H+ NiHL3+ | 4.00(7) | - | - |
| NiL(OH)+ + H+ NiL2+ | - | 8.38 c | - |
| Cu2+ + L CuL2+ | 22.31(2) | 20.34 c | 28.0 h |
| CuL2+ + H+ CuHL3+ | 2.49(5) | - | - |
| CuL(OH)+ + H+ CuL2+ | 9.8(1) | 10.4 c | - |
| Zn2+ + L ZnL2+ | 13.472(8) | 13.21 c | 19.1 i |
| ZnL2+ + H+ ZnHL3+ | 4.06(3) | - | 3.1 i |
| ZnL(OH)+ + H+ ZnL2+ | 7.16(4) | - | - |
| Cd2+ + L CdL2+ | 13.61(2) | 13.41 d | 19.2 i |
| CdL2+ + H+ CdHL3+ | 3.97(4) | - | 3.4 i |
| CdL(OH)+ + H+ CdL2+ | 9.25(7) | - | - |
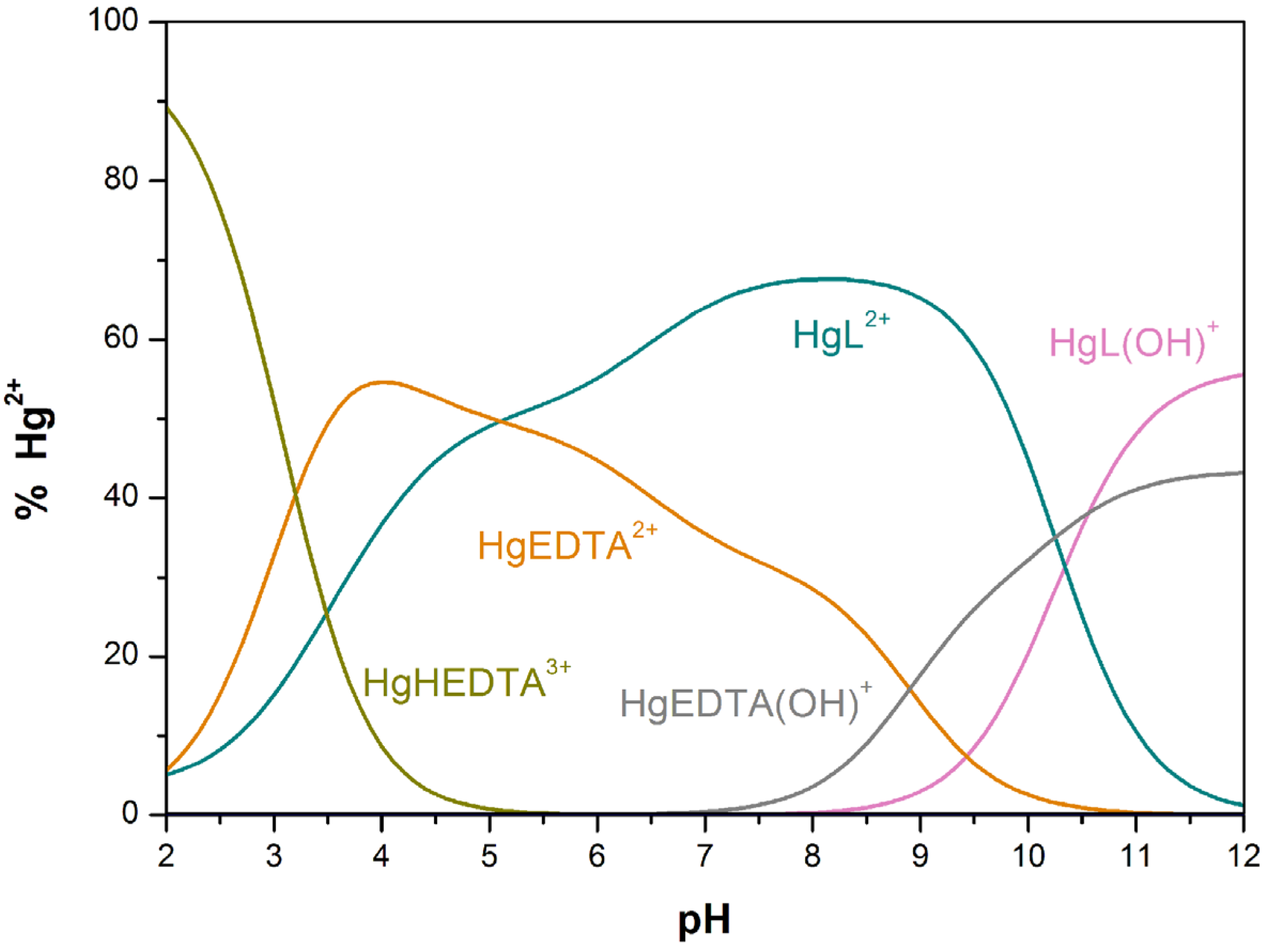
| Hg2+ + L HgL2+ | 23.74(5) | - | 28.5 j |
| HgL(OH)+ + H+ HgL2+ | 10.3(1) | - | - |
| Pb2+ + L PbL2+ | 12.44(2) | 12.28 d | 17.3 i |
| PbL2+ + H+ PbHL3+ | 4.44(4) | - | 3.8 i |
| PbL(OH)+ + H+ PbL2+ | 7.76(7) | - | - |
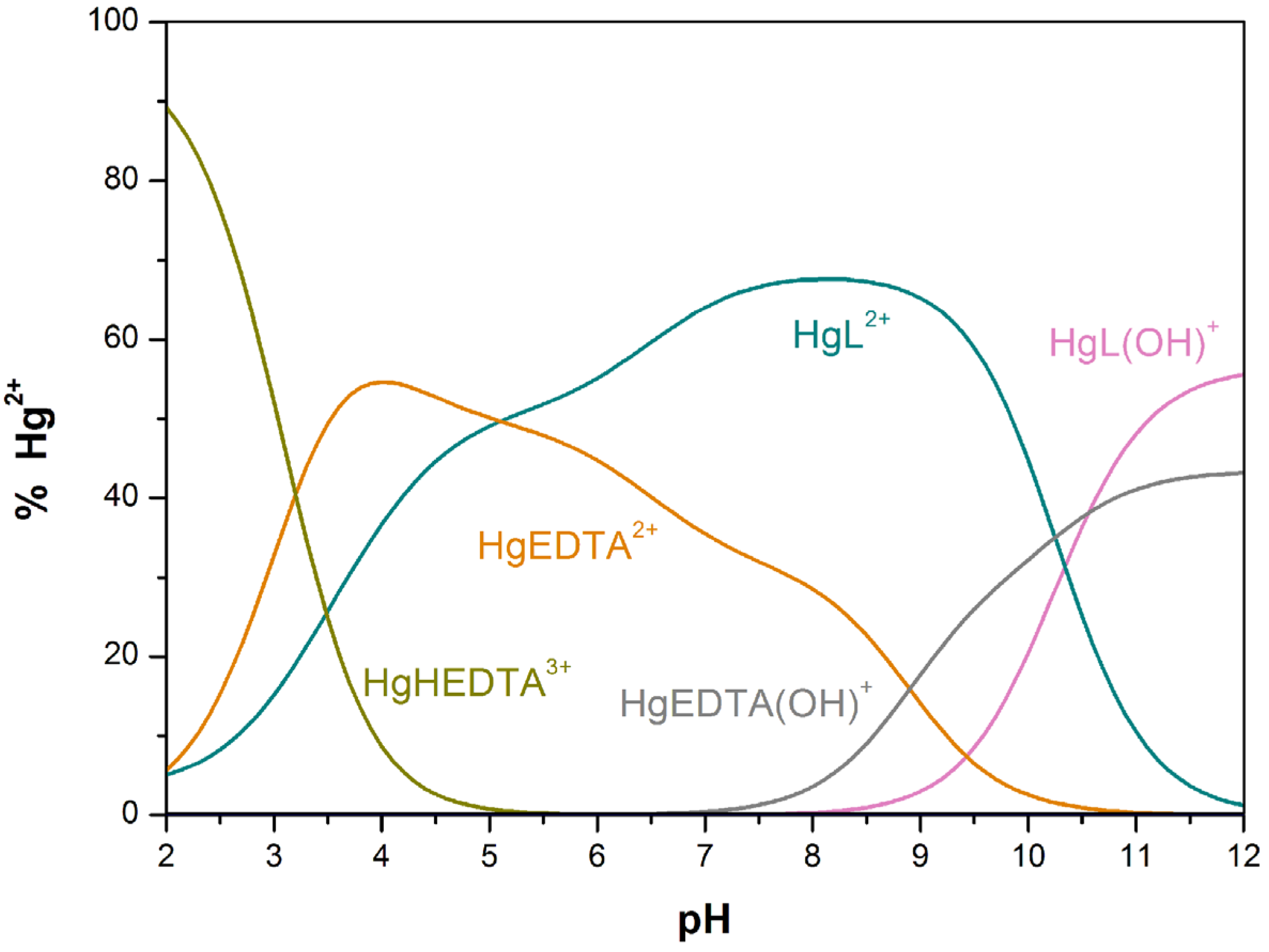
| Metal ion | [15]aneN4S | [15]aneN4O | [15]aneN5 |
|---|---|---|---|
| Mn2+ | 5.02 | 5.31 | 6.38 |
| Fe2+ | 6.85 | 5.74 | - |
| Co2+ | 10.32 | 9.07 | 12.25 |
| Ni2+ | 14.65 | 11.15 | 13.59 |
| Cu2+ | 19.01 | 16.69 | 23.49 |
| Zn2+ | 10.61 | 9.56 | 14.59 |
| Cd2+ | 10.32 | 9.76 | 14.69 |
| Hg2+ | 20.44 | - | 23.99 |
| Pb2+ | 9.3 | 8.63 | 12.79 |
2.4. Spectroscopic Studies
| Complex; (colour) | pH | UV-visible-near IR a λmax/nm (ε, M−1 cm−1) | µ (MB) |
|---|---|---|---|
| Co[15]aneN4S2+ (pink) | 6.99 | 1075 (4.2), 970 (sh., 4.9), 504 (67.6), 491 (sh., 99.0), 325 (sh., 2.61 × 103), 270 (sh., 2.61 × 103). | 4.9 |
| Ni[15]aneN4S2+ (yellow) | 6.98 | 1040 (2.12), 945 (23.0), 847 (sh., 27.2), 813 (sh., 26.0), 528 (19.3), 310 (sh., 1.49 × 103), 264 (1.91 × 104). | 3.1 |
| Cu[15]aneN4S2+ (purple) | 7.08 | 977 (23.8), 748 (sh., 16.3), 562 (45.6), 273 (1.17 × 103). | 1.8 |
 (equal to the difference between 10Dqxy and 35/4Dt). The octahedral field splitting parameter 10Dq and the values of the equatorial (Dqxy) and axial (Dqz) ligand field were calculated according to those assignments: 10Dq = 18975 cm−1, Dqxy = 1898 cm−1 and Dqz = 219 cm−1. These values, together with the ratio ν1/ν2 of 1.79 (ν1 and ν2 being the near IR and the visible absorption bands, respectively) and the magnetic moment of 3.1 MB, are characteristic of six coordinate nickel(II) environments, indicating a tetragonal (D4h) distorted octahedral geometry for this complex [43].
(equal to the difference between 10Dqxy and 35/4Dt). The octahedral field splitting parameter 10Dq and the values of the equatorial (Dqxy) and axial (Dqz) ligand field were calculated according to those assignments: 10Dq = 18975 cm−1, Dqxy = 1898 cm−1 and Dqz = 219 cm−1. These values, together with the ratio ν1/ν2 of 1.79 (ν1 and ν2 being the near IR and the visible absorption bands, respectively) and the magnetic moment of 3.1 MB, are characteristic of six coordinate nickel(II) environments, indicating a tetragonal (D4h) distorted octahedral geometry for this complex [43].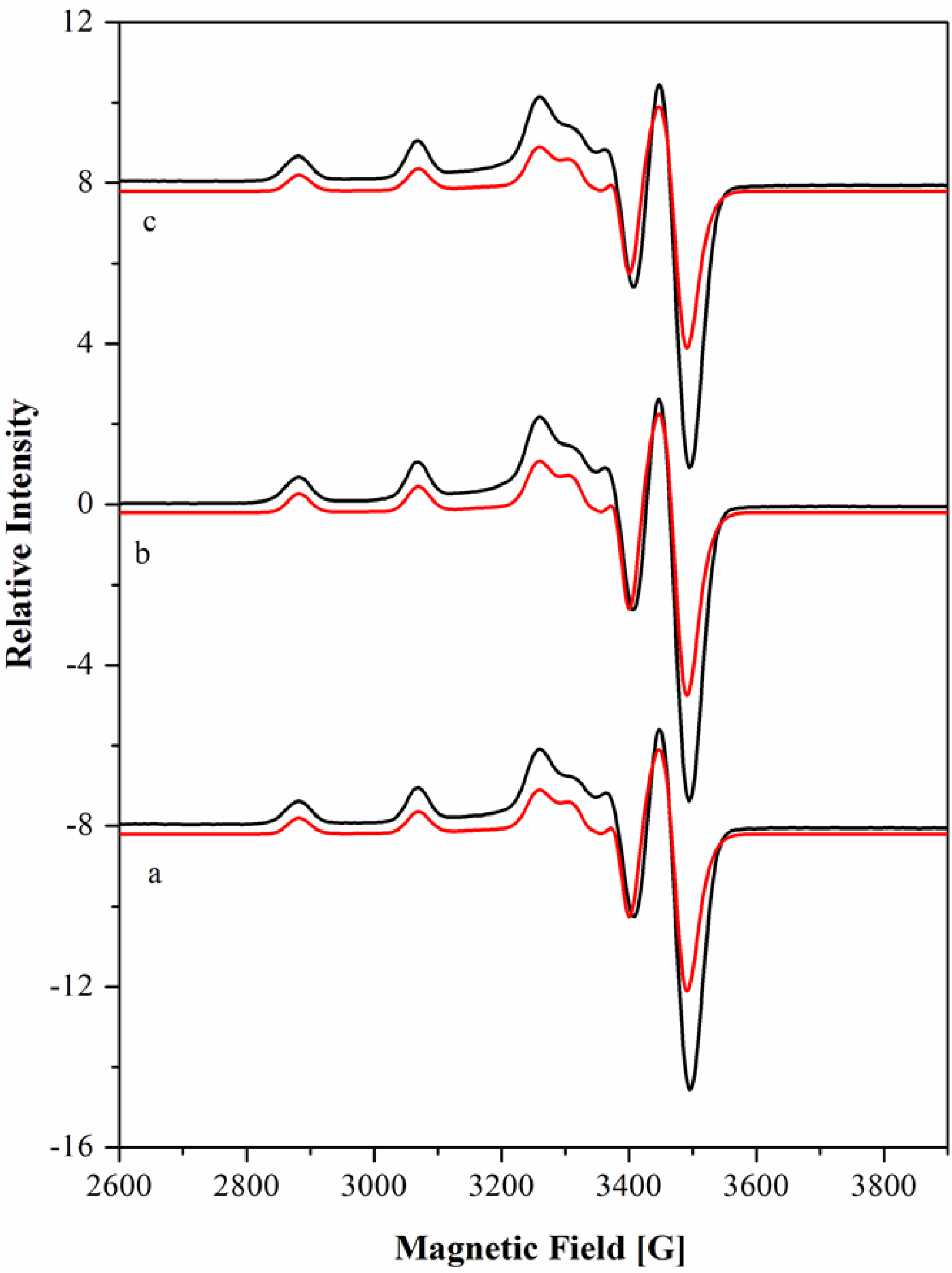
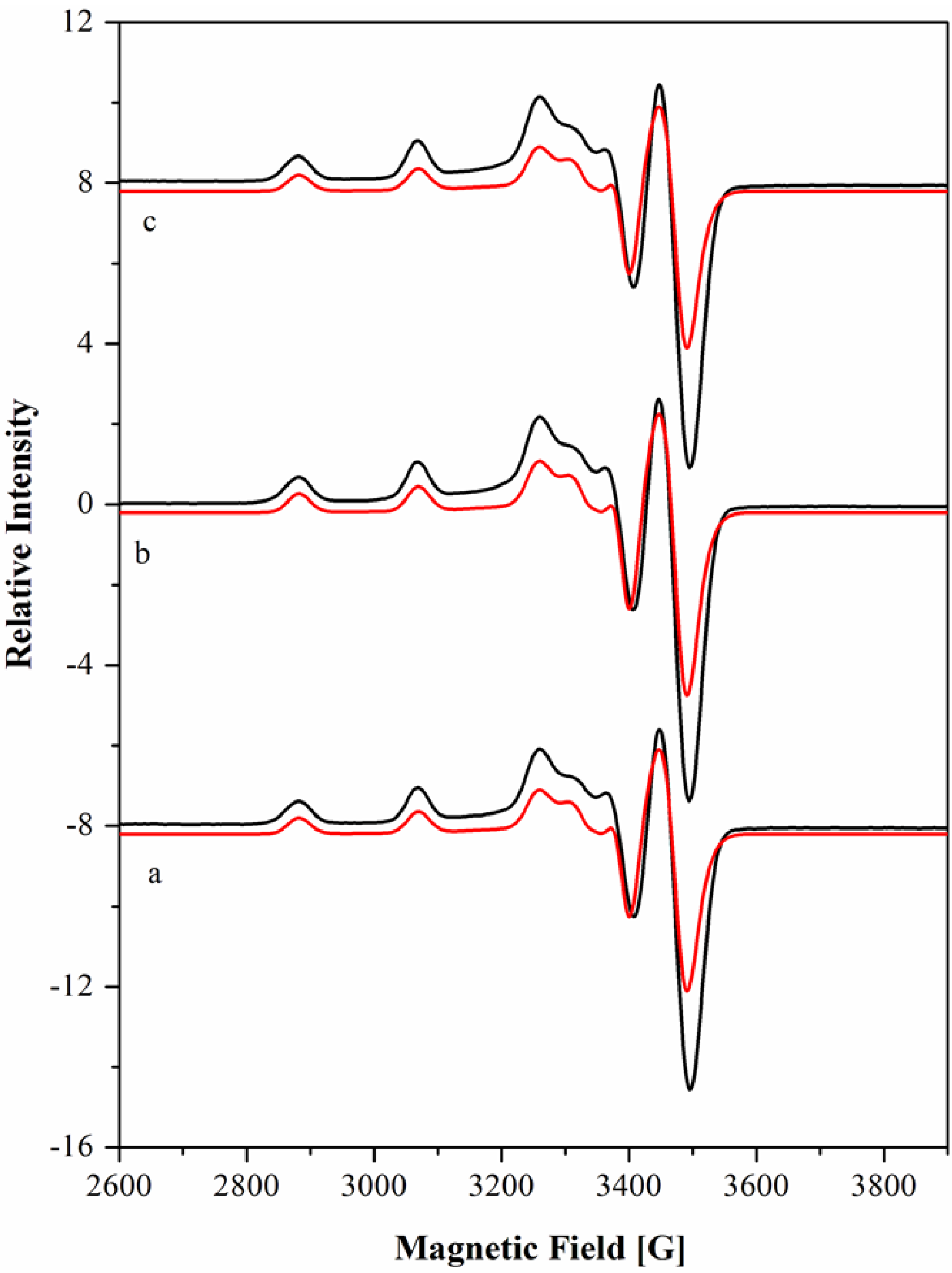
| Complex | pH | Visible band λmax/nm (εmolar, M−1 cm−1) | EPR parameters (Ai × 104 cm−1) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| gx | gy | gz | Ax | Ay | Az | ref. | |||
| 9.00 | 2.043 | 2.046 | 2.186 | 30.6 | 28.7 | 192.5 | - | ||
| Cu[15]aneN4S2+ | 7.40 | 565 (46) | 2.042 | 2.045 | 2.182 | 30.3 | 28.0 | 191.1 | - |
| 4.10 | 2.041 | 2.042 | 2.185 | 30.4 | 28.0 | 191.3 | - | ||
| Cu[15]aneN4O2+ | 7.41 | 580 (141) | 2.042 | 2.046 | 2.192 | 30.0 | 27.9 | 199.4 | [45] |
3. Experimental
3.1. General
3.1.1. Reagents
3.2. Synthesis of 1-Thia-4,7,10,13-tetraazacyclopentadecane ([15]aneN4S)
3.3. Potentiometric Studies
3.3.1. Reagents and Solutions
3.3.2. Measurements
3.3.3. Calculation of Equilibrium Constants
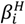 and
and 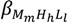 (being = [MmHhLl]/[M]m [H]h [L]l) were calculated by fitting the potentiometric data from protonation or complexation titrations with the HYPERQUAD program [34]. Species distribution diagrams were plotted from the calculated constants with the HySS program [37]. Only mononuclear species, ML, MHL and MH-1L were found for the metal complexes of [15]aneN4S (being βMH-1L = βMLOH × Kw). The hydrolysis constants of the metal ions were taken from literature and kept constant for the calculations. Differences, in log units, between the values of βMHL (or βMH-1L) and βML constants, provide the stepwise reaction constants. The species considered in a particular model were those that could be justified by the principles of coordination chemistry. The errors quoted are the standard deviations of the overall stability constants given directly by the program for the input data, which include all the experimental points of all titration curves, and were determined by the normal propagation rules for the stepwise constants.
(being = [MmHhLl]/[M]m [H]h [L]l) were calculated by fitting the potentiometric data from protonation or complexation titrations with the HYPERQUAD program [34]. Species distribution diagrams were plotted from the calculated constants with the HySS program [37]. Only mononuclear species, ML, MHL and MH-1L were found for the metal complexes of [15]aneN4S (being βMH-1L = βMLOH × Kw). The hydrolysis constants of the metal ions were taken from literature and kept constant for the calculations. Differences, in log units, between the values of βMHL (or βMH-1L) and βML constants, provide the stepwise reaction constants. The species considered in a particular model were those that could be justified by the principles of coordination chemistry. The errors quoted are the standard deviations of the overall stability constants given directly by the program for the input data, which include all the experimental points of all titration curves, and were determined by the normal propagation rules for the stepwise constants.3.4. Spectroscopic Studies
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Hayes, R.B. The carcinogenicity of metals in humans. Cancer Cause. Control 1997, 8, 371–385. [Google Scholar] [CrossRef]
- Järup, L. Hazards of heavy metal contamination. Br. Med. Bull. 2003, 68, 167–182. [Google Scholar] [CrossRef]
- Caussy, D.; Gochfeld, M.; Gurzau, E.; Neagu, C.; Ruedel, H. Lessons from case studies of metals: Investigating exposure, Bioavailability, and risk. Ecotoxicol. Environ. Saf. 2003, 56, 45–51. [Google Scholar] [CrossRef]
- Blanuša, M.; Varnai, V.M.; Piasek, M.; Kostial, K. Chelators as antidotes of metal toxicity: Therapeutic and experimental aspects. Curr. Med. Chem. 2005, 12, 2771–2794. [Google Scholar] [CrossRef]
- Flora, S.J.S.; Pachauri, V. Chelation in metal intoxication. Int. J. Environ. Res. Public Health 2010, 7, 2745–2788. [Google Scholar] [CrossRef]
- Sears, M.E. Chelation: Harnessing and enhancing heavy metal detoxification—A review. Sci. World J. 2013, 2013, 1–13. [Google Scholar] [CrossRef]
- Fernandes, A.S.; Cabral, M.F.; Costa, J.; Castro, M.; Delgado, R.; Drew, M.G.; Felix, V. Two macrocyclic pentaaza compounds containing pyridine evaluated as novel chelating agents in copper(II) and nickel(II) overload. J. Inorg. Biochem. 2011, 105, 410–419. [Google Scholar] [CrossRef]
- Andersen, O.; Aaseth, J. Molecular mechanisms of in vivo metal chelation: Implications for clinical treatment of metal intoxications. Environ. Health. Perspect. 2002, 110, 887–890. [Google Scholar] [CrossRef]
- Andersen, O. Chemical and biological considerations in the treatment of metal intoxications by chelating agents. Mini Rev. Med. Chem. 2004, 4, 11–21. [Google Scholar] [CrossRef]
- Aposhian, H.V.; Maiorino, R.M.; Gonzalez-Ramirez, D.; Zuniga-Charles, M.; Xu, Z.; Hurlbut, K.M.; Junco-Munoz, P.; Dart, R.C.; Aposhian, M.M. Mobilization of heavy metals by newer, Therapeutically useful chelating agents. Toxicology 1995, 97, 23–38. [Google Scholar] [CrossRef]
- Gonçalves, S.; Fernandes, A.S.; Oliveira, N.G.; Marques, J.; Costa, J.; Cabral, M.F.; Miranda, J.; Cipriano, M.; Guerreiro, P.S.; Castro, M. Cytotoxic effects of cadmium in mammary epithelial cells: Protective role of the macrocycle [15]pyN5. Food Chem. Toxicol. 2012, 50, 2180–2187. [Google Scholar] [CrossRef]
- Mewis, R.E.; Archibald, S. Biomedical applications of macrocyclic ligand complexes. Coord. Chem. Rev. 2010, 254, 1686–1712. [Google Scholar] [CrossRef]
- Hancock, R.D.; Martell, A.E. Ligand design for selective complexation of metal ions in aqueous solution. Chem. Rev. 1989, 89, 1875–1914. [Google Scholar] [CrossRef]
- Hancock, R.D.; Dobson, S.M.; Boeyens, J.C.A. Metal ion size selectivity of 1-Thia-4, 7-diazacyclononane (9-aneN2S), and other tridentate macrocycles. A study by molecular mechanics calculation, structure determination, and formation constant determination of complexes of 9-aneN2S. Inorganica Chim. Acta 1987, 133, 221–231. [Google Scholar] [CrossRef]
- Westerby, B.C.; Juntunen, K.L.; Leggett, G.H.; Pett, V.B.; Koenigbauer, M.J.; Purgett, M.D.; Taschner, M.J.; Ochrymowycz, L.A.; Rorabacher, D.B. Macrocyclic polyamino polythiaether ligands with NxS4-x and NxS5-x donor sets: Protonation constants, stability constants, and kinetics of complex formation with the aquocopper(II) ion. Inorg. Chem. 1991, 30, 2109–2120. [Google Scholar] [CrossRef]
- Arnaud-Neu, F.; Schwing-Weill, M.J.; Louis, R.; Weiss, R. Thermodynamic and spectroscopic properties in aqueous solutions of pentadentate macrocyclic complexes. Inorg. Chem. 1979, 18, 2956–2961. [Google Scholar] [CrossRef]
- Balakrishnan, K.P.; Kaden, T.A.; Siegfried, L.; Zuberbühler, A.D. Stabilities and redox properties of Cu(I) and Cu(II) complexes with macrocyclic ligands containing the N2S2 donor set. Helv. Chim. Acta 1984, 67, 1060–1069. [Google Scholar] [CrossRef]
- Walker, T.L.; Malasi, W.; Bhide, S.; Parker, T.; Zhang, D.; Freedman, A.; Modarelli, J.M.; Engle, J.T.; Ziegler, C.J.; Custer, P.; et al. Synthesis and characterization of 1,8-dithia-4,11-diazacyclotetradecane. Tetrahedron Lett. 2012, 53, 6548–6551. [Google Scholar] [CrossRef]
- Papini, G.; Alidori, S.; Lewis, J.S.; Reichert, D.E.; Pellei, M.; Lobbia, G.G.; Biddlecombe, G.B.; Anderson, C.J.; Santini, C. Synthesis and characterization of the copper(II) complexes of new N2S2-donor macrocyclic ligands: Synthesis and in vivo evaluation of the 64Cu complexes. Dalton Trans. 2009, 7, 177–184. [Google Scholar]
- Aquilanti, G.; Giorgetti, M.; Minicucci, M.; Papini, G.; Pellei, M.; Tegoni, M.; Trasatti, A.; Santini, C. A study on the coordinative versatility of new N,S-donor macrocyclic ligands: XAFS, and Cu2+ complexation thermodynamics in solution. Dalton Trans. 2011, 40, 2764–2777. [Google Scholar] [CrossRef]
- Kodama, M.; Koike, T.; Hoshiga, N.; Machida, R.; Kimura, E. Metal chelates of sulphur-containing polyamine macrocycles and oxygenation of the corresponding cobalt(II) complexes. J. Chem. Soc. Dalton Trans. 1984, 673–678. [Google Scholar]
- Vollhardt, K.P.C.; Schore, N.E. Organic Chemistry, 6th ed.; Freeman and Co.: New York, NY, USA, 2011. [Google Scholar]
- Tabushi, I.; Okino, H.; Kuroda, Y. Convenient synthesis of macrocyclic-compounds containing two of nitrogen, Oxygen or sulphur atoms. Tetrahedron Lett. 1976, 17, 4339–4342. [Google Scholar] [CrossRef]
- Steenland, M.W.A.; Westbroek, P.; Dierck, I.; Herman, G.G.; Lippens, W.; Temmerman, E.; Goeminne, A.M. Aqueous solution study of Cu(II) and Ni(II) complexes of macrocyclic oxa- and thia- containing trans-dioxo-tetraamines. Polyhedron 1999, 18, 3417–3424. [Google Scholar]
- Bencini, A.; Bianchi, A.; Garcia-España, E.; Micheloni, M.; Ramirez, J.A. Proton coordination by polyamine compounds in aqueous solution. Coord. Chem. Rev. 1999, 188, 97–156. [Google Scholar] [CrossRef]
- Cabral, M.F.; Delgado, R. Metal complexes of pentadentate macrocyclic ligands containing oxygen and nitrogen as donor atoms. Helv. Chim. Acta 1994, 77, 515–524. [Google Scholar] [CrossRef]
- Motekaitis, R.J.; Rogers, B.E.; Reichert, D.E.; Martell, A.E.; Welch, M.J. Stability and structure of activated macrocycles. Ligands with biological applications. Inorg. Chem. 1996, 35, 3821–3827. [Google Scholar] [CrossRef]
- Delgado, R.; Fraústo da Silva, J.J.R.; Amorim, M.T.S.; Cabral, M.F.; Chaves, S.; Costa, J. Dissociation constants of Brønsted acids in D2O and H2O: Studies on polyaza and polyoxa-polyaza macrocycles and a general correlation. Anal. Chim. Acta 1991, 245, 271–282. [Google Scholar]
- Hancock, R.D.; Wade, P.W.; Ngwenya, M.P.; de Sousa, A.S.; Damu, K.V. Ligand design for complexation in aqueous solution. 2. Chelate ring size as a basis for control of size-based selectivity for metal ions. Inorg. Chem. 1990, 29, 1968–1974. [Google Scholar] [CrossRef]
- Riley, D.P.; Henke, S.L.; Lennon, P.J.; Weiss, R.H.; Neumann, W.L.; Rivers, W.J., Jr.; Aston, K.W.; Sample, K.R.; Rahman, H.; Ling, C.S.; et al. Synthesis, characterization, and stability of manganese(II) C-substituted 1,4,7,10,13-pentaazacyclopentadecane complexes exhibiting superoxide dismutase activity. Inorg. Chem. 1996, 35, 5213–5231. [Google Scholar] [CrossRef]
- Kodama, M.; Kimura, E. Effects of axial ligation on molecular oxygen binding by donor atoms built in saturated macrocycles. Equilibrium and kinetic study with cobalt(II) complexes of macrocyclic pentaamines and oxatetraamine. Inorg. Chem. 1980, 19, 1871–1875. [Google Scholar] [CrossRef]
- Kodama, M.; Kimura, E.; Yamaguchi, S. Complexation of the macrocyclic hexa-amine ligand 1,4,7,10,13,16-hexa-azacyclo-octadecane(“18-azacrown-6”). J. Chem. Soc. Dalton Trans. 1980, 2536–2538. [Google Scholar] [CrossRef]
- Kodama, M.; Kimura, E. Equilibria of complex formation between several bivalent metal ions and macrocyclic tri- and penta-amines. J. Chem. Soc. Dalton Trans. 1978, 1081–1085. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Kodama, M.; Kimura, E. Effects of cyclization and ring size on complex formation between penta-amine ligands and copper(II). J. Chem. Soc. Dalton Trans. 1978, 104–110. [Google Scholar] [CrossRef]
- Costa, J.; Delgado, R.; Drew, M.G.B.; Félix, V. Design of selective macrocyclic ligands for the divalent first-row transition-metal ions. J. Chem. Soc. Dalton Trans. 1998, 1063–1071. [Google Scholar]
- Alderighi, L.; Gans, P.; Ienco, A.; Peters, D.; Sabatini, A.; Vacca, A. Hyperquad simulation and speciation (HySS): A utility program for the investigation of equilibria involving soluble and partially soluble species. Coord. Chem. Rev. 1999, 184, 311–318. [Google Scholar] [CrossRef]
- Evans, D.F. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J. Chem. Soc. 1959, 2003–2005. [Google Scholar] [CrossRef]
- Lever, A.B.P. Inorganic Electronic Spectroscopy, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 1984. [Google Scholar]
- Banci, L.; Bencini, A.; Benelli, C.; Gatteschi, D.; Zanchini, C. Spectral-structural correlations in high-spin cobalt(II) complexes. Sruct. Bond. 1982, 52, 37–86. [Google Scholar]
- Bertini, I.; Luchinat, C. High spin cobalt(II) as a probe for the investigation of metalloproteins. Adv. Inorg. Biochem. 1984, 6, 71–111. [Google Scholar]
- Martin, L.Y.; Sperati, C.R.; Busch, D.H. The spectrochemical properties of tetragonal complexes of high spin nickel(II) containing macrocyclic ligands. J. Am. Chem. Soc. 1977, 99, 2968–2981. [Google Scholar] [CrossRef]
- Sacconi, L.; Mani, F.; Bencini, A. Nickel. In Comprehensive Coordination Chemistry; Wilkinson, G., Gillard, R.D., McCleverty, J.A., Eds.; Pergamon Press: Oxford, UK, 1987; pp. 1–137. [Google Scholar]
- Neese, F. Electronic Structure and Spectroscopy of Novel Copper Chromophores in Biology. Diploma Thesis, University of Konstanz, Konstanz, Germany, June 1993. [Google Scholar]
- Fernandes, A.S.; Gaspar, J.; Cabral, M.F.; Caneiras, C.; Guedes, R.; Rueff, J.; Castro, M.; Costa, J.; Oliveira, N.G. Macrocyclic copper(II) complexes: Superoxide scavenging activity, Structural studies and cytotoxicity evaluation. J. Inorg. Biochem. 2007, 101, 849–858. [Google Scholar] [CrossRef]
- Li, Y. X-Ray structures and spectroscopic studies of diaqua- and dichlorocopper(II) complexes of 15 crown 5 and 4' substituted benzo-15-Crown-5 with a 3dx(x=z2) ground state doublet. Bull. Chem. Soc. Jpn. 1996, 69, 2513–2523. [Google Scholar] [CrossRef]
- Barbaro, P.; Bianchini, C.; Capannesi, G.; di Luca, L.; Laschi, F.; Petroni, D.; Salvadori, P.A.; Vacca, A.; Vizza, F. Synthesis and characterization of the tetraazamacrocycle 4,10-dimethyl-1,4,7,10-tetraazacyclododecane-1,7-diacetic acid (H2Me2DO2A) and of its neutral copper(II) complex [Cu(Me2DO2A)]. A new 64Cu-labeled macrocyclic complex for positron emission tomography imaging. J. Chem. Soc. Dalton Trans. 2000, 2393–2401. [Google Scholar]
- Hathaway, B.J. Copper. Coord. Chem. Rev. 1983, 52, 87–169. [Google Scholar] [CrossRef]
- Gersmann, H.R.; Swalen, J.D. Electron paramagnetic resonance spectra of copper complexes. J. Chem. Phys. 1962, 36, 3221–3233. [Google Scholar] [CrossRef]
- Yokoi, H.; Sai, M.; Isobe, T.; Ohsawa, S. ESR studies of the copper(II) complexes of amino acids. Bull. Chem. Soc. Jpn. 1972, 45, 2189–2195. [Google Scholar] [CrossRef]
- Lau, P.W.; Lin, W.C. Electron spin resonance and electronic structure of some metalloporphyrins. J. Inorg. Nucl. Chem. 1975, 37, 2389–2398. [Google Scholar]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals, 3rd ed.; Pergamon Press: Oxford, UK, 1988. [Google Scholar]
- Schwarzenbach, G.; Flaschka, W. Complexometric Titrations, 2nd ed.; Methuen & Co: London, UK, 1969. [Google Scholar]
- Rossotti, F.J.C.; Rossotti, H. Potentiometric titrations using Gran plots: A textbook omission. J. Chem. Educ. 1965, 42, 375–378. [Google Scholar] [CrossRef]
- Delgado, R.; do Carmo Figueira, M.; Quintino, S. Redox method for the determination of stability constants of some trivalent metal complexes. Talanta 1997, 45, 451–462. [Google Scholar] [CrossRef]
- Pettit, L.D.; Powell, H.K.J. IUPAC Stability Constants Database; Academic Software: Timble, Otley, Yorks, UK, 2003. [Google Scholar]
- Frassineti, C.; Ghelli, S.; Gans, P.; Sabatini, A.; Moruzzi, M.S.; Vacca, A. Nuclear magnetic resonance as a tool for determining protonation constants of natural polyprotic bases in solution. Anal. Biochem. 1995, 231, 374–382. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Torres, N.; Gonçalves, S.; Fernandes, A.S.; Machado, J.F.; De Brito, M.J.V.; Oliveira, N.G.; Castro, M.; Costa, J.; Cabral, M.F. [15]aneN4S: Synthesis, Thermodynamic Studies and Potential Applications in Chelation Therapy. Molecules 2014, 19, 550-567. https://doi.org/10.3390/molecules19010550
Torres N, Gonçalves S, Fernandes AS, Machado JF, De Brito MJV, Oliveira NG, Castro M, Costa J, Cabral MF. [15]aneN4S: Synthesis, Thermodynamic Studies and Potential Applications in Chelation Therapy. Molecules. 2014; 19(1):550-567. https://doi.org/10.3390/molecules19010550
Chicago/Turabian StyleTorres, Nuno, Sandrina Gonçalves, Ana S. Fernandes, J. Franco Machado, Maria J. Villa De Brito, Nuno G. Oliveira, Matilde Castro, Judite Costa, and Maria F. Cabral. 2014. "[15]aneN4S: Synthesis, Thermodynamic Studies and Potential Applications in Chelation Therapy" Molecules 19, no. 1: 550-567. https://doi.org/10.3390/molecules19010550
APA StyleTorres, N., Gonçalves, S., Fernandes, A. S., Machado, J. F., De Brito, M. J. V., Oliveira, N. G., Castro, M., Costa, J., & Cabral, M. F. (2014). [15]aneN4S: Synthesis, Thermodynamic Studies and Potential Applications in Chelation Therapy. Molecules, 19(1), 550-567. https://doi.org/10.3390/molecules19010550