Enantioselective Synthesis of 2,2-Disubstituted Terminal Epoxides via Catalytic Asymmetric Corey-Chaykovsky Epoxidation of Ketones

Abstract
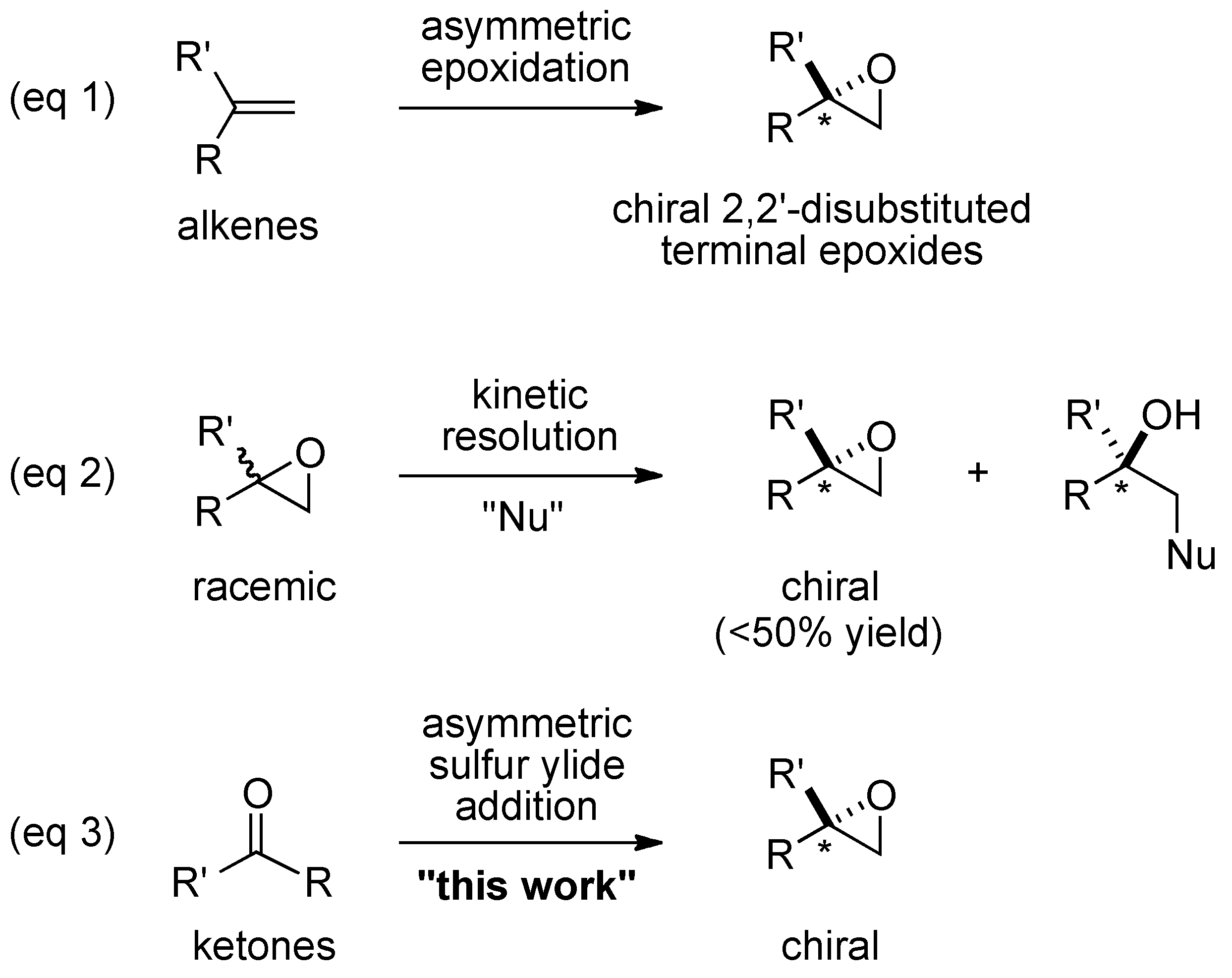
:1. Introduction
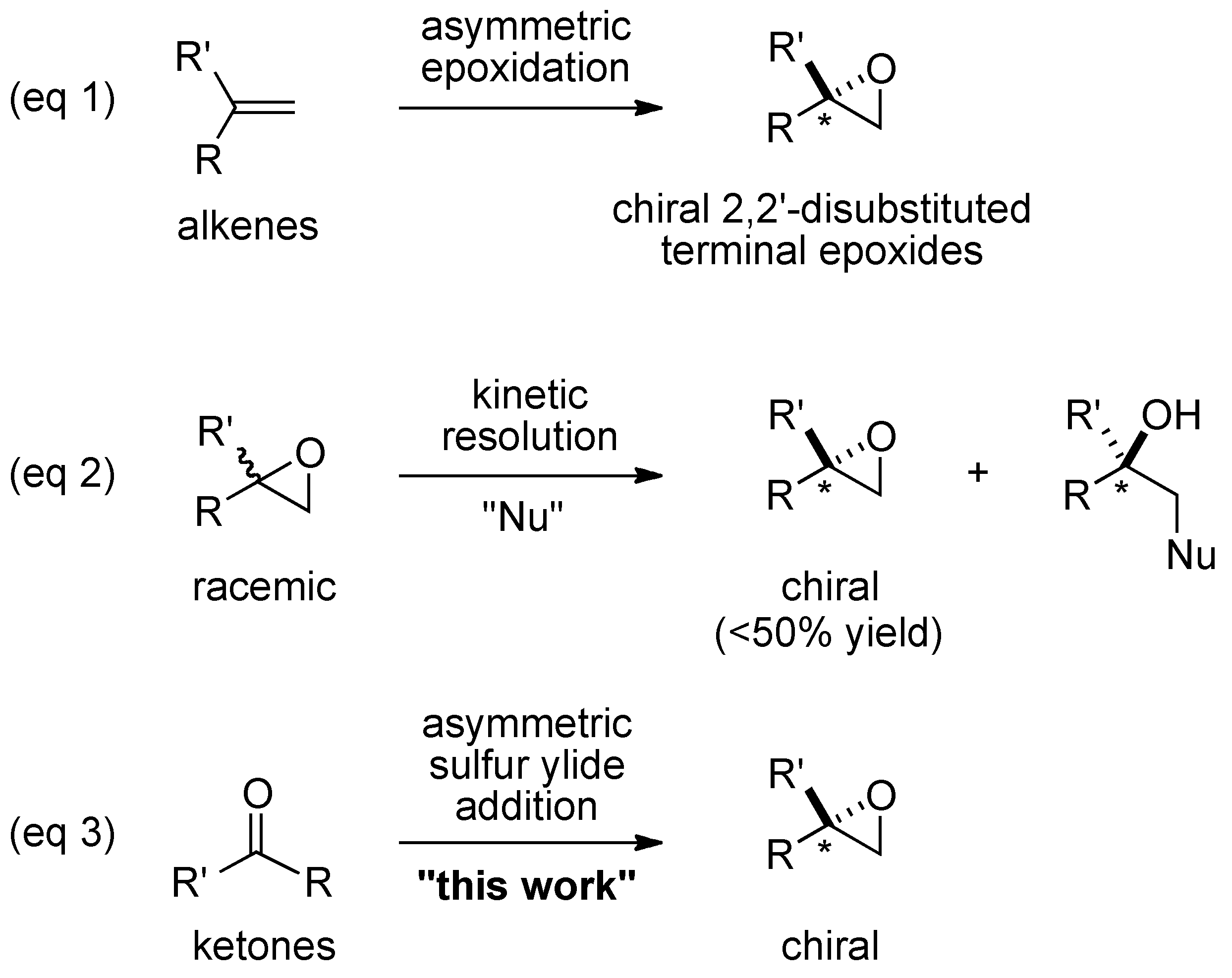

2. Results and Discussion
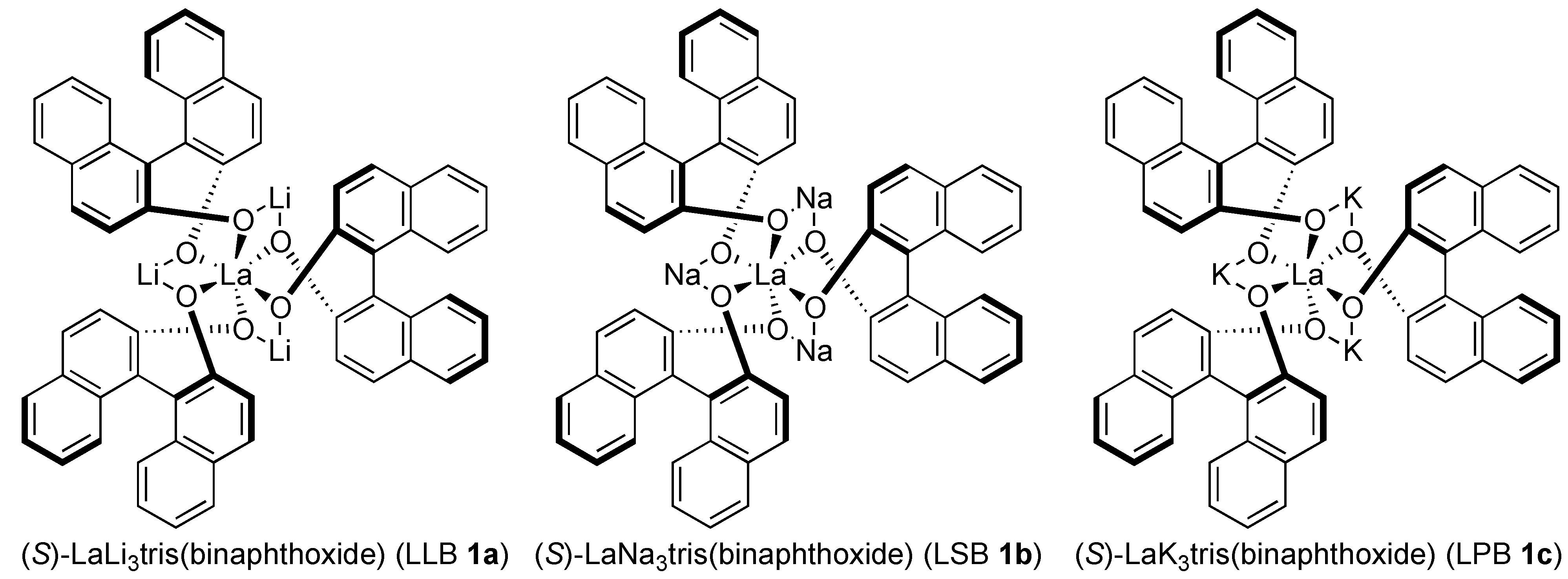
2.1. Optimization Studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | REMB catalyst | R: (mol%) | 5 | time (h) | % yield a | % ee |
|---|---|---|---|---|---|---|
| 1 b | LLB 1a | none | 48 | 79 | 15 | |
| 2 | LLB 1a | none | 12 | 80 | 72 | |
| 3 | LSB 1b | none | 12 | 25 | 14 | |
| 4 | LPB 1c | none | 12 | 17 | 52 | |
| 5 | LLB 1a | Ph- (5) | 5a | 12 | 77 | 80 |
| 6 | LLB 1a | 4-Cl-C6H4- (5) | 5b | 12 | 99 | 74 |
| 7 | LLB 1a | C6F5- (5) | 5c | 12 | 61 | 48 |
| 8 | LLB 1a | n-butyl (5) | 5d | 12 | 87 | 75 |
| 9 | LLB 1a | cyclohexyl (5) | 5e | 12 | 99 | 76 |
| 10 | LLB 1a | 2,4,6-Me3-C6H2- (5) | 5f | 12 | 97 | 75 |
| 11 | LLB 1a | 4-MeO-C6H4- (5) | 5g | 12 | 82 | 77 |
| 12 | LLB 1a | 2,6-(MeO)2-C6H3- (5) | 5h | 12 | 84 | 93 |
| 13 | LLB 1a | 2,4,6-(MeO)3-C6H2- (5) | 5i | 12 | 98 c | 96 |
| 14 | LLB 1a | 2,4,6-(MeO)3-C6H2- (10) | 5i | 12 | 94 | 95 |
| 15 | LLB 1a | 2,4,6-(MeO)3-C6H2- (15) | 5i | 12 | 92 | 92 |
2.2. Substrate Scope and Limitations

| Entry | Ketone: R | 3 | Epoxide 4 | Cat. (x mol%) | Time (h) | % Yield b | % ee |
|---|---|---|---|---|---|---|---|
| 1 | Ph | 3a | 4a | 5 | 12 | 98 | 96 |
| 2 | 2-naphthyl | 3b | 4b | 5 | 12 | 97 | 96 |
| 3 | 2-naphthyl | 3b | 4b | 2.5 | 18 | 96 | 94 |
| 4 | 2-naphthyl | 3b | 4b | 1 | 60 | 96 | 92 |
| 5 | 4-Cl-C6H4 | 3c | 4c | 5 | 12 | >99 | 94 |
| 6 | 3-Cl-C6H4 | 3d | 4d | 5 | 12 | >99 | 94 |
| 7 | 2-Cl-C6H4 | 3e | 4e | 5 | 12 | 96 | 95 |
| 8 | 4-F-C6H4 | 3f | 4f | 5 | 12 | 94 | 97 |
| 9 | 4-EtO2C-C6H4 | 3g | 4g | 5 | 12 | 94 | 94 |
| 10c | 4-Me-C6H4 | 3h | 4h | 5 | 12 | 97 | 92 |
| 11 | 3-pyridyl | 3i | 4i | 5 | 12 | 97 | 92 |
| 12 | PhCH2CH2- | 3j | 4j | 5 | 12 | 99 e | 93 |
| 13 | n-octyl | 3k | 4k | 5 | 12 | >99 | 93 |
| 14 c | cyclohexyl | 3l | 4l | 5 | 12 | 88 d | 96 |
| 15 | 4-EtO2C-(CH2)3- | 3m | 4m | 5 | 12 | >99 | 91 |

| Entry | Ketone: R | R′ | 3 | Epoxide 4 | Time (h) | % Yield b | % ee |
|---|---|---|---|---|---|---|---|
| 1 | Ph | Et | 3n | 4n | 18 | 91 | 88 |
| 2 | 4-Cl-C6H4 | Et | 3o | 4o | 18 | 94 | 87 |
| 3 | 4-Br-C6H4 | Et | 3p | 4p | 18 | 92 | 85 |
| 4 | 3-Cl-C6H4 | Et | 3q | 4q | 18 | 96 | 81 |
| 5 | 2-F-C6H4 | Et | 3r | 4r | 18 | 88 | 67 |
| 6 | 3-pyridyl | Et | 3s | 4s | 18 | 89 | 73 |
| 7 | 4-Cl-C6H4 | nPr | 3t | 4t | 18 | 90 | 73 |
| 8 | Ph | iPr | 3u | 4u | 36 | 60 | 70 |
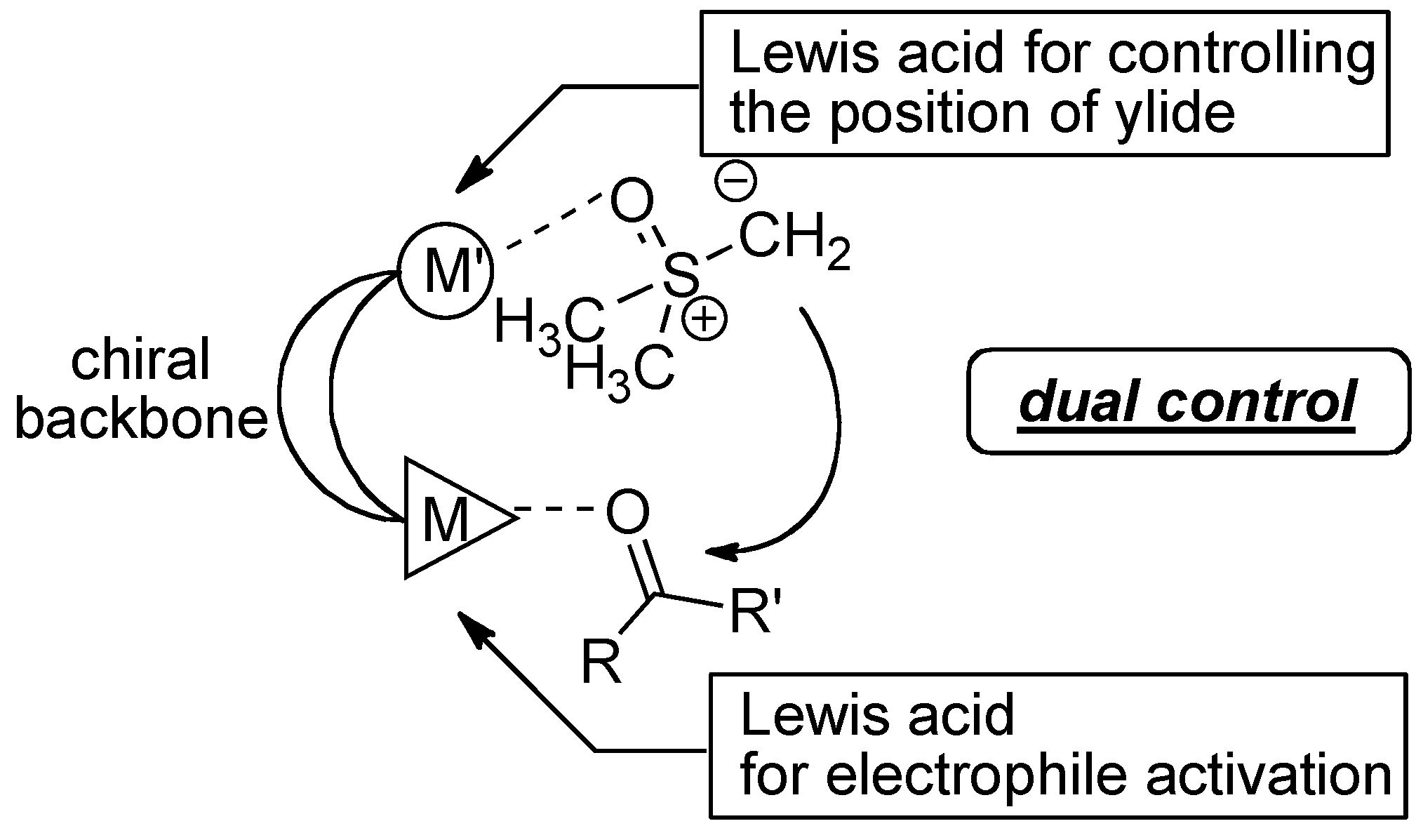
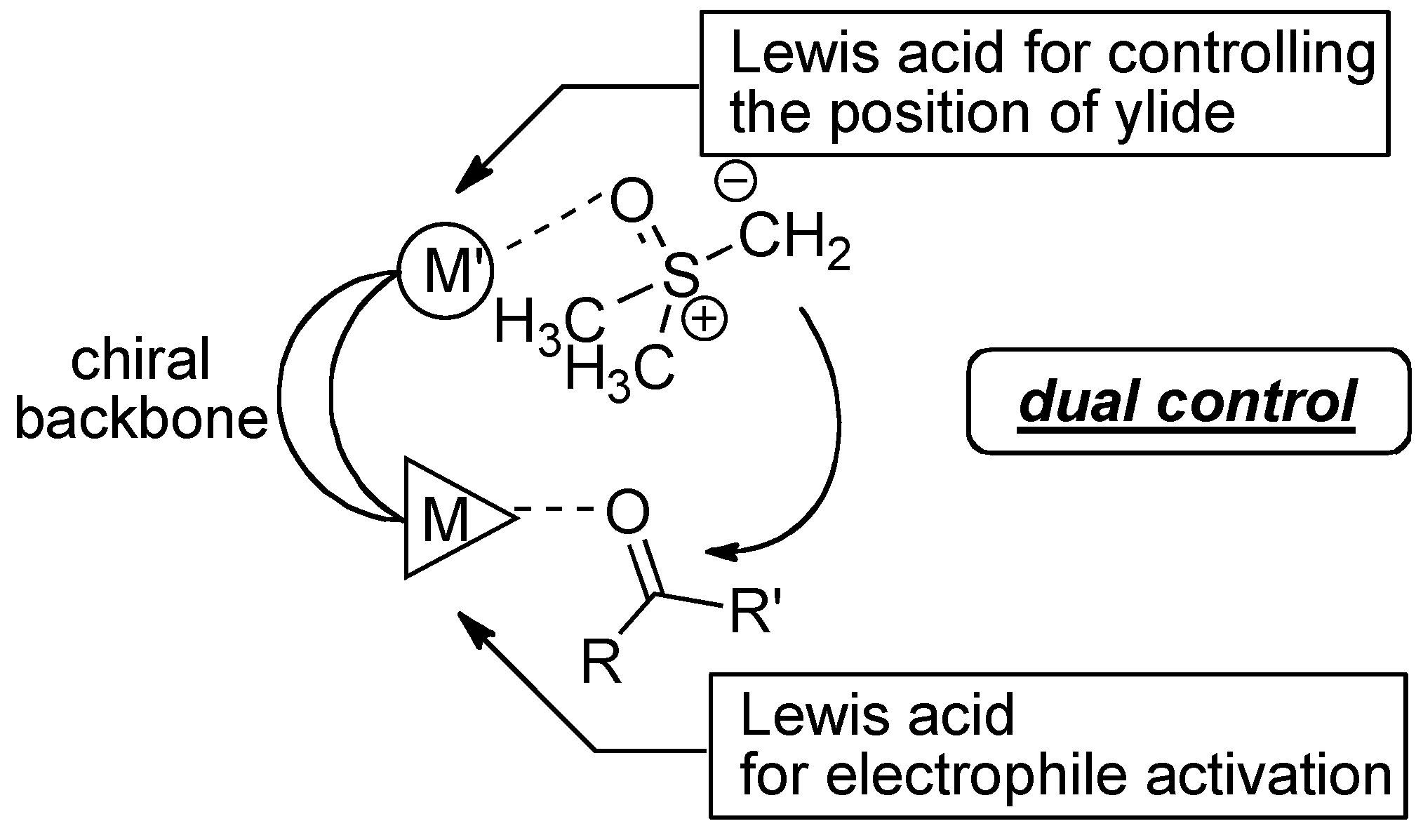
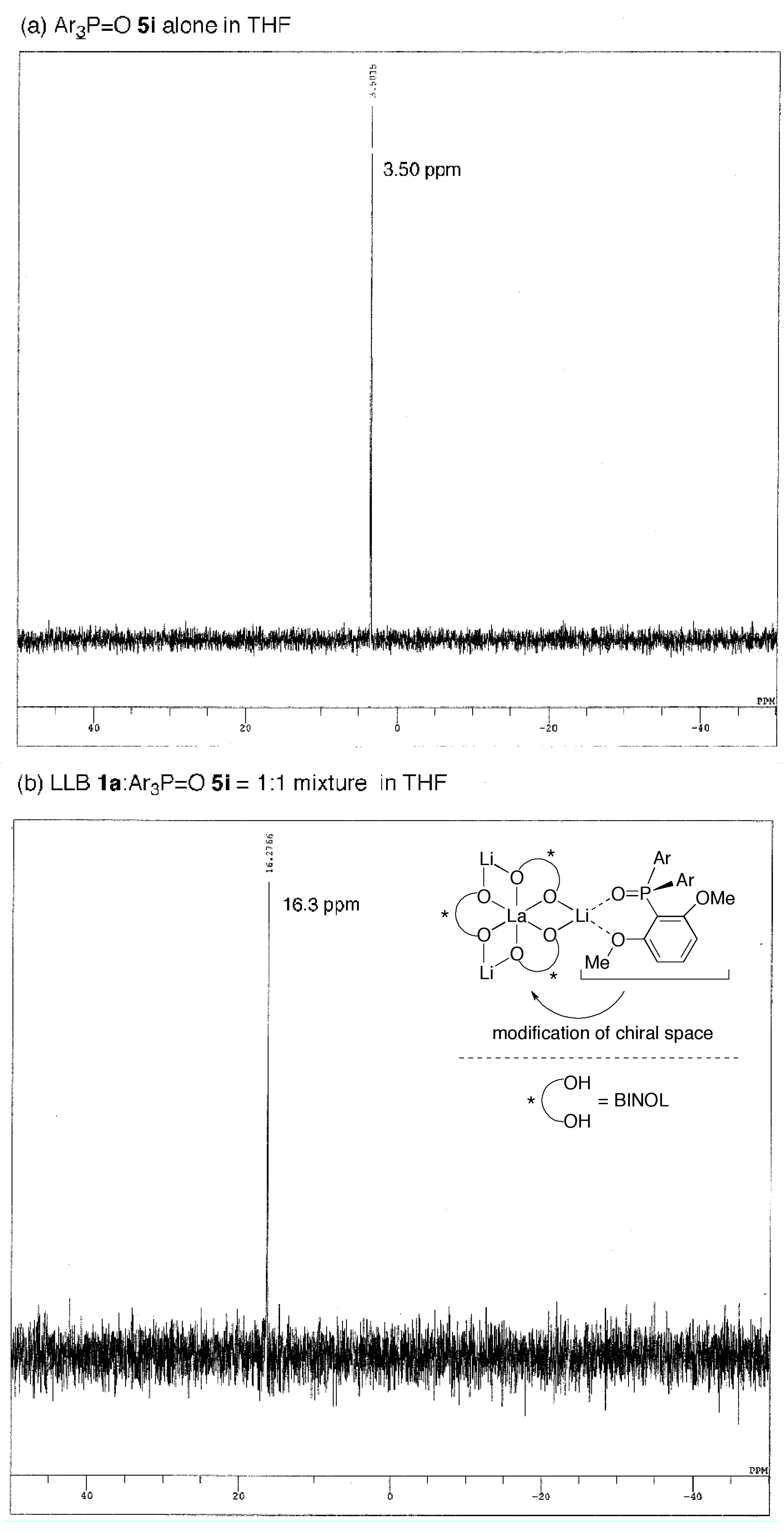
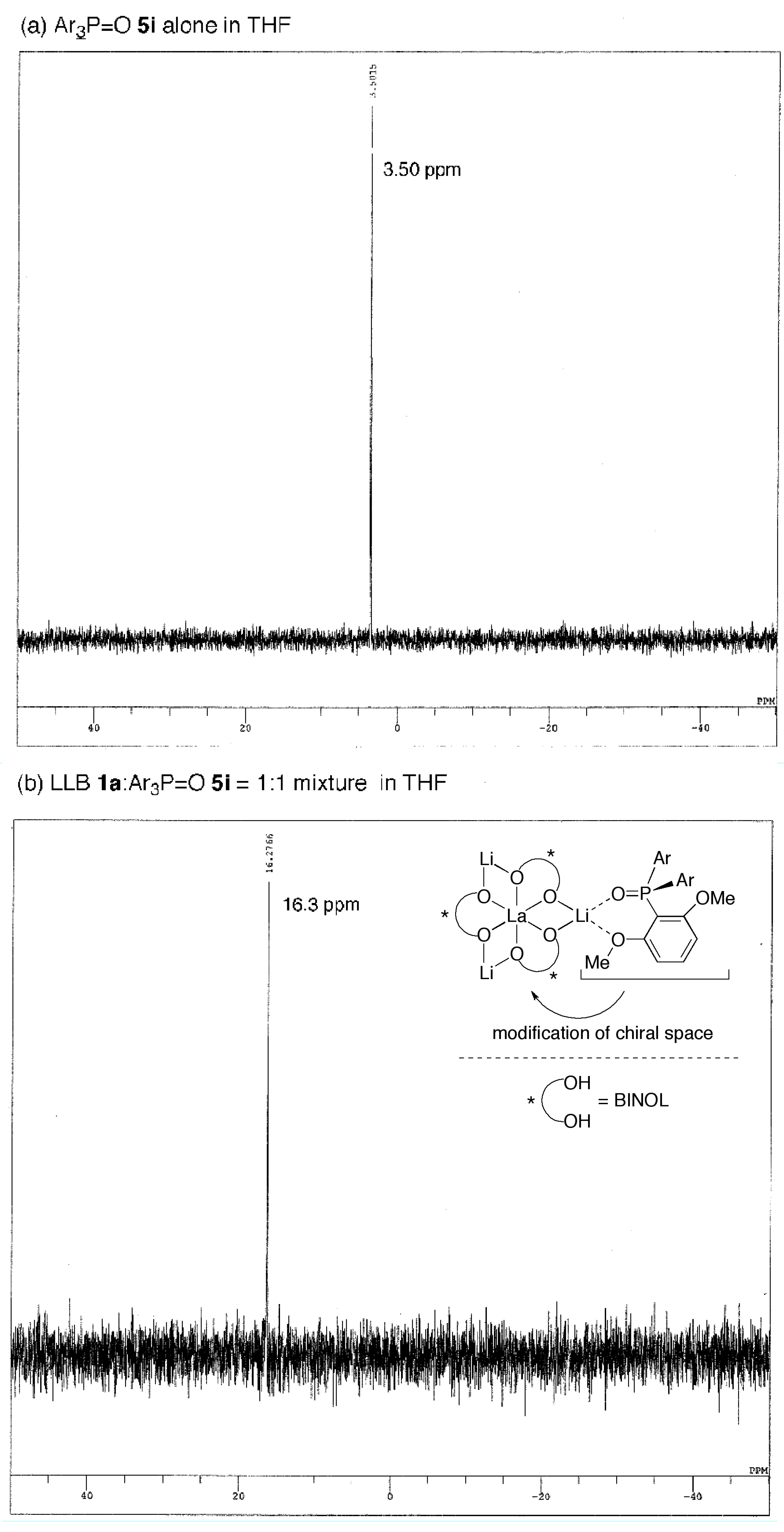
2.3. Postulated Role of Phosphine Oxide Additive
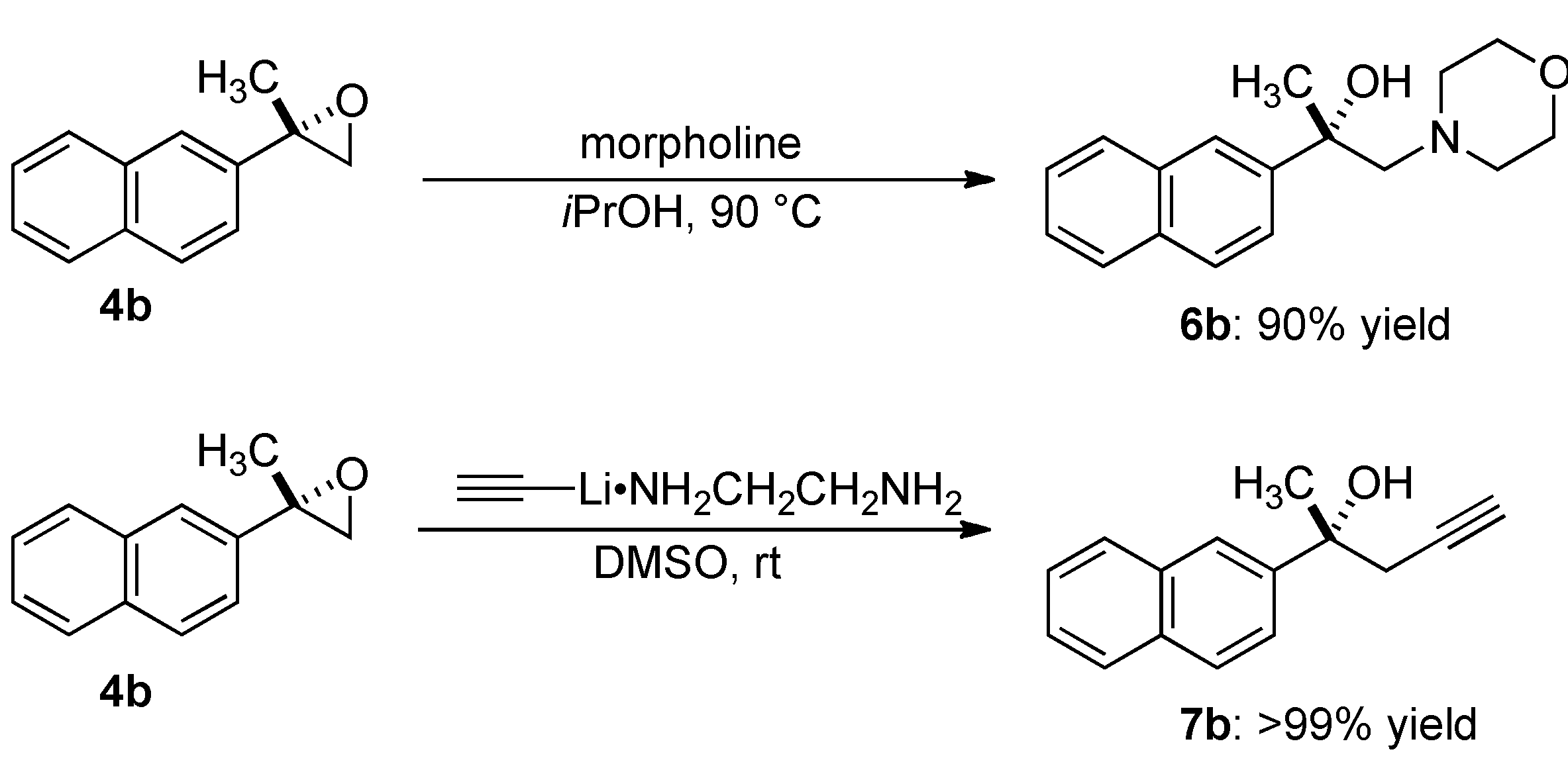
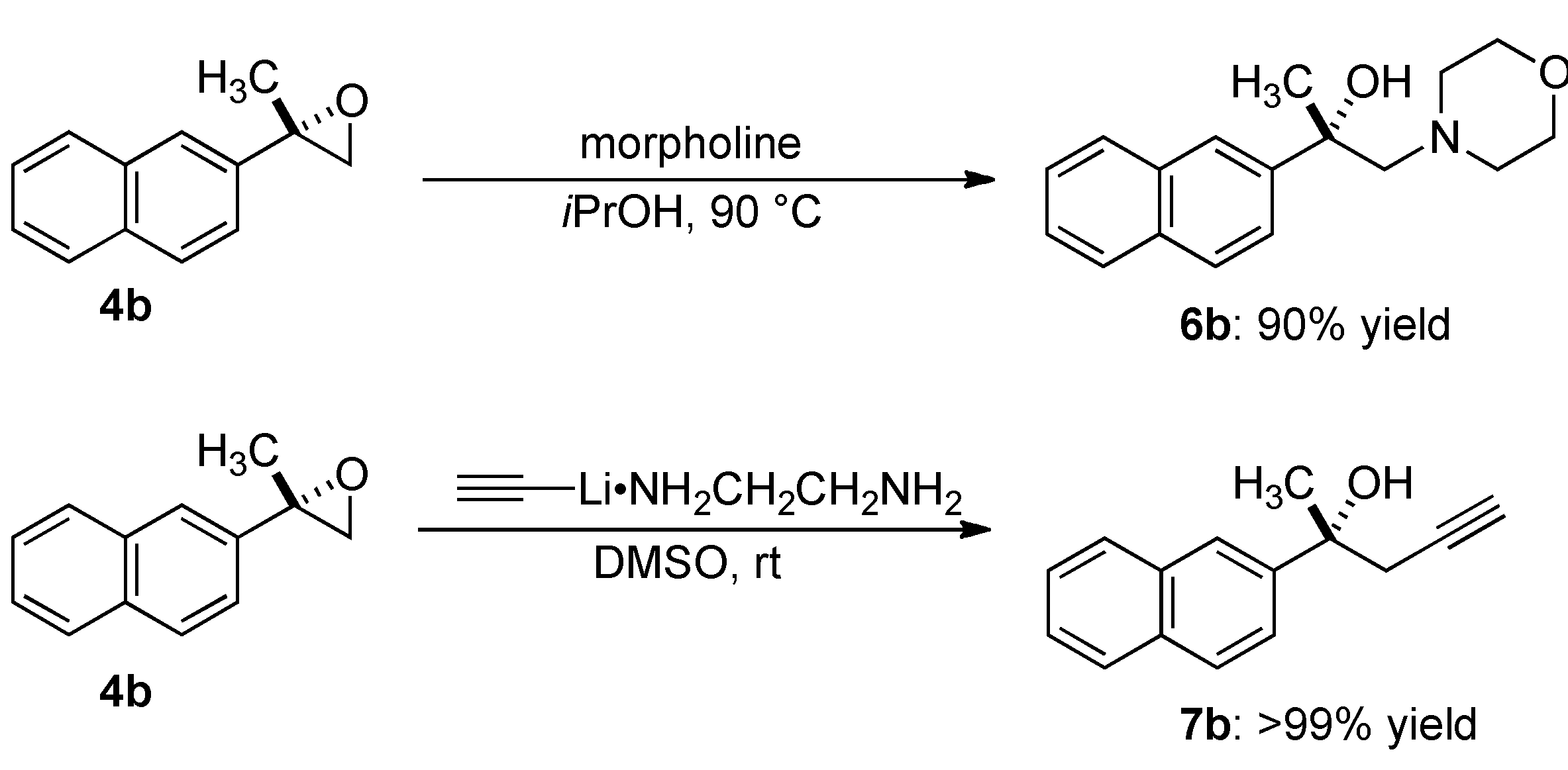
2.4. Transformation of Epoxide
3. Experimental
3.1. General
3.2. Preparation of (S)-La-Li3-(binaphthoxide)3 (1a) Catalyst Solution
3.3. Preparation of Dimethyloxosulfonium Methylide (2) Solution
3.4. General Procedure for Catalytic Asymmetric 1,4-Additions of β-Keto Esters to Nitroalkenes under Solvent-Free Conditions
3.4.1. (2S)-2-Methyl-2-phenyloxirane (4a)
3.4.2. (2S)-2-Methyl-2-naphthalen-2-yloxirane (4b)
3.4.3. (2S)-2-(4-Chlorophenyl)-2-methyloxirane (4c) [36]
3.4.4. (2S)-2-(3-chlorophenyl)-2-methyloxirane (4d)
3.4.5. (2S)-2-(2-Chlorophenyl)-2-methyloxirane (4e)
3.4.6. (2S)-2-(4-Fluorophenyl)-2-methyloxirane (4f) [36]
3.4.7. Ethyl 4-[(2S)-2-methyloxiran-2-yl]benzoate (4g)
3.4.8. (2S)-2-Methyl-2-(4-methylphenyl)oxirane (4h)
3.4.9. 3-[(2S)-2-Methyloxiran-2-yl]pyridine (4i)
3.4.10. (2S)-2-methyl-2-(2-phenylethyl)oxirane (4j) [10]
3.4.11. (2S)-2-Methyl-2-octyloxirane (4k) [37]
3.4.12. (2S)-2-Cyclohexyl-2-methyloxirane (4l) [10]
3.4.13. Ethyl 4-[(2S)-2-methyloxiran-2-yl]butanoate (4m)
3.4.14. (2S)-2-Ethyl-2-phenyloxirane (4n) [3]
3.4.15. (2S)-2-(4-Chlorophenyl)-2-ethyloxirane (4o)
3.4.16. (2S)-2-(4-Bromophenyl)-2-ethyloxirane (4p)
3.4.17. (2S)-2-(3-Chlorophenyl)-2-ethyloxirane (4q)
3.4.18. (2S)-2-Ethyl-2-(2-fluorophenyl)oxirane (4r)
3.4.19. 3-[(2S)-2-Ethyloxiran-2-yl]pyridine (4s)
3.4.20. (2S)-2-(4-Chlorophenyl)-2-propyloxirane (4t)
3.4.21. (2S)-2-Phenyl-2-(propan-2-yl)oxirane (4u)
3.5. Regioselective Ring-Oepning of Epoxide
3.5.1. (2S)-1-Morpholin-4-yl-2-naphthalen-2-ylpropan-2-ol (6b)
3.5.2. (2R)-2-Naphthalen-2-ylpent-4-yn-2-ol (7b)
4. Conclusions
Acknowledgements
References and Notes
- Xia, Q.-H.; Ge, H.-Q.; Ye, C.-P.; Liu, Z.-M.; Su, K.-X. Advances in homogeneous and heterogeneous catalytic asymmetric epoxidation. Chem. Rev. 2005, 105, 1603–1662. [Google Scholar]
- Aggarwal, V.K.; Winn, C.L. Catalytic, asymmetric sulfur ylide-mediated epoxidation of carbonyl compounds: Scope, selectivity, and applications in synthes. Acc. Chem. Res. 2004, 37, 611–620. [Google Scholar] [CrossRef]
- Dexter, A.F.; Lakner, F.J.; Campbell, R.A.; Hager, L.P. Highly enantioselective epoxidation of 1,l-disubstituted alkenes catalyzed by chloroperoxidase. J. Am. Chem. Soc. 1995, 117, 6412–6413. [Google Scholar]
- Lakner, F.J.; Hager, L.P. Chloroperoxidase as enantioselective epoxidation catalyst: An efficient synthesis of (R)-(−)-mevalonolactone. J. Org. Chem. 1996, 61, 3923–3925. [Google Scholar] [CrossRef]
- Jacobsen, E.N.; Wu, M.H. Epoxidation of alkenes other than allylic alcohols. In Comprehensive Asymmetric Catalysis; Jacobsen, E.N., Pfaltz, A., Yamamoto, H., Eds.; Springer: Berlin, Germany, 1999; p. 649. [Google Scholar]
- Katsuki, T. Asymmetric epoxidation of unfunctionalized olefins and related reactions. In Catalytic Asymmetric Synthesis, 2nd; Ojima, I., Ed.; Wiley-VCH: New York, NY, USA, 2000; p. 287. [Google Scholar]
- Shi, Y. Organocatalytic asymmetric epoxidation of olefins by chiral ketones. Acc. Chem. Res. 2004, 37, 488–496. [Google Scholar] [CrossRef]
- Wang, B.; Wong, O.A.; Zhao, M.-X.; Shi, Y. Asymmetric epoxidation of 1,1-disubstituted terminal olefins by chiral dioxirane via a planar-like transition state. J. Org. Chem. 2008, 73, 9539–9543. [Google Scholar] [CrossRef]
- Jacobsen, E.N. Asymmetric catalysis of epoxide ring-opening reactions. Acc. Chem. Res. 2000, 33, 421–431. [Google Scholar] [CrossRef]
- Lebel, H.; Jacobsen, E.N. Chromium catalyzed kinetic resolution of 2,2-disubstituted epoxides. Tetrahedron Lett. 1999, 40, 7303–7306. [Google Scholar]
- Steinreiber, A.; Faber, K. Microbial epoxide hydrolases for preparative biotransformations. Curr. Opin. Biotechnol. 2001, 12, 552–558. [Google Scholar]
- Archelas, A.; Furstoss, R. Synthetic applications of epoxide hydrolases. Curr. Opin. Chem. Biol. 2001, 5, 112–119. [Google Scholar]
- Katsuki, T. Epoxidation of allylic alcohols. In Comprehensive Asymmetric Catalysis; Jacobsen, E.N., Pfaltz, A., Yamamoto, H., Eds.; Springer: Berlin, Germany, 1999; p. 621. [Google Scholar]
- Corey, E.J.; Chaykovsky, M. Dimethyloxosulfonium methylide ((CH3)2SOCH2) and dimethylsulfonium methylide ((CH3)2SOCH2). Formation and application to organic synthesi. J. Am. Chem. Soc. 1965, 87, 1353–1364. [Google Scholar]
- Sone, T.; Yamaguchi, A.; Matsunaga, S.; Shibasaki, M. Catalytic asymmetric synthesis of 2,2-disubstituted terminal epoxides via dimethyloxosulfonium methylide addition to ketones. J. Am. Chem. Soc. 2008, 130, 10078–10079. [Google Scholar]
- Sone, T.; Gang, L.; Matsunaga, S.; Shibasaki, M. Catalytic asymmetric synthesis of 2,2-disubstituted oxetanes from ketones by using a one-pot sequential addition of sulfur ylide. Angew. Chem. Int. Ed. 2009, 48, 1677–1680. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, W. Enantioselective synthesis of oxiranes by the reactions of dimethylsulfonium methylide and aromatic aldehydes and ketones in the presence of chiral micelle. Tetrahedron: Asymmetry 1997, 8, 2723–2725. [Google Scholar]
- Shibasaki, M.; Kanai, M.; Matsunaga, S.; Kumagai, N. Recent progress in asymmetric bifunctional catalysis using multimetallic systems. Acc. Chem. Res. 2009, 42, 1117–1127. [Google Scholar] [CrossRef]
- Shibasaki, M.; Matsunaga, S. Bifunctional asymmetric catalysis based on dinuclear Schiff base complexes. J. Synth. Org. Chem. Jpn. 2009, 68, 1142–1149. [Google Scholar] [CrossRef]
- Matsunaga, S.; Shibasaki, M. Multimetallic bifunctional asymmetric catalysis based on proximity effect control. Bull. Chem. Soc. Jpn. 2008, 81, 60–75. [Google Scholar]
- Matsunaga, S.; Kumagai, N. Strategies for constructing diverse chiral environments in multimetallic bifunctional asymmetric catalysis. Synlett 2008, 1583–1602. [Google Scholar]
- Shibasaki, M.; Yoshikawa, N. Lanthanide complexes in multifunctional asymmetric catalysis. Chem. Rev. 2002, 102, 2187–2210. [Google Scholar]
- Yamagiwa, N.; Qin, H.; Matsunaga, S.; Shibasaki, M. Lewis acid-Lewis acid heterobimetallic cooperative catalysis: Mechanistic studies and application in enantioselective Aza-Michael reaction. J. Am. Chem. Soc. 2005, 127, 13419–13427. [Google Scholar] [CrossRef]
- Mihara, H.; Sohtome, Y.; Matsunaga, S.; Shibasaki, M. Chiral-catalyst-based convergent synthesis of HIV protease inhibitor GRL-06579A. Chem. Asian J. 2008, 3, 359–366. [Google Scholar] [CrossRef]
- Sohtome, Y.; Kato, Y.; Handa, S.; Aoyama, N.; Nagawa, K.; Matsunaga, S.; Shibasaki, M. Stereodivergent catalytic doubly diastereoselective nitroaldol reactions using heterobimetallic complexes. Org. Lett. 2008, 10, 2231–2234. [Google Scholar]
- Kakei, H.; Sone, T.; Sohtome, Y.; Matsunaga, S.; Shibasaki, M. Catalytic asymmetric cyclopropanation of enones with dimethyloxosulfonium methylide promoted by a La-Li3-(Biphenyldiolate)3+ NaI complex. J. Am. Chem. Soc. 2007, 129, 13410–13411. [Google Scholar] [CrossRef]
- Kino, R.; Daikai, K.; Kawanami, T.; Furuno, H.; Inanaga, J. Remarkable effect of tris(4-fluorophenyl)phosphine oxide on the stabilization of chiral lanthanum complex catalysts. A new and practical protocol for the highly enantioselective epoxidation of conjugated enone. Org. Biomol. Chem. 2004, 2, 1822–1824. [Google Scholar]
- Tian, J.; Yamagiwa, N.; Matsunaga, S.; Shibasaki, M. An asymmetric cyanation reaction and sequential asymmetric Cyanation-Nitroaldol reaction using a [YLi3{tris(binaphthoxide)}] single catalyst component: Catalyst tuning with achiral additives. Angew. Chem. Int. Ed. 2002, 41, 3636–3638. [Google Scholar] [CrossRef]
- Yamagiwa, N.; Tian, J.; Matsunaga, S.; Shibasaki, M. Catalytic asymmetric cyano-ethoxycarbonylation reaction of aldehydes using a YLi3Tris(binaphthoxide) (YLB) complex: Mechanism and roles of achiral additives. J. Am. Chem. Soc. 2005, 127, 3413–3422. [Google Scholar]
- Hara, K.; Park, S.-Y.; Yamagiwa, N.; Matsunaga, S.; Shibasaki, M. Catalytic asymmetric epoxidation of α,β-unsaturated phosphane oxides with a Y(O-iPr)3/biphenyldiol complex. Chem. Asian J. 2008, 3, 1500–1504. [Google Scholar]
- Furutachi, M.; Mouri, S.; Matsunaga, S.; Shibasaki, M. A heterobimetallic Ni/La-salan complex for catalytic asymmetric decarboxylative 1,4-addition of malonic acid half-thioester. Chem. Asian J. 2010, 5, 2351–2354. [Google Scholar] [CrossRef]
- Wooten, A.J.; Carroll, P.J.; Walsh, P.J. Insight into substrate binding in Shibasaki’s Li3(thf)n(binolate)3Ln complexes and implications in catalysis. J. Am. Chem. Soc. 2008, 130, 7407–7419. [Google Scholar] [CrossRef]
- Wooten, A.J.; Carroll, P.J.; Walsh, P.J. Evidence for substrate binding by the lanthanide centers in [Li3(thf)n(binolate)3Ln]: Solution and solid-state characterization of seven- and eight-coordinate [Li3(sol)n(binolate)3Ln(S)m] adducts. Angew. Chem. Int. Ed. 2006, 45, 2549–2552. [Google Scholar] [CrossRef]
- Aspinall, H.C.; Bickley, J.F.; Dwyer, J.L.M.; Greeves, N.; Kelly, R.V.; Steiner, A. Pinwheel-shaped heterobimetallic lanthanide alkali metal binaphtholates: Ionic size matters. Organometallics 2000, 19, 5416–5423. [Google Scholar]
- Di Bari, L.; Lelli, M.; Pintacuda, G.; Pescitelli, G.; Marchetti, F.; Salvadori, P. Solution versus solid-state structure of ytterbium heterobimetallic catalysts. J. Am. Chem. Soc. 2003, 125, 5549–5558. [Google Scholar]
- Archelas, A.; Furstoss, R. Absolute configuration of α-methylstyrene oxide: The correct absolute configuration/optical rotation correlation. J. Org. Chem. 1999, 64, 6112–6114. [Google Scholar] [CrossRef]
- Osprian, I.; Stampfer, W.; Faber, K. Selectivity enhancement of epoxide hydrolase catalyzed resolution of 2,2-disubstituted oxiranes by substrate modificatio. J. Chem. Soc. Perkin Trans. 1 2000, 3779–3785. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sone, T.; Yamaguchi, A.; Matsunaga, S.; Shibasaki, M. Enantioselective Synthesis of 2,2-Disubstituted Terminal Epoxides via Catalytic Asymmetric Corey-Chaykovsky Epoxidation of Ketones. Molecules 2012, 17, 1617-1634. https://doi.org/10.3390/molecules17021617
Sone T, Yamaguchi A, Matsunaga S, Shibasaki M. Enantioselective Synthesis of 2,2-Disubstituted Terminal Epoxides via Catalytic Asymmetric Corey-Chaykovsky Epoxidation of Ketones. Molecules. 2012; 17(2):1617-1634. https://doi.org/10.3390/molecules17021617
Chicago/Turabian StyleSone, Toshihiko, Akitake Yamaguchi, Shigeki Matsunaga, and Masakatsu Shibasaki. 2012. "Enantioselective Synthesis of 2,2-Disubstituted Terminal Epoxides via Catalytic Asymmetric Corey-Chaykovsky Epoxidation of Ketones" Molecules 17, no. 2: 1617-1634. https://doi.org/10.3390/molecules17021617