3.2. Materials
3′,5′-Di-O-(tert
-butyldimetylsilyl)thymidine (
1a) was synthesized as described previously [
24]. Thymidine (41.3 mmol; 10.02 g) was dissolved in DMF (35 mL). Imidazole (264 mmol, 18.0 g) in DMF (65 mL) and
tert-butyldimethylsilyl chloride (131 mmol; 19.87 g) in DMF (25 mL) were added. The mixture was stirred at r.t. for 70 h and then equilibrated between water (50 mL) and diethyl ether (50 mL). The organic phase was washed twice with water, dried over Na
2SO
4 and concentrated to white solid (87%, 16.93 g). The crude product was subjected to desilylation (
cf. preparation of compd.
2a) without further purification. The
1H- and
13C-NMR spectra were identical to those reported in literature [
25,
26]. Positive ion ESI-MS:
m/z obsd. 471.28 [M+H]
+, 493.27 [M+Na]
+, 509.24 [M+K]
+; calcd. 471.27 [M+H]
+, 493.25 [M+Na]
+, 509.23 [M+K]
+.
N4-Benzoyl-5′-O-(4,4′-dimethoxytrityl)-3′-O-(tert-butyldimethylsilyl)-2′-deoxycytidine (
1b). Commercially available
N6-benzoyl-5′-
O-(4,4′-dimethoxytrityl)-2′-deoxycytidine (1.42 mmol, 0.90 g) was dissolved in DMF (5 mL).
tert-Butyldimethylsilyl chloride (0.33 g, 2.35 mmol, 1.5 equiv.) and imidazole (0.29 g, 4.70 mmol, 3 equiv.) were added and the reaction was allowed to proceed for 15 h at r.t. The mixture was equilibrated between water and EtOAc. The organic phase was dried on Na
2SO
4 and concentrated to dryness, giving
1b in 92% yield (1.31 mmol, 0.98 g). The
1H- and
13C-NMR spectra were identical with those reported in literature [
27]. Negative ion ESI-MS:
m/z obsd. 746.32 [M−H]
−; calcd. 746.33 [M−H]
−.
N6-Benzoyl-5′-O-(4,4′-dimethoxytrityl)-3′-O-(tert-butyldimethylsilyl)-2′-deoxyadenosine (1c) was obtained in 89% yield (1.55 mmol, 1.20 g) from commercial N6-benzoyl-5′-O-(4,4′-dimethoxytrityl)-2′-deoxyadenosine as described above for compd. 1b. 1H-NMR (500 MHz, CDCl3): δ = 0.00 (s, 6H, SiMe2), 0.84 (s, 9H, SiCMe3), 2.24–2.29 (m, 1H, H2′), 2.35–2.44 (m, 1H, H2′), 3.18 (d, J = 11.2 Hz, 1H, H5′), 3.29 (d, J = 11.2 Hz, 1H, H5′), 3.68 (s, 6H, OMe), 3.96–4.00 (m, 1H, H4′), 4.71–4.77 (m, 1H, H3′), 6.38 (m, 1H, H1′), 6.74 (d, J = 9.7 Hz, 4H, DMTr), 7.20–7.40 (m, 9H, DMTr), 7.60–8.00 (m, 5H, Bz), 8.24 (s, 1H, H2), 8.56 (s, 1H, H8), 9.52 (s, 1H, NH). 13C-NMR (126 MHz, CDCl3) δ = −4.6 (SiMe), −3.9 (SiMe), 17.3 (SiCMe3), 24.8 (SiCMe3), 38.8 (C2′), 54.6 (OMe), 62.9 (C5′), 71.7 (C3′), 84.1 (C4′), 85.7 (DMTr), 86.0 (C1′), 112.7 (DMTr), 116.9 (DMTr), 126.4 (C5), 127.4 (DMTr), 127.6 (Bz), 127.8 (DMTr), 128.3 (Bz), 129.6 (DMTr), 132.1 (Bz), 133.6 (Bz), 135.3 (DMTr), 142.4 (C8), 144.7 (DMTr), 149.5 (C4), 151.4 (C2), 151.5 (C6), 158.3 (DMTr), 164. 9 (C=O). Negative ion ESI-MS: m/z obsd. 770.3509 [M−H]−; calcd. 770.3374 [M−H]−.
3′-O-(tert-Butyldimethylsilyl)thymidine (
2a). A 1:1:4 (
v/v/v) mixture of TFA, water and THF (30 mL) was added dropwise on an ice-bath to compd.
1a dissolved in a minimal volume of THF. After 1.5 h, another portion (30 mL) of the same mixture was added. The progress of desilylation was monitored by TLC and the reaction was quenched after 5.5 h by equilibration between diethyl ether and aq. NaHCO
3 (sat). The organic phase was washed with aq. NaHCO
3, dried over Na
2SO
4 and concentrated to white solid foam (81%, 10.38 g). The
1H-NMR spectrum (500 MHz, CDCl
3) was identical to that reported previously [
28].
13C-NMR (126 MHz, CDCl
3): δ = −4.9 (SiMe), −4.8 (SiMe), 12.4 (C5-Me), 17.9 (Si
CMe
3), 25.7 (SiC(
CH
3)
3), 40.7 (C2′), 61.8 (C5′), 71.9 (C3′), 86.2 (C4′), 87.8 (C1′), 110.8 (C5), 137.0 (C6), 150.6 (C2), 164.2 (C4). Negative ion ESI-MS:
m/z obsd. 355.15 [M−H]
−, 391.13 [M+Cl]
−, calcd. 355.1689 [M−H]
−, 391.15 [M+Cl]
−.
N4-Benzoyl-3′-O-(tert-butyldimethylsilyl)-2′-deoxycytidine (
2b). Compd.
1b (1.31 mmol, 0.98 g) was dissolved in DCM (5 mL) containing 3% dichloroacetic acid (150 μL) and MeOH (2 mL) was added. After 2 h stirring at r.t., DCM/aq. NaHCO
3 work-up was carried out and the organic phase was dried over Na
2SO
4. The solvent was removed by evaporation, and the residue was purified by a silica gel column chromatography (a stepwise gradient of 1–10% MeOH in DCM) to obtain
2b in 90% yield as white solid foam (1.17 mmol, 0.52 g). The
1H- and
13C-NMR spectra were identical with those reported in literature [
19]. Negative ion ESI-MS:
m/z obsd. 444.20 [M−H]
−; calcd. 444.20 [M−H]
−.
N6-Benzoyl-3′-O-(tert-butyldimethylsilyl)-2′-deoxyadenosine (
2c) was obtained in 84% yield (1.32 mmol, 0.62 g) from
1c as described above for
2b. The
1H-NMR spectrum was identical with that reported in literature [
29].
13C-NMR (126 MHz, CD
3CN): δ = −4.9 (Si
Me2), 17.3 (Si-
CMe
3), 24.8 (Si-C
Me3), 40.0 (C2′), 61.6 (C5′), 72.5 (C3′), 85.2 (C4′), 88.7 (C1′), 124.3 (C5), 127.8 (Bz), 128.2 (Bz), 132.2 (Bz), 133.6 (Bz), 142.6 (C8), 149.6 (C4), 151.1 (C2&C6), 165.0 (Bz). High resolution negative ion ESI-MS:
m/z obsd. 468.2067 [M−H]
−; calcd. 468.2067 [M−H]
−.
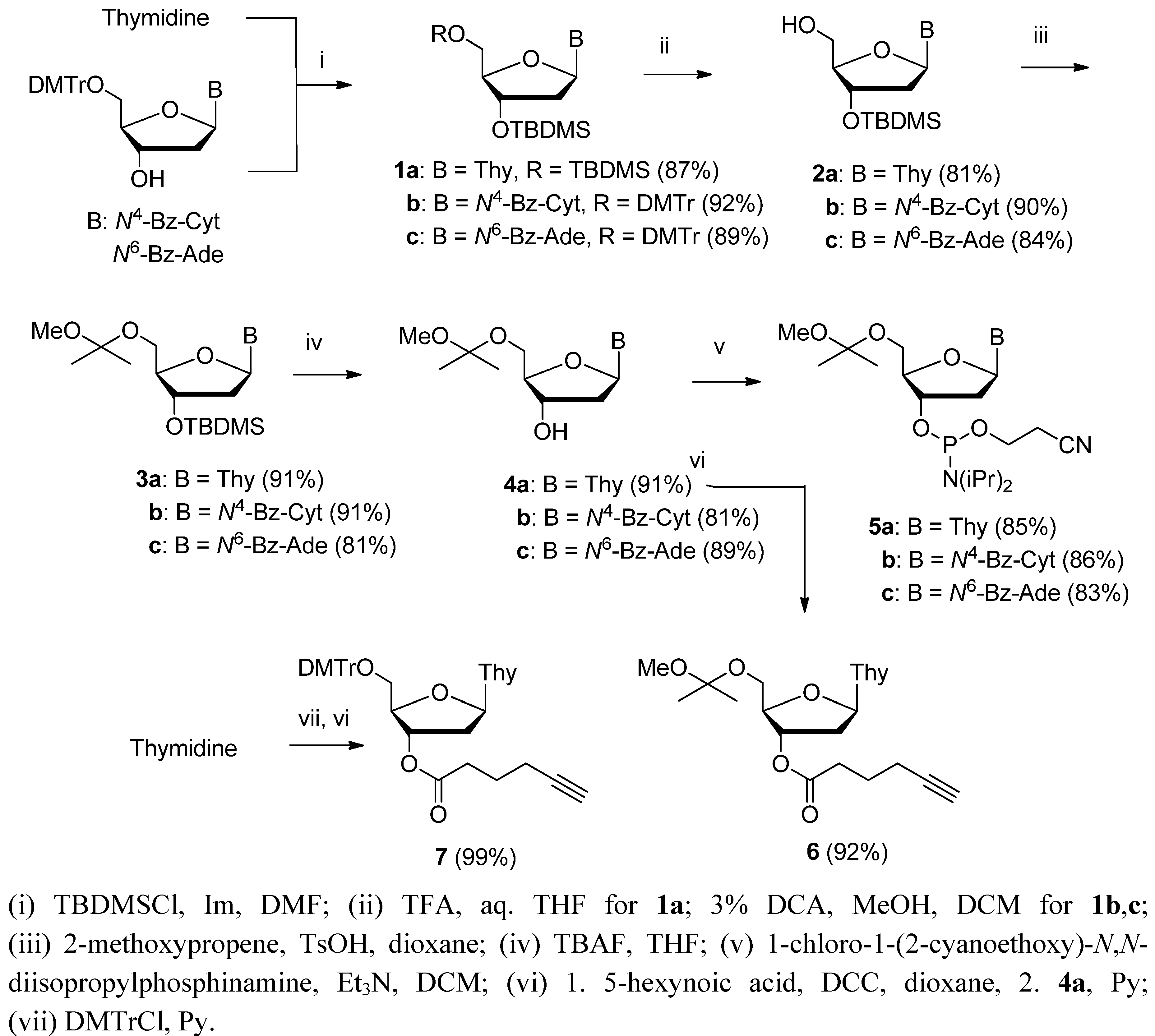
5′-O-(1-Methoxy-1-methylethyl)-3′-O-(tert-butyldimethylsilyl)thymidine (3a). Compound 2a (0.42 mmol, 150 mg) was dissolved in 1,4-dioxane (5 mL) and 11 equiv. of 2-methoxypropene (4.47 mmol, 0.400 mL) and 0.02 equiv. of p-toluenesulfonic acid monohydrate (4.2 mmol, 0.80 g) in dioxane (4 mL) were added. In 1 h, all the starting material had disappeared. The crude product was purified by silica gel column chromatography using EtOAc containing 1% petroleum ether and Et3N as an eluent. The yield was 91%. 1H-NMR (500 MHz, CDCl3): δ = −0.01 (s, 6H, SiMe2), 0.81 (s, 9H, SiCMe3), 1.31 (s, 6H, OC(OMe)Me2), 1.84 (s, 3H, C5-Me), 1.95–2.06 (m, 1H, H2′), 2.15–2.25 (m, 1H, H2′), 3.14 (s, 3H, OC(OMe)Me2), 3.47–3.92 (m, 3H, H4′&2 × H5′), 4.32–4.36 (m, 1H, H3′), 6.24–6.26 (m, 1H, H1′), 7.46 (s, 1H, H6), 10.00 (s, 1H, NH); 13C NMR (126 MHz, CDCl3): δ = −4.9 and −4.8 (SiMe2), 12.4 (C5-Me), 17.9 (Si-CMe3), 24.3 (C5′-OC(OMe)Me2), 25.6 (Si-CMe3), 41.2 (C2′), 48.6 (C5′-OC(OMe)Me2), 60.3 (C5′), 72.2 (C3′), 84.7 (C4′), 86.2 (C1′), 100.2 (C5′-OC(OMe)Me2), 110.6 (C5),135.3 (C6), 150.6 (C2), 164.2 (C4). High resolution positive ion ESI-MS: m/z obsd. 451.2241 [M+Na]+; calcd. 451.2240 [M+Na]+.
N4-Benzoyl-5′-O-(1-methoxy-1-methylethyl)-3′-O-(tert-butyldimethylsilyl)-2′-deoxycytidine (3b) was obtained in 91% yield (1.06 mmol, 0.55 g) from compd. 2b, as described above for compd. 3a. 1H-NMR (400 MHz, CDCl3): δ = −0.07 (s, 6H, SiMe2), 0.82 (s, 9H, SiCMe3), 1.33 and 1.35 (s, 2 × 3H, OC(OMe)Me2), 2.11–2.17 (m, 1H, H2′), 2.43–2.50 (m, 1H, H2′), 3.17 (s, 3H, OC(OMe)Me2), 3.53 (dd, J = 10.9 and 2.6 Hz, 1H, H5′), 3.70 (dd, J = 10.9 and 3.0, 1H, H5′′), 3.97–4.01 (m, 1H, H4′), 4.30–4.34 (m, 1H, H3′), 6.17–6.20 (m, 1H, H1′), 7.44 (t, J = 7.7 Hz, 3H, Bz&H5), 7.53 (t, J = 7.7 Hz, 1H, Bz), 7.85 (d, J = 7.7 Hz, 2H, Bz); 8.45 (d, J = 7.5 Hz, 1H, H6), 9.22 (br s, 1H, NH); 13C-NMR (101 MHz, CDCl3): δ = −4.9 and −4.6 (SiMe2), 17.9 (Si-CMe3), 24.3 (C5′-OC(OMe)Me2), 25.6 (Si-CMe3), 42.1 (C2′), 48.7 (C5′-OC(OMe)Me2), 59.1 (C5′), 70.3 (C3′), 86.3 (C4′), 86.9 (C1′), 96.0 (C5), 100.3 (C5′-OC(OMe)Me2), 127.7 (Bz), 128.8 (Bz), 133.0 (Bz), 133.2 (Bz), 144.7 (C6), 154.7 (C2), 162.3 (C4), 167.0 (Bz). High resolution negative ion ESI-MS: m/z obsd. 516.2564 [M−H]−; calcd. 516.2530 [M−H]−.
N6-Benzoyl-5′-O-(1-methoxy-1-methylethyl)-3′-O-(tert-butyldimethylsilyl)-2′-deoxyadenosine (3c) was obtained in 81% yield (1.07 mmol, 0.58 g) from compd. 2c, as described above for compd. 3a. 1H-NMR (500 MHz, CD3CN): δ = 0.00 (s, 6H, SiMe2), 0.80 (s, 9H, SiCMe3), 1.14 (s, 6H, OC(OMe)Me2, 2.27–2.37 (m, 1H, H2′), 2.67–2.73 (m, 1H, H2′), 2.92 (s, 3H, OC(OMe)Me2), 3.36–4.03 (m, 3H, H4′&2 × H5′), 4.56–4.62 (m, 1H, H3′), 6.32 (br s, 1H, H1′), 7.37 (t, J = 7.7 Hz, 2H, Bz), 7.47 (t, J = 7.7 Hz, 1H, Bz), 7.86 (d, J = 7.7 Hz, 2H, Bz), 8.24 (s, 1H, H2), 8.50 (s, 1H, H8), 9.44 (s, 1H, NH); 13C-NMR (126 MHz, CD3CN): δ = −6.0 (SiMe2), 17.3 (Si-CMe3), 23.2 and 23.4 (C5′-OC(OMe)Me2), 24.8 (Si-CMe3), 39.7 (C2′), 47.5 (C5′-OC(OMe)Me2), 60.0 (C5′), 72.0 (C3′), 83.9 (C4′), 86.1 (C1′), 99.6 (C5′-OC(OMe)Me2), 124.2 (C5), 127.8 (Bz), 128.2 (Bz), 132.1 (Bz), 133.5 (Bz), 141.9 (C8), 149.4 (C4), 151.5 (C2&C6), 164.9 (Bz). Hugh resolution negative ion ESI-MS: m/z obsd. 540.2663 [M−H]−, calcd. 540.2642 [M−H]−.
5′-O-(1-Methoxy-1-methylethyl)thymidine (4a). Compound 3a (0.38 mmol, 165 mg) was treated with 2 equiv. of tetrabutylammonium fluoride (0.75 mmol, 0.197 g) in THF (4 mL). After stirring for 4.5 h at r.t., the mixture was equilibrated between EtOAc and aq. NaHCO3, the organic phase was evaporated to dryness and the residue was purified by silica gel column chromatography using DCM containing 1-5% MeOH as eluent. Yield 91% (111 mg). 1H-NMR (500 MHz, CDCl3): δ = 1.39 and 1.41 (2 × s, 2 × 3H, C5′-OC(OMe)Me2), 1.93 (s, 3H, C5-Me), 2.14–2.19 (m, 1H, H2′), 2.38–2.41 (m, 1H, H2′), 3.23 (s, 3H, C5′-OC(OMe)Me2), 3.64 (d, J = 10.6 Hz, 1H, H5′), 3.69 (d, J = 10.6 Hz, 1H, H5′), 4.15 (s, 1H, H4′), 4.48 (m, 1H, H3′), 6.41 (dd, J = 6.3 and 6.3 Hz, 1H, H1′), 7.61 (s, 1H, H6), 9.50 (br s, 1H NH); 13C-NMR (126 MHz, CDCl3): δ = 12.3 (C5-Me), 24.2 (C5′-OC(OMe)Me2), 40.7 (C2′), 48.6 (C5′-OC(OMe)Me2), 60.8 (C5′), 72.2 (C3′), 84.8 (C4′), 85.8 (C1′), 100.2 (C5′-OC(OMe)Me2), 110.6 (C5), 135.6 (C6), 150.4 (C2), 163.8 (C4). High resolution negative ion ESI-MS: m/z obsd. 313.1424 [M−H]−, calcd. 313.1400 [M−H]−.
N4-Benzoyl-5′-O-(1-methoxy-1-methylethyl)-2′-deoxycytidine (4b) was obtained in 81% yield (0.87 mmol, 0.35 g) from 3b as described above for compd. 4a. 1H-NMR (400 MHz, CDCl3): δ = 1.28 and 1.31 (2 × s, 2 × 3H, C5′-OC(OMe)Me2), 2.21–2.28 (m, 1H, H2′), 2.65–2.73 (m, 1H, H2′), 3.23 (s, 3H, C5′-OC(OMe)Me2), 3.67 (dd, J = 10.9 and 2.9 Hz, 1H, H5′), 3.76 (dd, J = 3.0 Hz, 1H, H5′′), 4.20–4.24 (m, 1H, H4′), 4.46–4.49 (m, 1H, H3′), 4.30–4.41 (m, 1H, H3′), 6.31–6.34 (m, 1H, H1′), 7.40–7.56 (m, 3H, Bz&H5), 7.61 (t, J = 7.4 Hz, 1H, Bz) 7.93 (d, J = 7.4 Hz, 2H, Bz), 8.48 (d, J = 7.5 Hz, 1H, H6); 9.60 (s, 1H NH); 13C-NMR (101 MHz, CDCl3): δ = 24.3 (C5′-OC(OMe)Me2), 42.1 (C2′), 48.8 (C5′-OC(OMe)Me2), 60.1 (C5′), 70.6 (C3′), 86.4 (C4′), 87.3 (C1′), 98.2 (C5), 100.3 (C5′-OC(OMe)Me2), 127.7 (Bz), 128.9 (Bz), 133.1 (Bz), 133.6 (Bz), 145.0 (C6), 155.1 (C2), 162.4 (C4), 167.0 (Bz). High resolution negative ion ESI-MS: m/z obsd. 402.1692 [M−H]−; calcd. 402.1665 [M−H]−.
N6-Benzoyl-5′-O-(1-methoxy-1-methylethyl)-2′-deoxyadenosine (4c) was obtained in 89% yield (0.94 mmol, 0.40 g) from 3c as described above for compd. 4a. 1H-NMR (500 MHz, CD3CN): δ = 1.26 (2s, 6H, C5′-OC(OMe)Me2), 2.40–2.50 (m, 1H, H2′), 2.70–2.79 (m, 1H, H2′), 3.04 (s, 3H, C5′-OC(OMe)Me2), 3.40–4.10 (m, 3H, H4′&2 × H5′), 4.56 (m, 1H, H3′), 6.45–6.48 (m, 1H, H1′), 7.50 (t, J = 7.6 Hz, 2H, Bz), 7.60 (t, J = 7.6 Hz, 1H, Bz), 7.99 (d, J = 7.6 Hz), 8.42 (s, 1H, H2), 8.63 (s, 1H, H8); 9.69 (s, 1H, NH); 13C-NMR (126 MHz, CD3CN): δ = 23.3 and 23.5 (C5′-OC(OMe)Me2), 39.6 (C2′), 47.5 (C5′-OC(OMe)Me2), 60.3 (C5′), 70.8 (C3′), 83.8 (C4′), 85.9 (C1′), 100.3 (C5′-OC(OMe)Me2), 124.0 (C5), 127.8 (Bz), 128.3 (Bz), 132.2 (Bz), 133.2 (Bz), 141.8 (C8 ), 149.4 (C4), 151.5 (C2), 151.6 (C6), 165.1 (Bz). High resolution negative ion ESI-MS: m/z obsd. 426.1783 [M−H]−; calcd. 426.1777 [M−H]−.
5′-O-(1-Methoxy-1-methylethyl)thymidine 3′-(2-cyanoethyl-N,N-diisopropyl)phosphoramidite (5a). Glassware and stirrer bar were dried for 15 hours in an oven (120 °C). All chemicals and reagents were placed in a box under nitrogen. Compd. 4a (4.27 mmol, 0.40 g) was dissolved in dry DCM (20 mL). Triethylamine (5 equiv., 6.37 mmol, 0.885 mL) and 1-chloro-1-(2-cyanoethoxy)-N,N-diisopropylphosphinamine (1.1 equiv., 0.330 mL) were added and the mixture was stirred for 2.5 h at r.t. under nitrogen. The mixture was then loaded onto a silica gel column and the product was eluted with a 50:49:1 mixture of EtOAc, petroleum ether and Et3N. Compd. 5a was obtained as a white foam (yield 85%), which was stored at −18 °C. For the mixture of RP and SP diastereomers: 1H-NMR (500 MHz, CD3CN): δ = 1.17 (d, J = 6.8 Hz, 12H, iPr), 1.33 and 1.34 (2 × s, 6H, C5′-OC(OMe)Me2), 1.84 (s, 3H, C5-Me), 2.20–2.45 (m, 2H, 2 × H2′), 2.67 (t, J = 5.9 Hz, 2H, OCH2CH2CN), 3.15 and 3.17 (2 × s, 3H, C5′-OC(OMe)Me2), 3.53–4.25 (m, 7H, 2 × H5′& H4′&OCH2CH2CN&2 × NCHMe2), 4.48–4.59 (m, 1H, H3′), 6.24 (m, 1H, H1′), 7.52 and 7.54 (2 × s, 1H, H6), 9.80 (br s, 1H, NH); 13C-NMR (126 MHz, CD3CN): δ = 11.3 (C5-Me), 19.7 (OCH2CH2CN), 23.4 (C5′-OC(OMe)Me2), 23.6 (NCHMe2), 38.7 (C2′), 42.6 (C5′-OC(OMe)Me2), 47.8 (NCHMe2), 57.9 (OCH2CH2CN), 60.2 (C5′), 73.4 (C3′), 81.7 (C4′), 84.1 (C1′), 99.8 (C5′-OC(OMe)Me2), 109.9 (C5), 118.1 (OCH2CH2CN), 135.6 (C6), 150.6 (C2), 163.8 (C4); 31P-NMR (CD3CN): δ = 149.3 and 148.9. Positive ion ESI-MS: m/z obsd. 537.25 [M+Na]+; calcd. 537.25 [M+Na]+.
N4-Benzoyl-5′-O-(1-methoxy-1-methylethyl)-2′-deoxycytidine 3′-(2-cyanoethyl-N,N-diisopropyl)-phosphoramidite (5b) was obtained in 86% yield (0.75 mmol, 0.45 g) from 4b as described above for compd. 5a. For the mixture of RP and SP diastereomers: 1H-NMR (500 MHz, CDCl3): δ = 1.20 (d, J = 6.6 Hz, 12H, iPr), 1.40 (2 × s, 6H, C5′-OC(OMe)Me2), 2.20–2.45 (m, 2H, 2 × H2′), 2.64 (t,J = 5.9 Hz, 2H, OCH2CH2CN), 3.22 (2 × s, 3H, C5′-OC(OMe)Me2), 3.45–4.45 (m, 7H, 2 × H5′&H4′&OCH2CH2CN&2 × NCHMe2), 4.51–4.60 (m, 1H, H3′), 6.25–6.35 (m, 1H, H1′), 7.46 (m, 3H, Bz&H5), 7.57 (m, 1H, Bz), 7.95 (m, 2H, Bz), 8.42 (m, 1H, H6), 9.60 (br s, 1H, NH); 13C-NMR (126 MHz, CDCl3): 18.5 (OCH2CH2CN), 22.4 (C5′-OC(OMe)Me2), 22.6 (NCHMe2), 38.9 (C2′), 41.4 (C5′-OC(OMe)Me2), 47.0 (NCHMe2), 57.0 (OCH2CH2CN), 58.2 (C5′), 71.1 (C3′), 85.2 (C4′), 86.0 (C1′), 95.0 (C5), 98.5 (C5′-OC(OMe)Me2), 116.2 (OCH2CH2CN), 126.2 (Bz), 127.2 (Bz), 131.3 (Bz), 131.4 (Bz), 143.3 (C6), 152.8 (C2), 156.6 (C4), 150.8 (C2), 165.5 (Bz); 31P-NMR (CDCl3): δ = 148.5 and 149.0. Negative ion ESI-MS: m/z obsd. 602.28 [M−H]−; calcd. 602.27 [M−H]−.
N6-Benzoyl-5′-O-(1-methoxy-1-methylethyl)-2′-deoxyadenosine 3′-(2-cyanoethyl-N,N-diisopropyl)-phosphoramidite (5c) was obtained in 83% yield (0.70 mmol, 0.44 g) from 4c as described above for compd. 5a. For the mixture of RP and SP diastereomers: 1H-NMR (500 MHz, CDCl3): δ = 1.18 (d, J = 6.6 Hz, 12H, iPr), 1.39 and 1.41 (2 × s, 6H, C5′-OC(OMe)Me2), 2.20–2.45 (m, 2H, 2 × H2′), 2.66 (t,J = 5.9 Hz, 2H, OCH2CH2CN), 3.22 and 3.24 (2 × s, 3H, C5′-OC(OMe)Me2), 3.43–4.25 (m, 7H, 2 × H5′&H4′&OCH2CH2CN&2 × NCHMe2), 4.53–4.59 (m, 1H, H3′), 6.28 (m, 1H, H1′), 7.46 (m, 2H, Bz), 7.58 (m, 1H, Bz), 7.96 (m, 2H, Bz), 8.28 (s, 1H, H2), 8.46 (s, 1H, H8), 9.30 (br s, 1H, NH); 13C-NMR (126 MHz, CDCl3): 18.6 (OCH2CH2CN), 22.5 (C5′-OC(OMe)Me2), 22.7 (NCHMe2), 39.3 (C2′), 41.4 (C5′-OC(OMe)Me2), 47.0 (NCHMe2), 56.9 (OCH2CH2CN), 58.0 (C5′), 70.9 (C3′), 83.9 (C4′), 85.3 (C1′), 98.5 (C5′-OC(OMe)Me2), 115.9 (OCH2CH2CN), 122.0 (C5), 126.1 (Bz), 127.0 (Bz), 131.1 (Bz), 131.4 (Bz), 142.8 (C8), 148.2 (C4), 150.2 (C6), 150.8 (C2), 163.8 (Bz); 31P-NMR (CDCl3): δ = 148.5 and 148.9. Negative ion ESI-MS: m/z obsd. 626.30 [M−H]−, calcd. 626.29 [M−H]−.
5′-O-(1-Methoxy-1-methylethyl)-3′-O-(hex-5-ynoyl)thymidine (6). Dicyclohexylcarbodiimide (0.722 mmol, 0.149 g) was dissolved in dry dioxane (2 mL) and the solution obtained was added dropwise on an ice-bath to 5-hexynoic acid (1.44 mmol, 0.162 g, 0.160 mL) in dioxane (1 mL). After stirring for 2 h at r.t., the precipitated dicyclohexylurea was filtrated off and the volatiles were removed under reduced pressure, giving hex-5-ynoic anhydride as a slightly yellow liquid. Compd. 4a (0.35 mmol, 0.111 g) was dissolved in dry pyridine (5 mL), hex-5-ynoic anhydride and a catalytic amount of dimethylaminopyridine were added, and the reaction was allowed to proceed 17 h at rt. The mixture was concentrated under reduced pressure and the residue was purified by silica gel chromatography applying gradient elution with 1–5% MeOH in DCM containing 1% triethylamine. Compound 6 was obtained as white foam in 92% yield (0.32 mmol, 0.132 g). 1H-NMR (500 MHz, CDCl3): δ = 1.41 and 1.42 (2 × s, 2 × 3H, C5′-OC(OMe)Me2), 1.84–1.89 (m, 2H, H3 of hex-5-ynoyl), 1.95 (s, 3H, C5-Me), 2.02–2.04 (m, 1H, H2′), 2.18–2.24 (m, 1H, H2′), 2.28–2.31 (m, 2H, H2 of hex-5-ynoyl), 2.39–2.43 (m, 2H, H4 of hex-5-ynoyl), 2.52 (t, J = 7.4 Hz, 1H, H6 of hex-5-ynoyl), 3.25 (s, 3H, C5′-OC(OMe)Me2), 3.70–3.99 (m, 2H, H5′), 4.21 (m, 1H, H4′), 5.30–5.32 (m, 1H, H3′), 6.43 (dd, J = 6.3 Hz, 1H, H1′), 7.62 (s, 1H, H6), 9.50 (s, 1H NH); 13C-NMR (126 MHz, CDCl3): δ = 12.5 (C5-Me), 17.7 (C4 of hex-5-ynoyl), 23.3 (C3 of hex-5-ynoyl), 24.4 (C5′-OC(OMe)Me2), 32.7 (C2 of hex-5-ynoyl), 37.9 (C2’), 48.8 (C5′-OC(OMe)Me2), 61.1 (C5′), 69.5 (C6 of hex-5-ynoyl), 75.4 (C3′), 82.9 (C5 of hex-5-ynoyl), 83.9 (C4′), 84.6 (C1′), 100.4 (C5′-OC(OMe)Me2), 111.2 (C5), 135.2 (C6), 150.8 (C2), 164.0 (C4), 172.7 (C=O of hex-5-ynoyl). High resolution negative ion ESI-MS: m/z obsd. 407.1841 [M−H]−, calcd. 407.1818 [M−H]−.
5′-O-(4,4′-Dimethoxytrityl)-3′-O-(hex-5-ynoyl)thymidine (
7). Thymidine was converted to 5′-
O-(4,4′-dimethoxytrityl)thymidine as described in literature [
30] and acylated with hex-5-ynoic anhydride as described above for compound
6. The yield of acylation was 99%.
1H- and
13C-NMR spectroscopic data was identical with that given by Oyelere
et al. [
31]. High resolution negative ion ESI-MS:
m/z obsd. 637.2534 [M−H]
−, calcd. 637.2550 [M−H]
−.
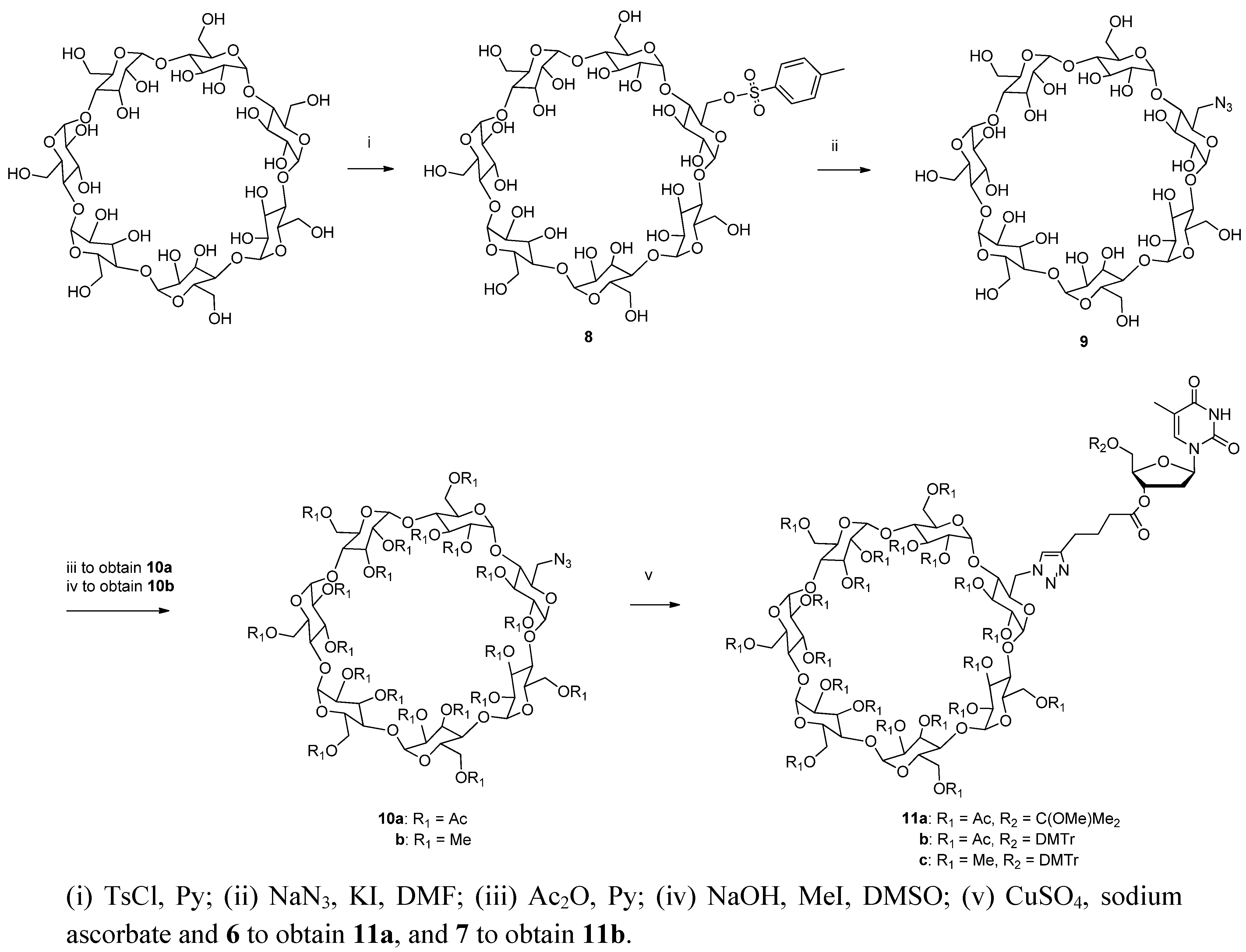
6-O-(p
-Toluenesulfonyl)-β-cyclodextrin (
8). Monotosylated β-cyclodextrin was prepared by the method of Kuzuya
et al. [
19] The
1H- and
13C-NMR spectra of the product were identical to those reported previously in literature [
32]. Negative ion ESI-MS:
m/z obsd 1287.41 [M−H]
−, calcd. 1287.37 [M−H]
−.
6-Azido-6-deoxy-β-cyclodextrin (
9) was obtained by displacing the
p-toluenesulfonyloxy group from compd.
8 with azide ion as reported by Petter
et al. [
32]. The
1H- and
13C-NMR chemical shifts were identical with those reported previously [
32]. Negative ion ESI-MS:
m/z obsd. 1158.36 [M−H]
−, calcd. 1158.37 [M−H]
−.
Peracetylated 6-azido-6-deoxy-β-cyclodextrin (10a). The azido-functionalized β-cyclodextrin (9) was peracetylated with acetic anhydride in pyridine at r.t., using 4 quiv. of acetic anhydride compared to the amount of free OH groups. The product was purified by silica gel chromatography applying a stepwise gradient elution with 1–5% MeOH in DCM containing 1% Et3N. The yield was 73%. 1H-NMR (500 MHz, DMSO-d6) 2.05 (br s, 60H, Ac), 3.65 (d, J = 10 Hz, 1H, CHHN3), 3.77 (d, J = 10 Hz, CHHN3), 3.85–3.90 (br s, 7H, H4), 4.03–4.12 (m, 7H, H5), 4,18–4.28 (m, 6H, H6b), 4.40–4.50 (m, 6H, H6a), 4.65–4.75 (m, 7H, H2), 5.00–5.15 (m, 7H, H3), 5.18–5.23 (m, 7H, H1); 13C-NMR (126 MHz, DMSO-d6): 169.5 (Ac), 97.2 (C1), 77.3 (C4), 71.0 (C5), 70.8 (C3), 70.0 (C2), 62.6 (C6), 50.8 (CH2N3), 21.0 (Ac). Positive ion ESI-MS: m/z obsd. 2022.56 [M+Na]+, calcd. 2022.58 [M+Na]+.
Permethylated 6-azido-6-deoxy-β-cyclodextrin (
10b). Permethylation of the azido-functionalized β-cyclodextrin was achieved by treating
9 over a weekend with a modest excess of methyl iodide (1.25 equiv.) and sodium hydroxide (1.25 equiv.) compared to the amount of free hydroxyl functions, as described previously by Schomburg
et al. [
20,
21]. The yield was 91%. The
1H-NMR spectrum was identical with that reported earlier [
33].
13C-NMR (126 MHz, DMSO-
d6): 96.8 (C1), 80.6 (C3), 80.2 (C2), 78.4 (C4), 70.2 (
C5-CH
2N
3), 69.4 (C6-N
3), 59.8 (O6-Me), 57.0 (O2-Me&O3-Me), 50.4 (
CH
2N
3). Negative ion ESI-MS:
m/z obsd. 1474.76 [M+Cl]
−, calcd. 1475.66 [M+Cl]
−.
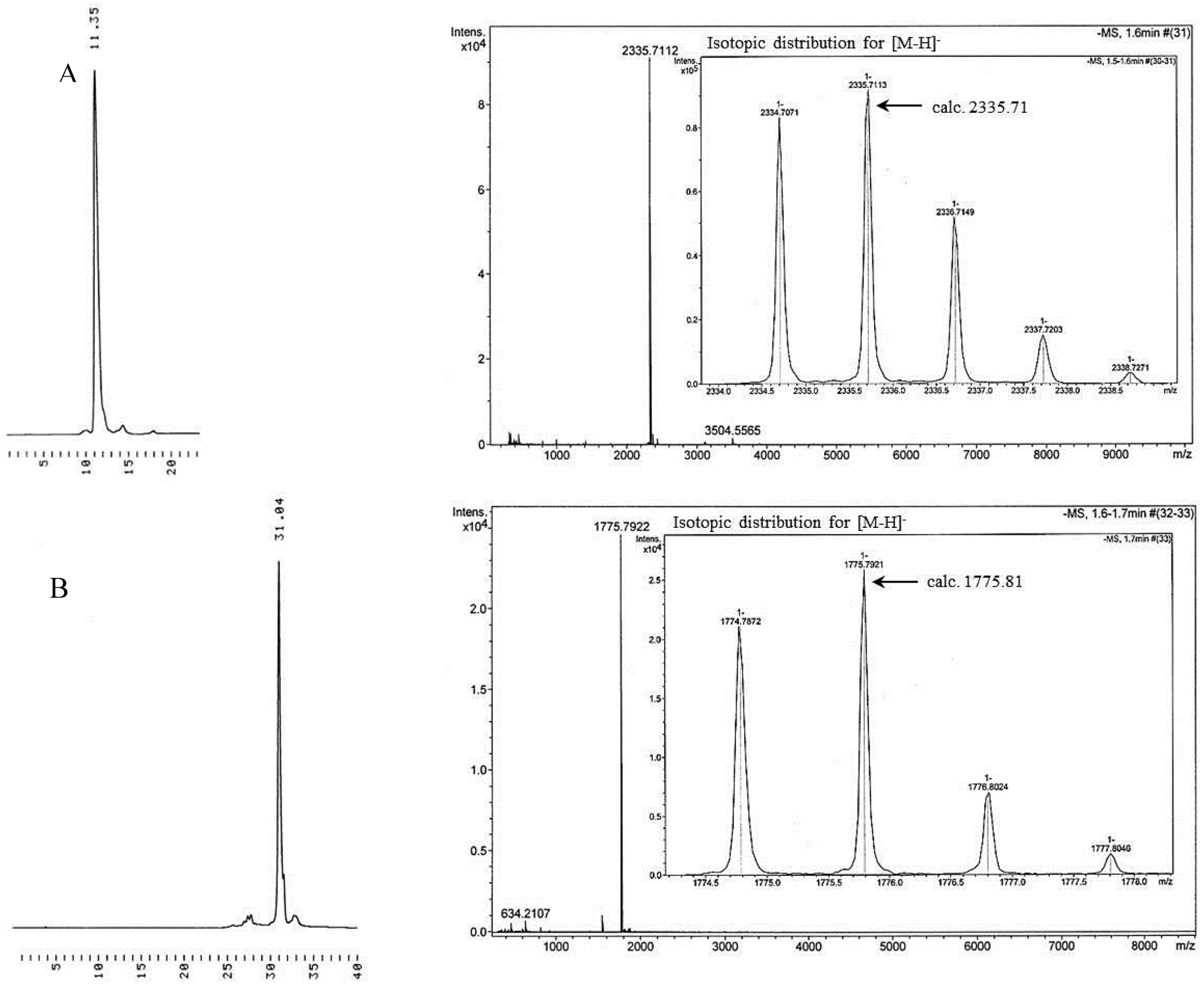
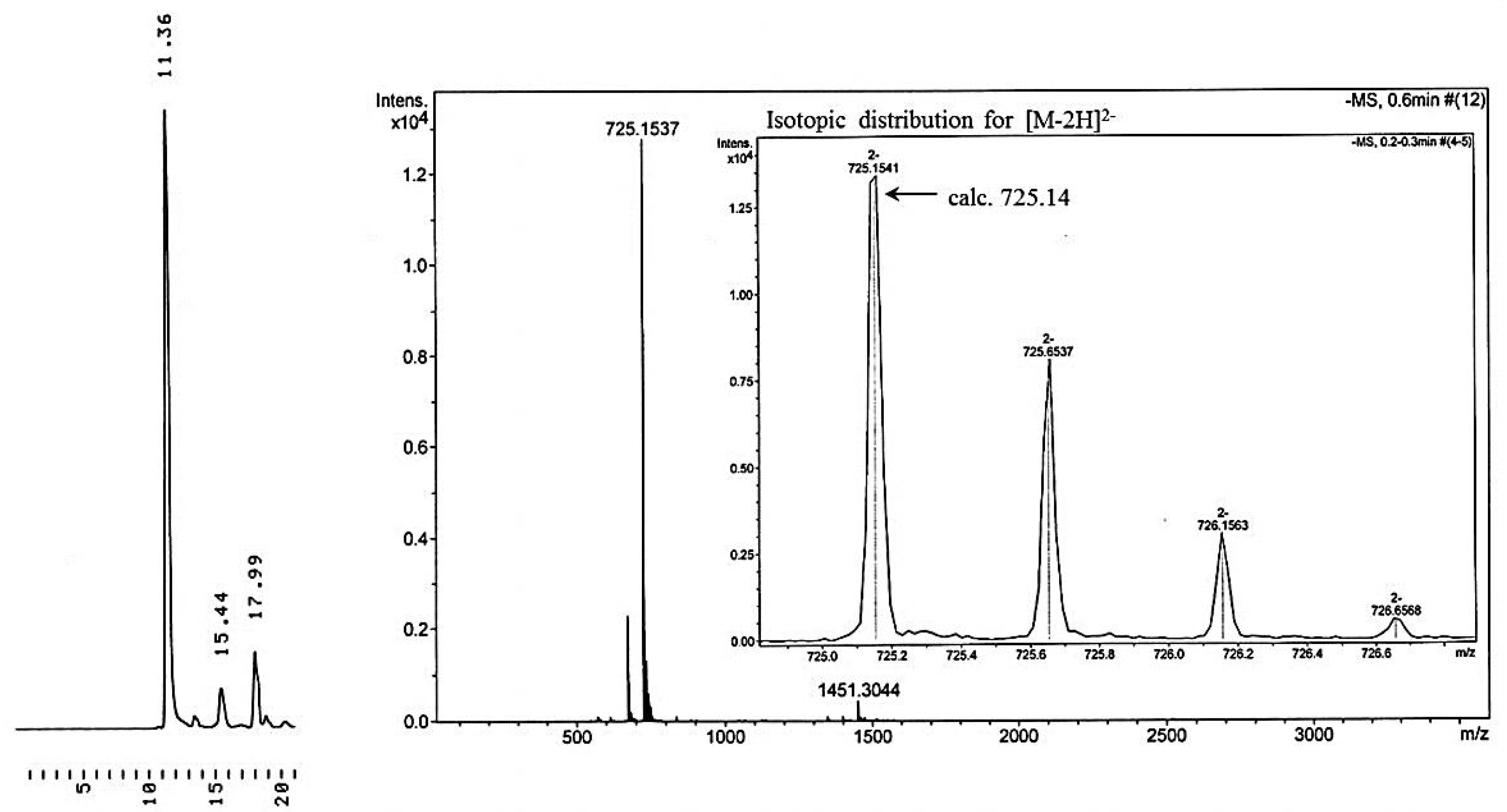
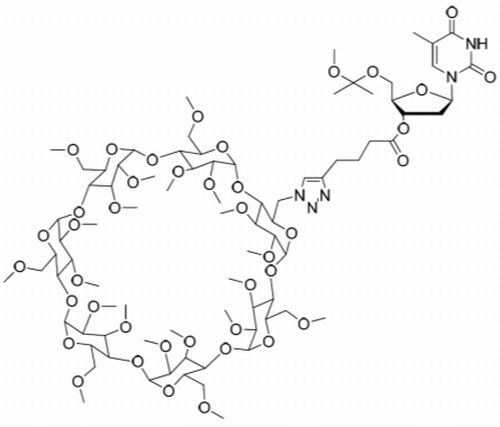
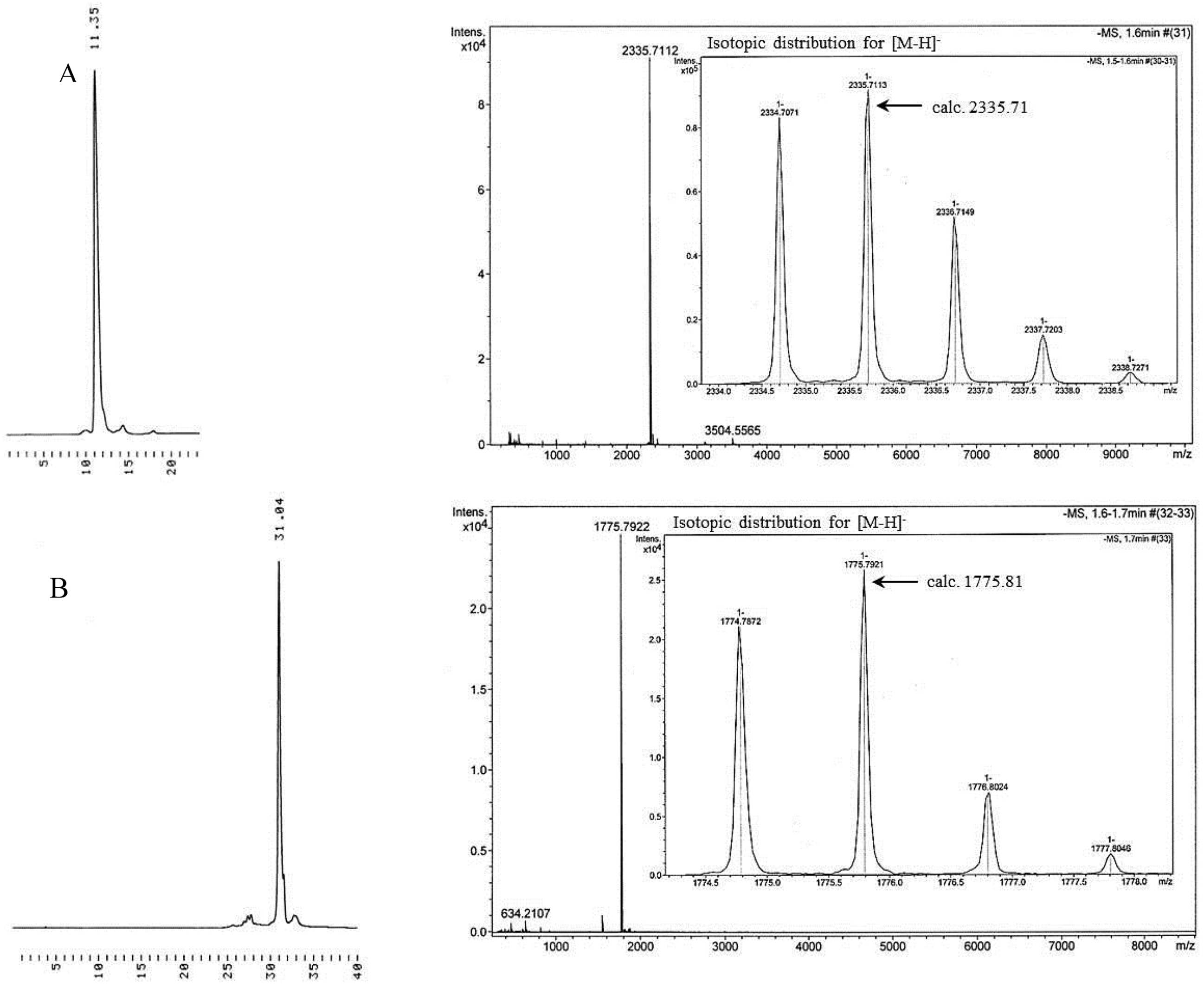
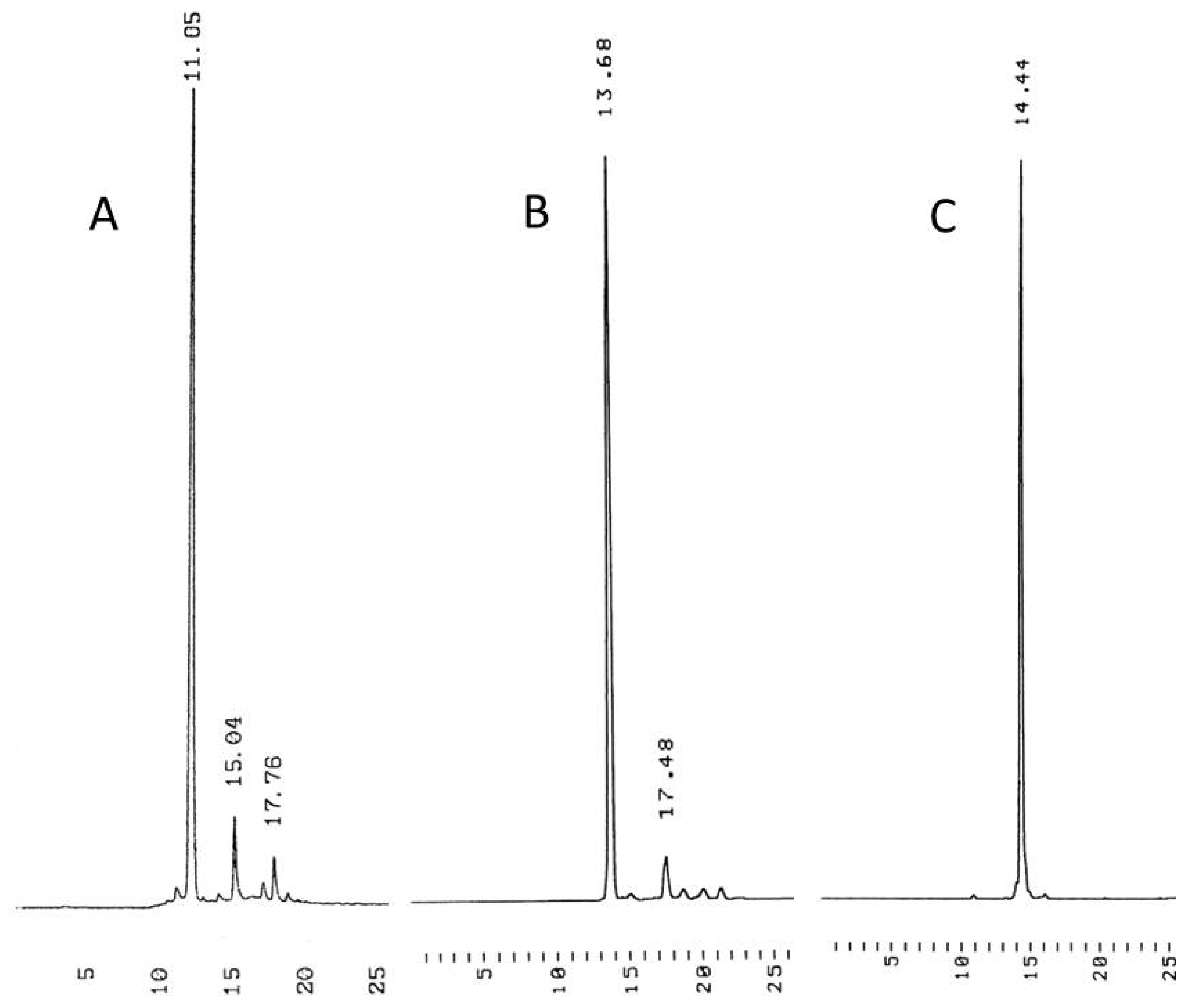
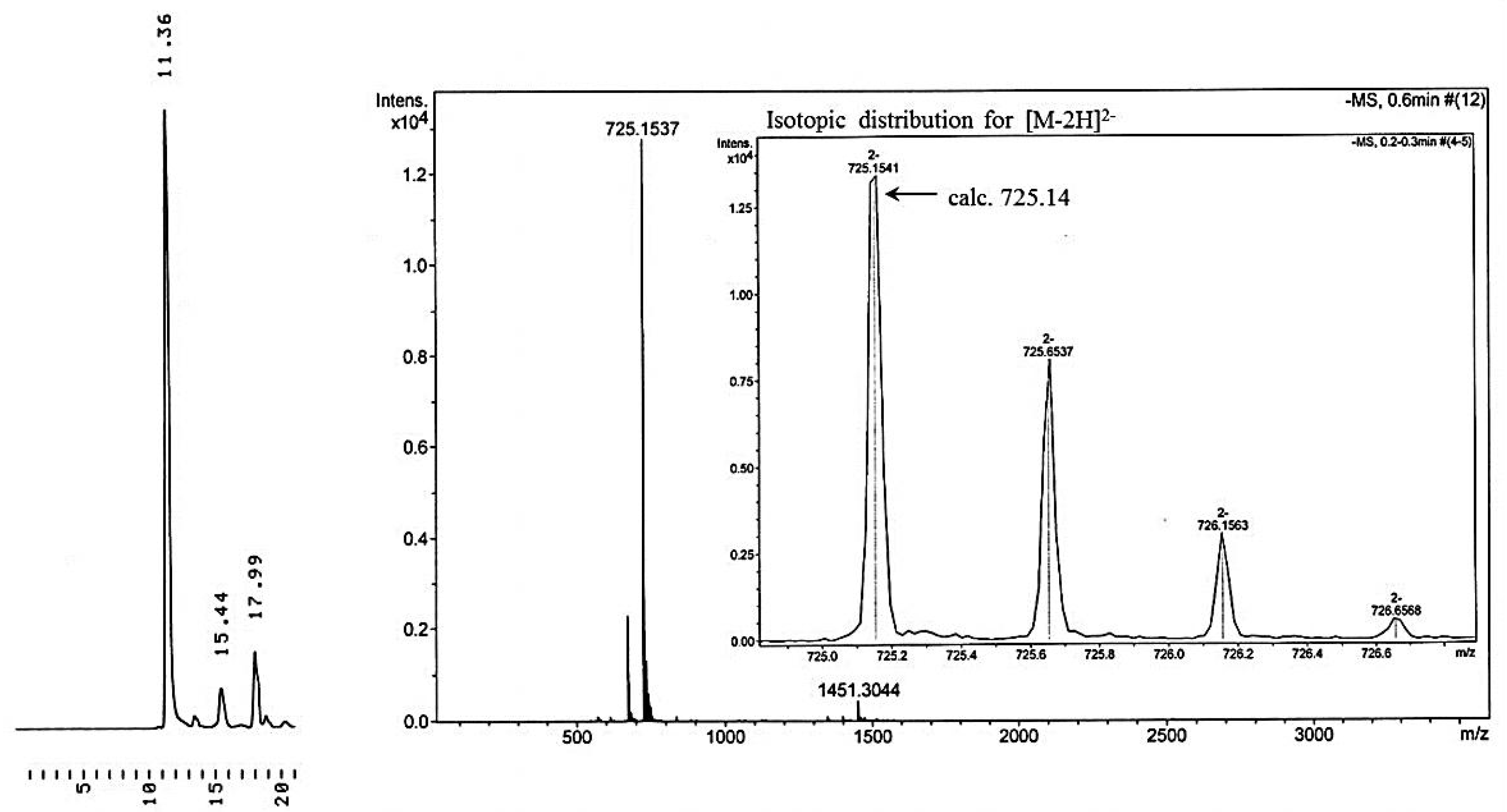
Immobilization of 3′-O-(hex-5-ynoyl)thymidines to cyclodextrin supports. 3′-O-(hex-5-ynoyl)thymidines 6 and 7 were immobilized to the azido-functionalized supports 10a,b by Cu+-promoted 1,3-dipolar cyclo addition. Accordingly, the appropriate nucleoside, either 6 (3.03 mmol, 1.25 g) or 7 (1.03 mmol, 0.66 g) in dioxane (15 mL) was mixed with either 10a or 10b in the same solvent. Aq. CuSO4 (50 mmol·L−1) and sodium ascorbate (0.1 mmol·L−1) were added and the reaction was allowed to proceed for 3 d at r.t. Aq. CuSO4.5H2O and aq. sodium ascorbate were added daily to ensure quantitative formation of the 1,2,3-triazole linkages. The products 11a–c were obtained in about 50% yield as white foams by silica gel chromatography applying first a stepwise gradient from 4% to 34% cyclohexane in EtOAc containing 1% triethylamine, and then a stepwise gradient of 5% to 10% MeOH in DCM containing 1% triethylamine. The identity and homogeneity of the products were verified by ESI-MS and HPLC, respectively.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}