Evaluation of the Antioxidant and Anti-glication Effects of the Hexane Extract from Piper auritum Leaves in Vitro and Beneficial Activity on Oxidative Stress and Advanced Glycation End-Product-Mediated Renal Injury in Streptozotocin-Treated Diabetic Rats

Abstract
:1. Introduction
2. Results and Discussion
2.1. Total Phenolic Content
2.2. Antioxidant Activity in Vitro
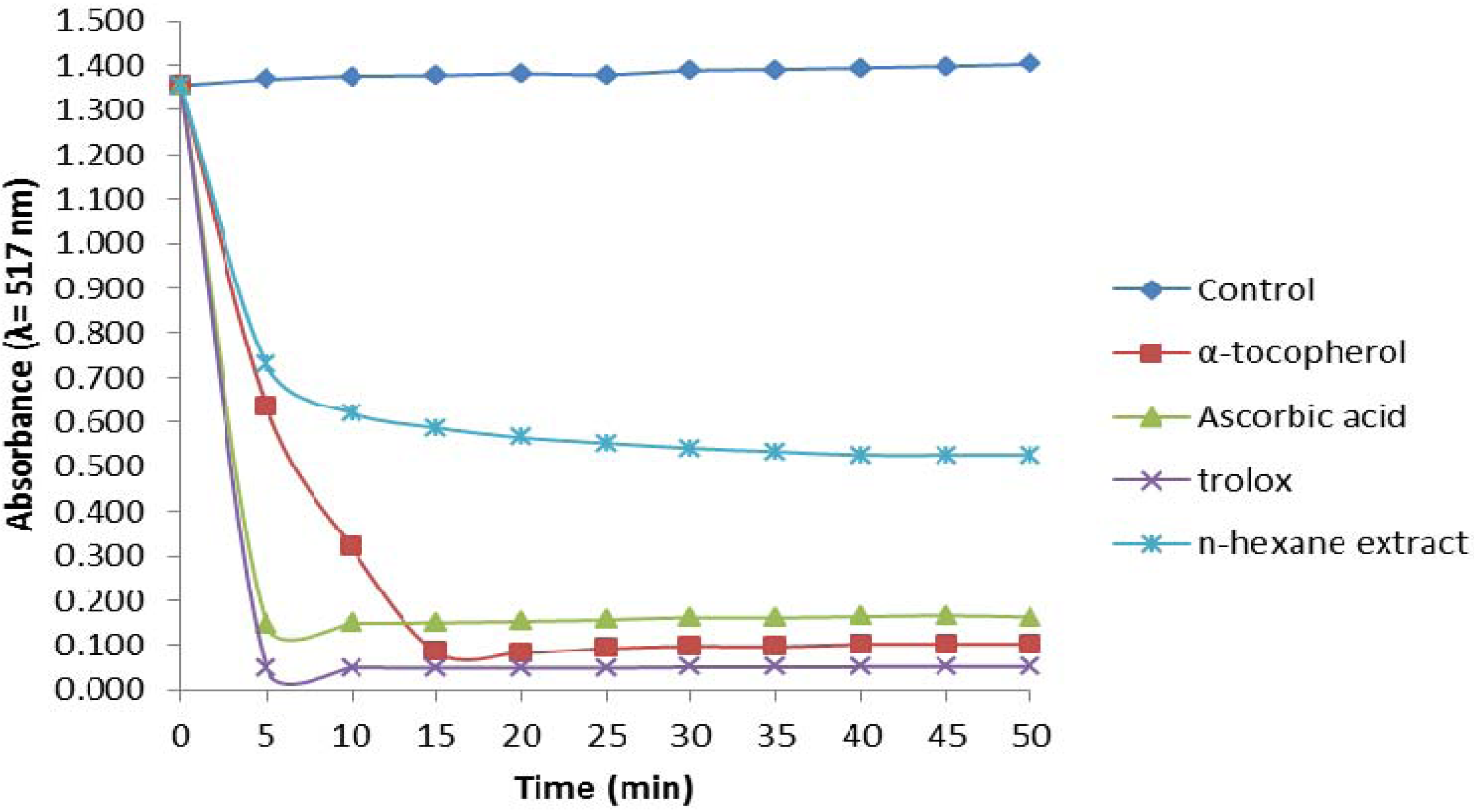
2.2.1. 1,1-Diphenyl-2-picrylhydrazyl (DPPH) Assay
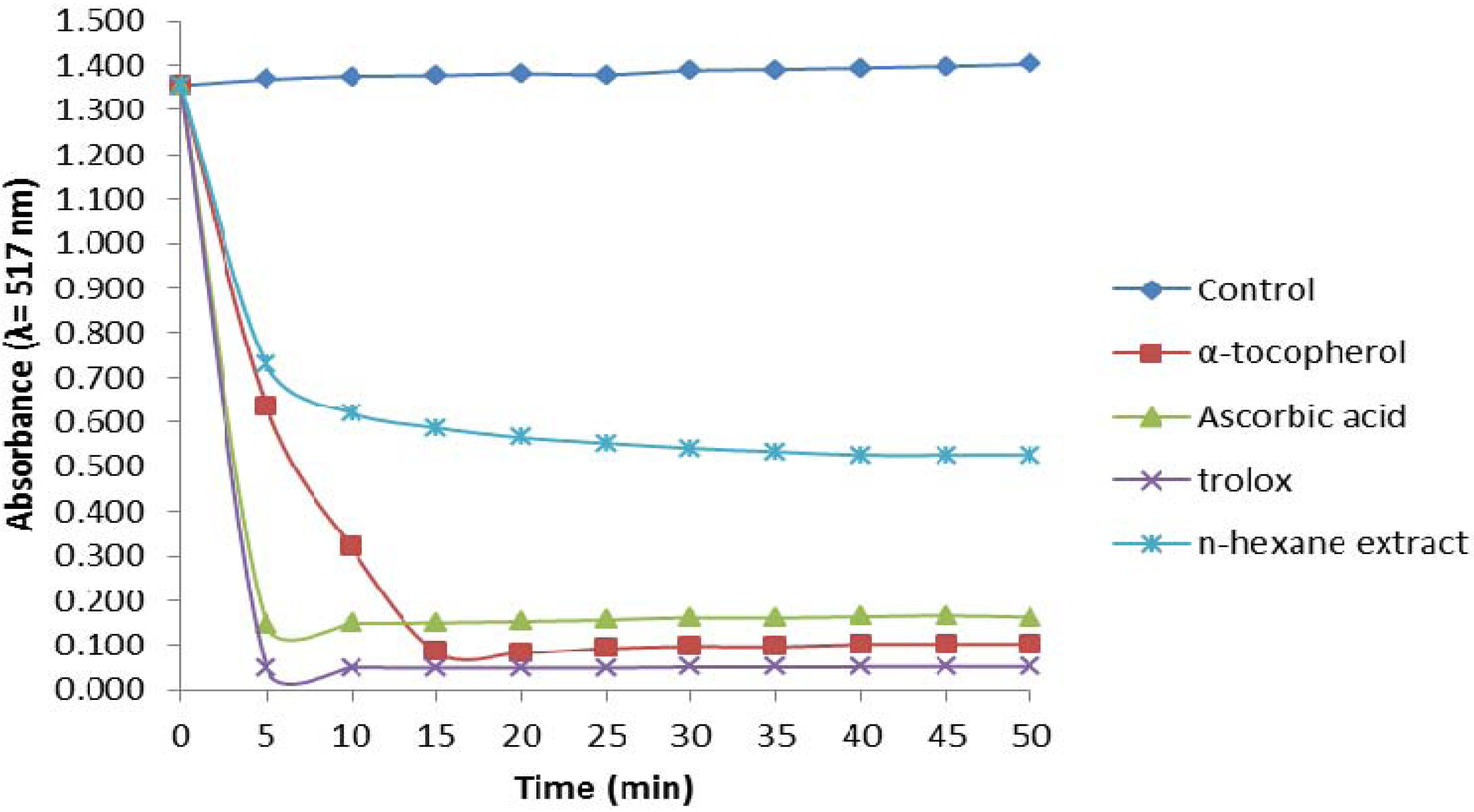
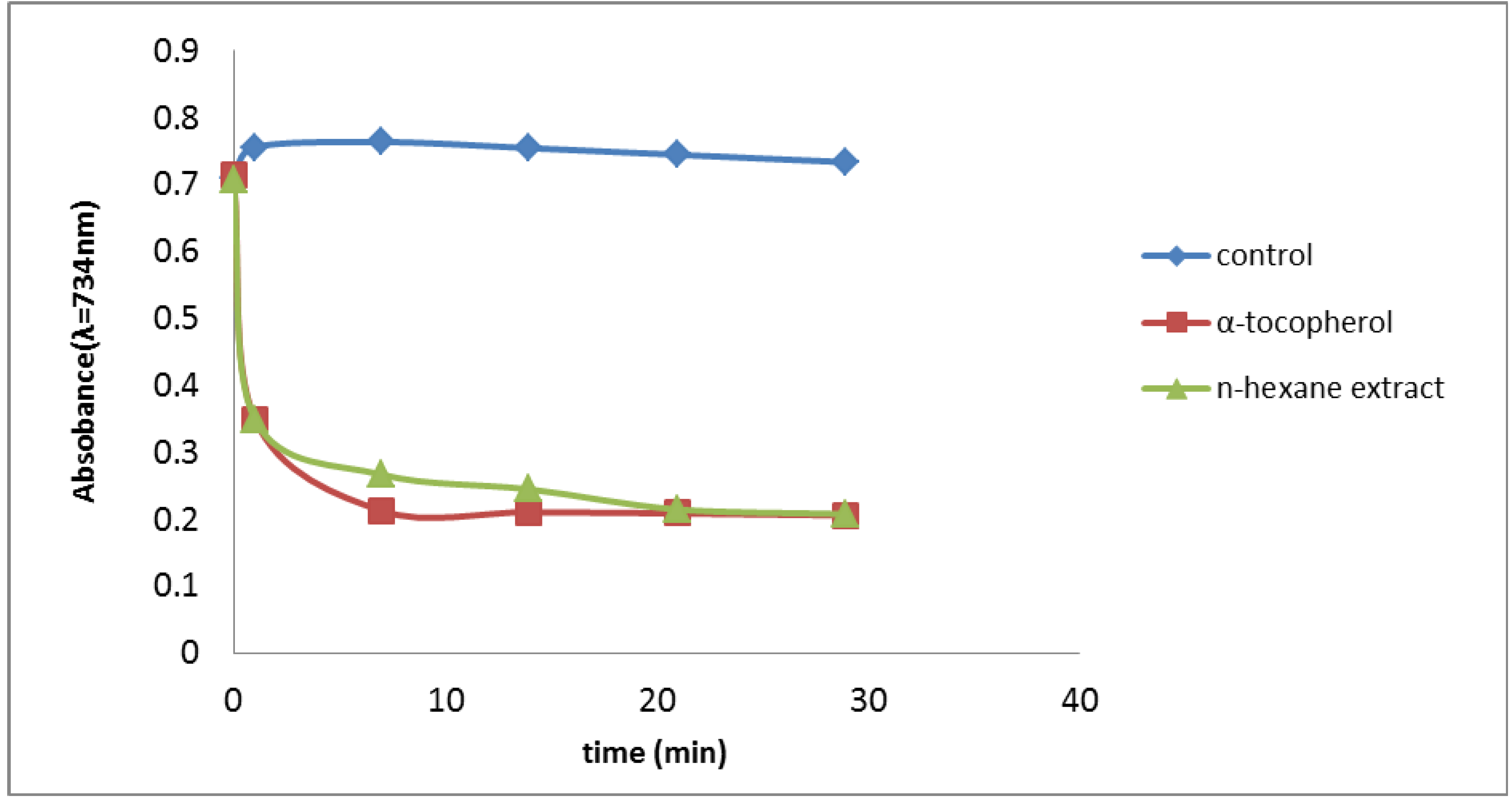
2.2.2. Trolox Equivalent Antioxidant Capacity (TEAC) Assay
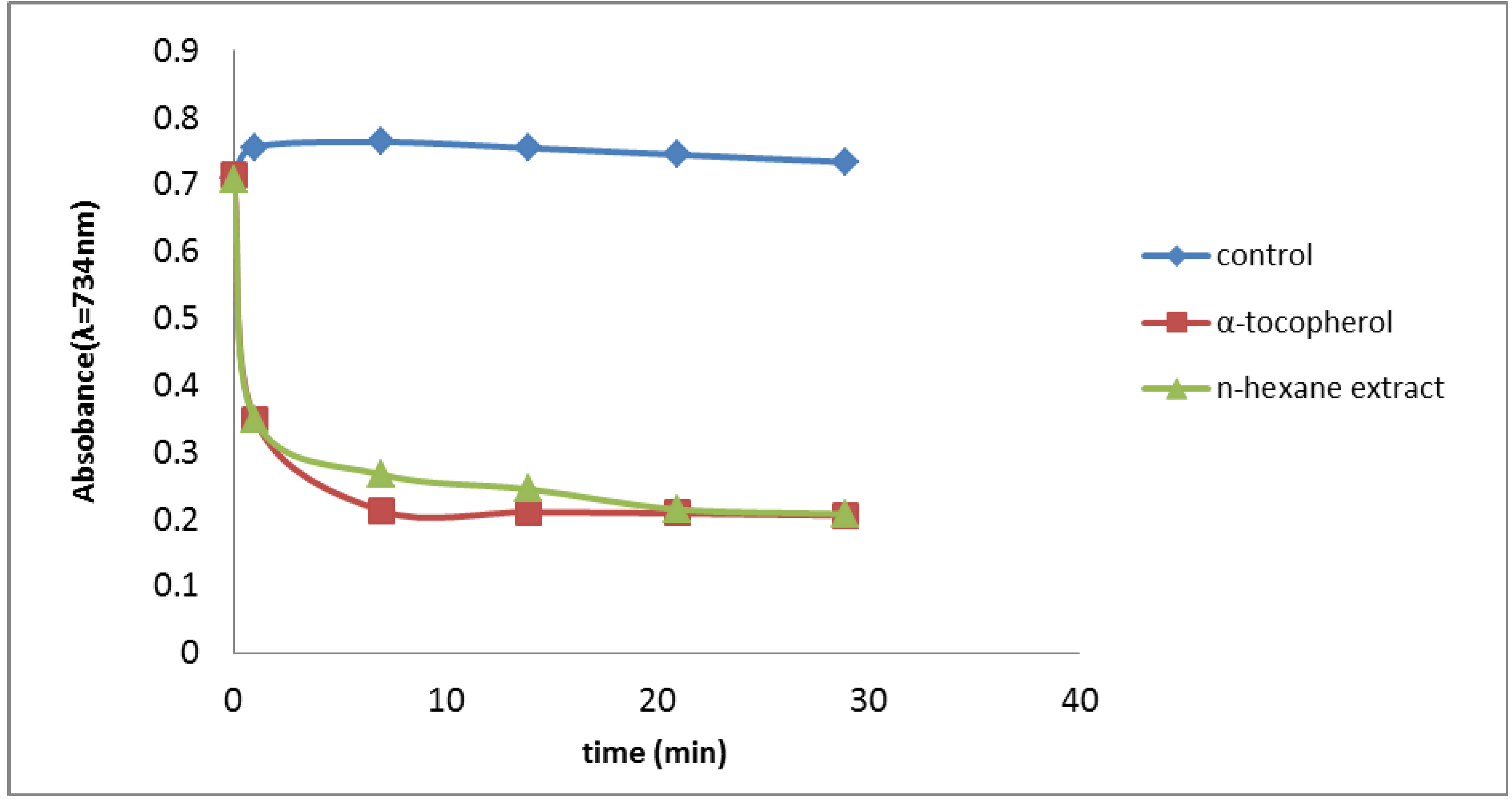
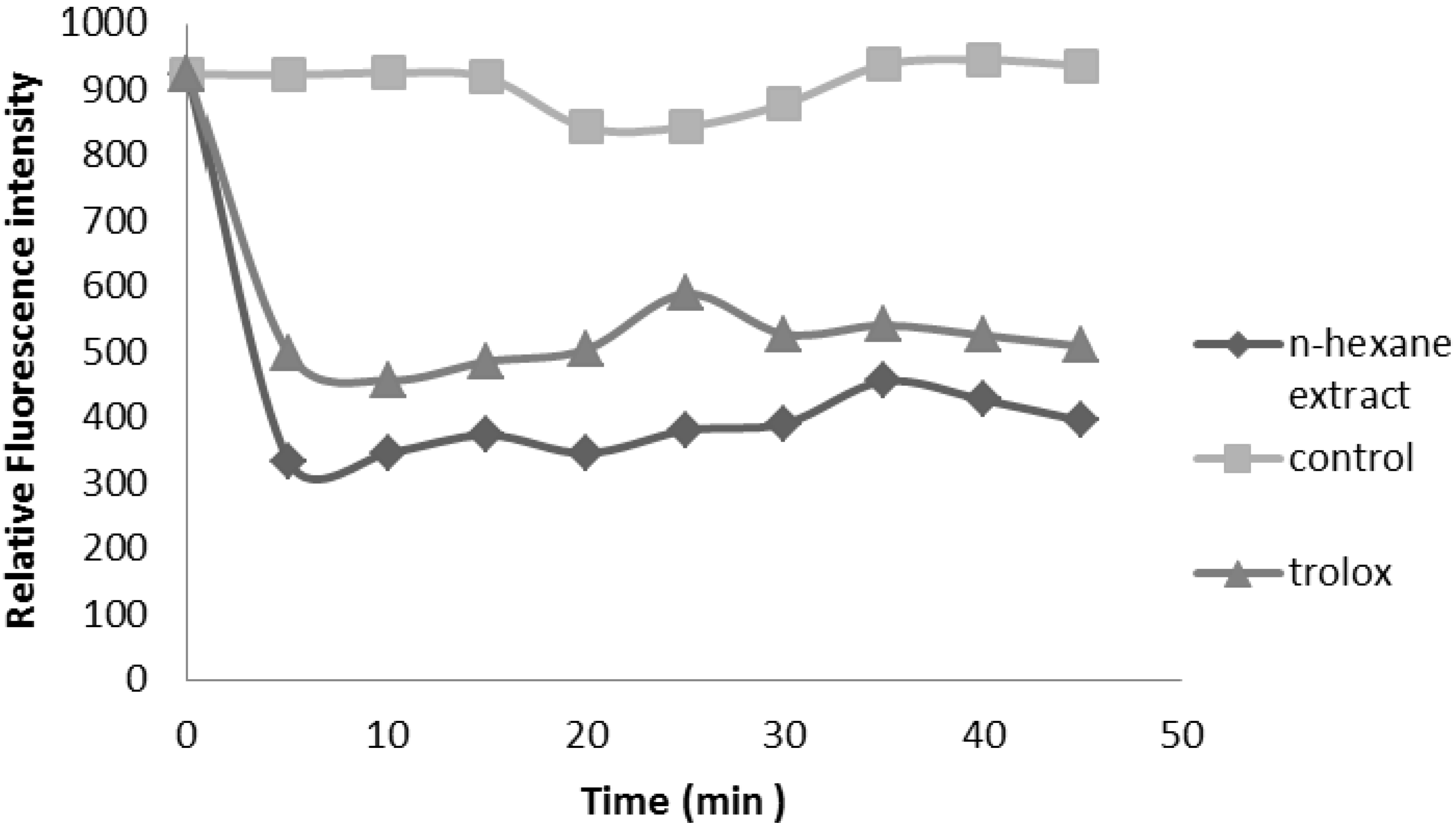
2.2.3. Oxygen Radical Absorbance Capacity (ORAC) Assay

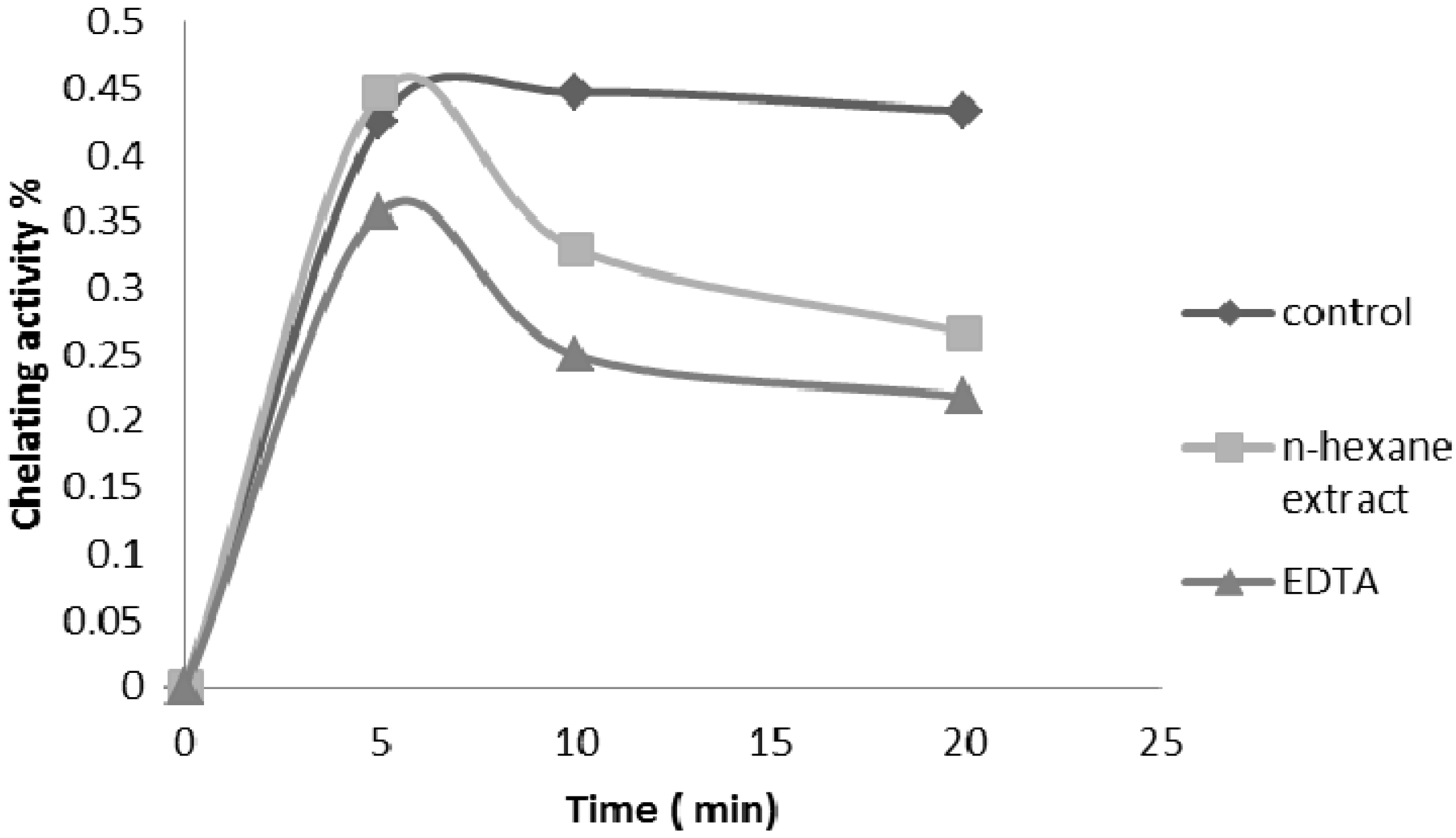
2.2.4. Ferrous Ion Chelating Ability
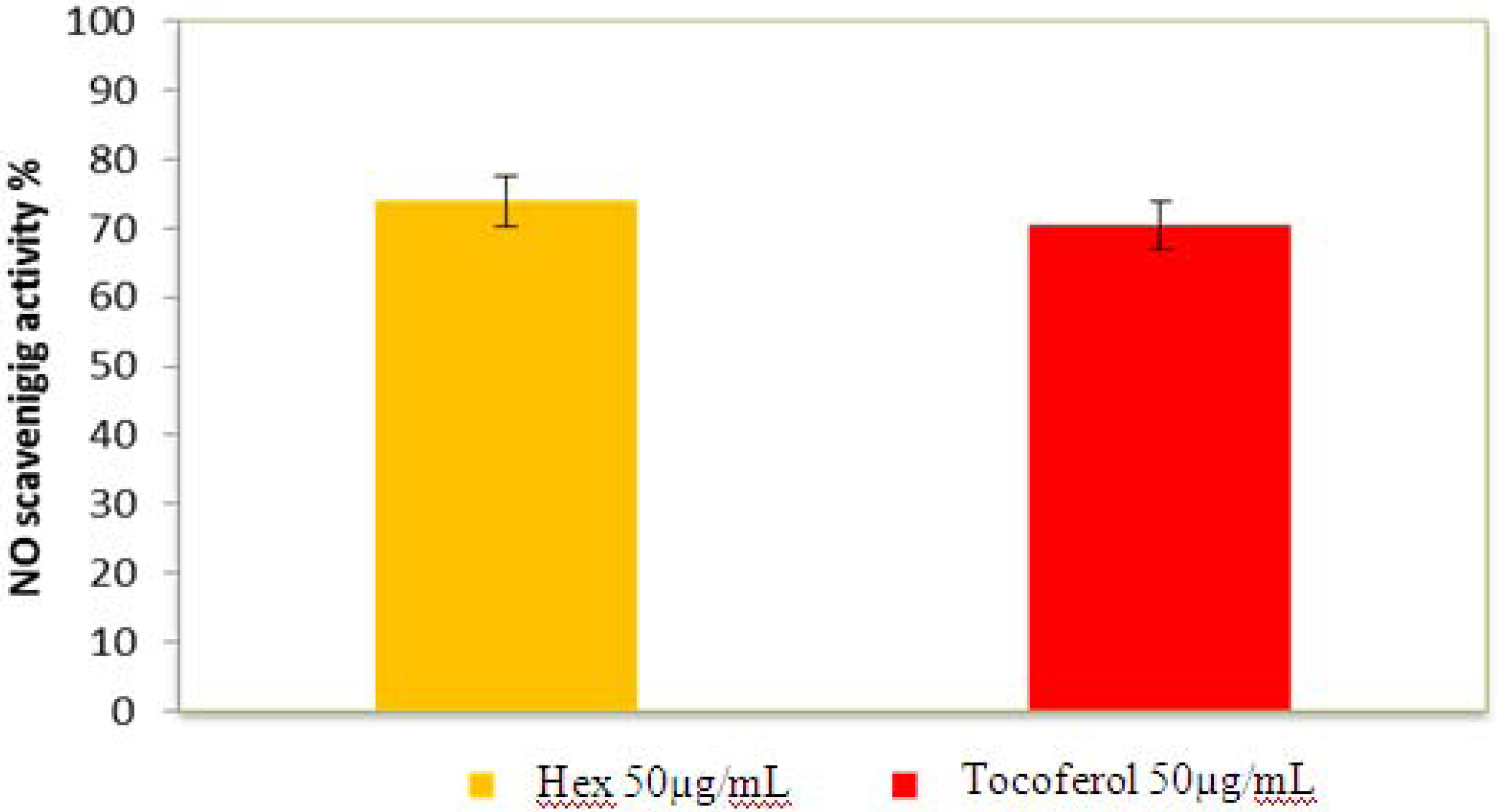
2.2.5. Nitric Oxide Radical Scavenging Assay
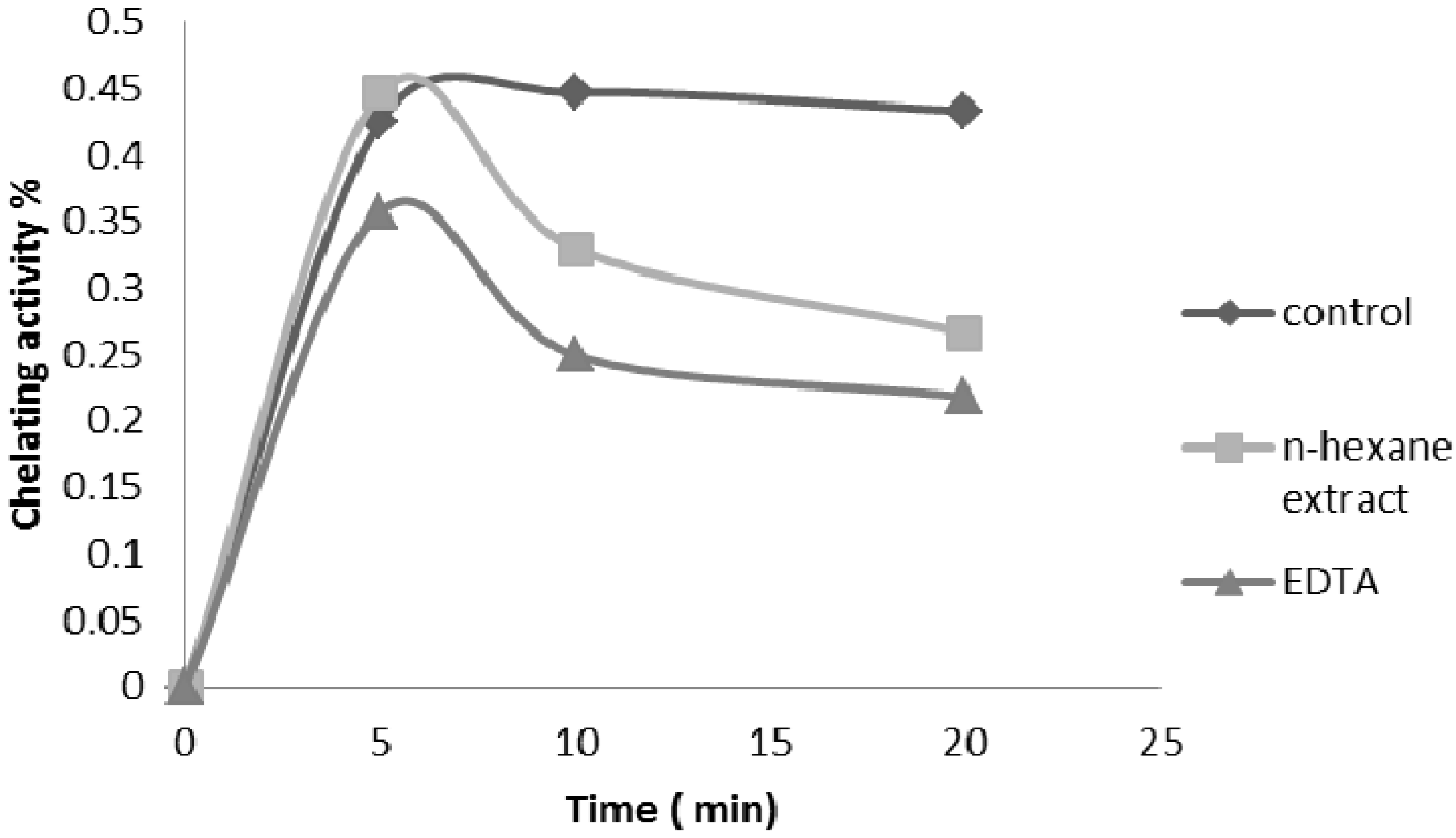
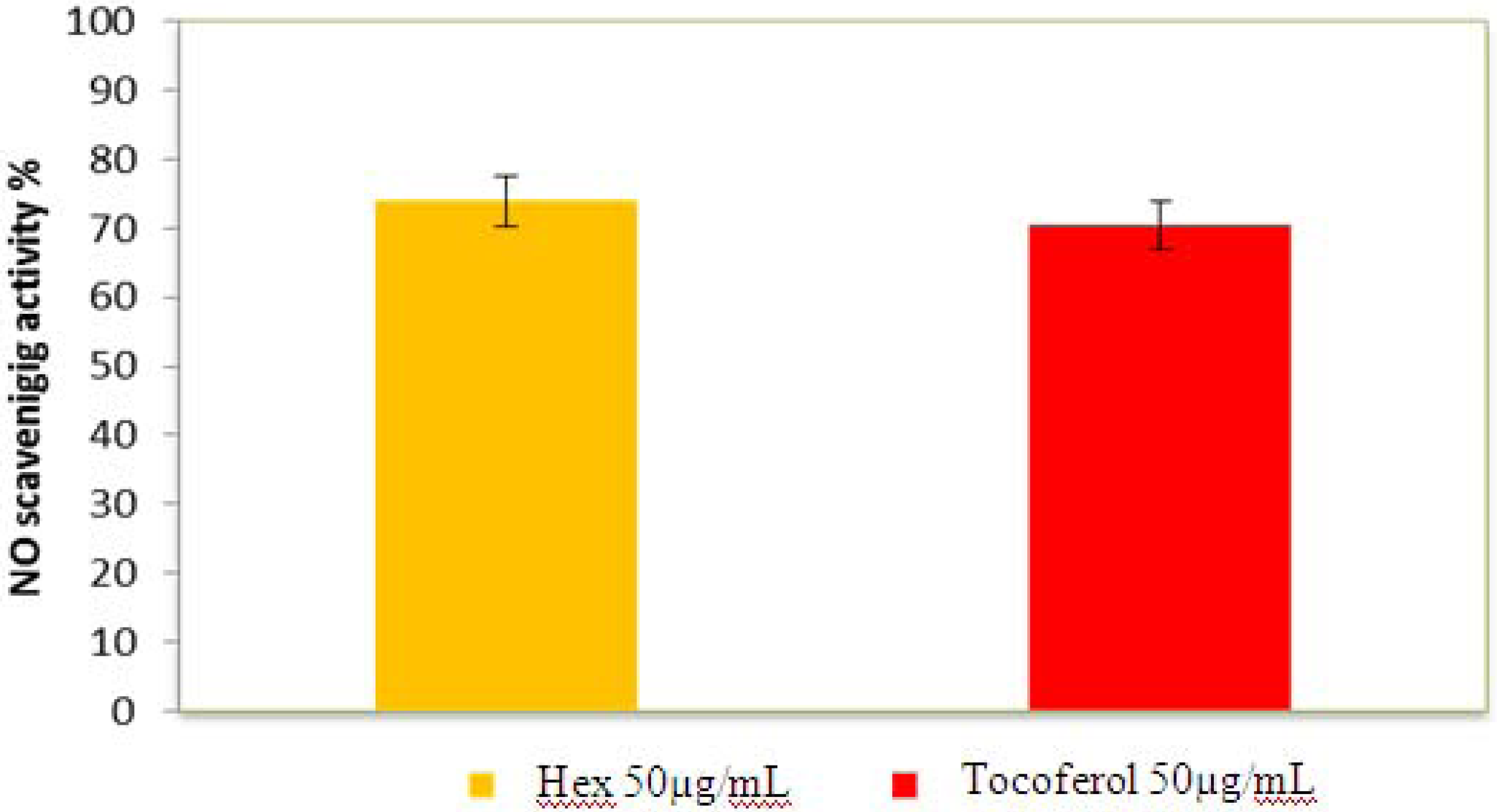
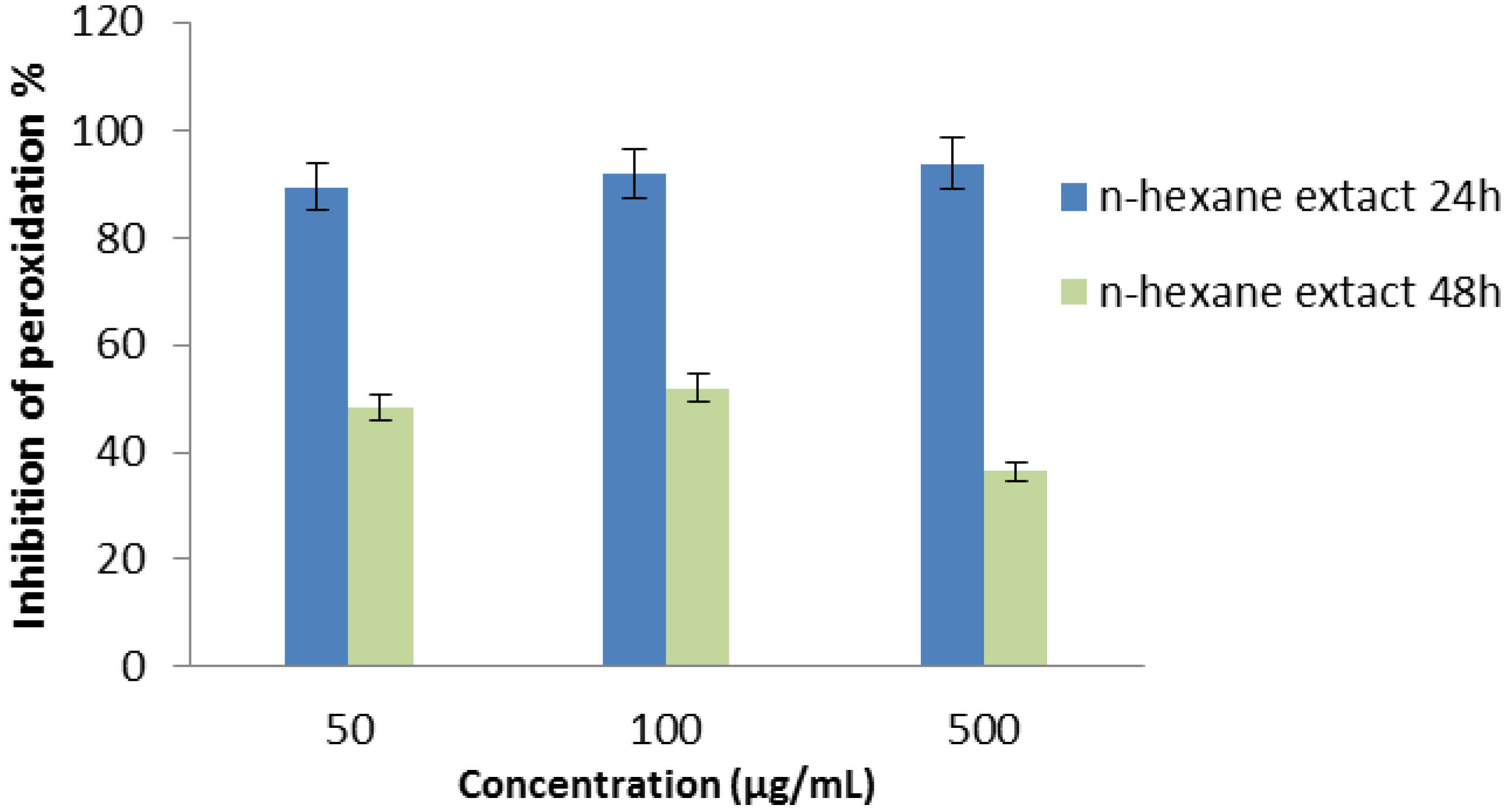
2.2.6. β-Carotene Bleaching (BCB) Assay
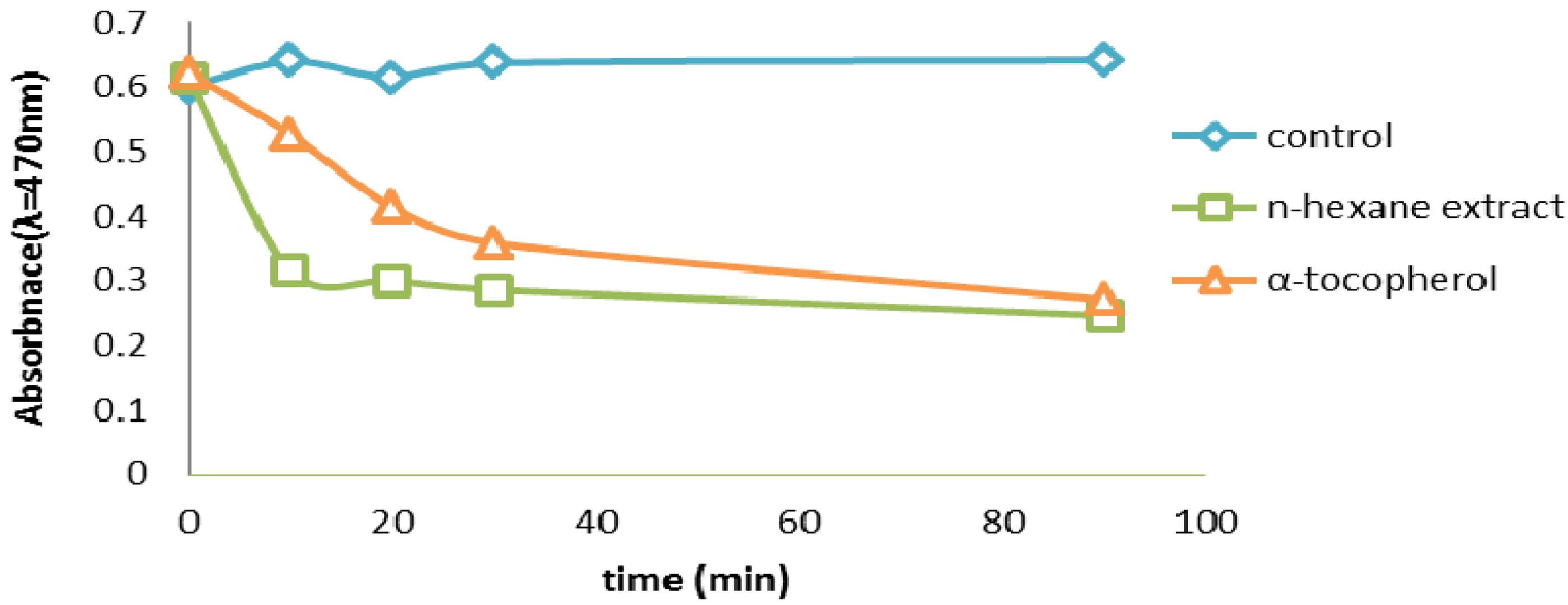
2.2.7. Thiocyanate Method
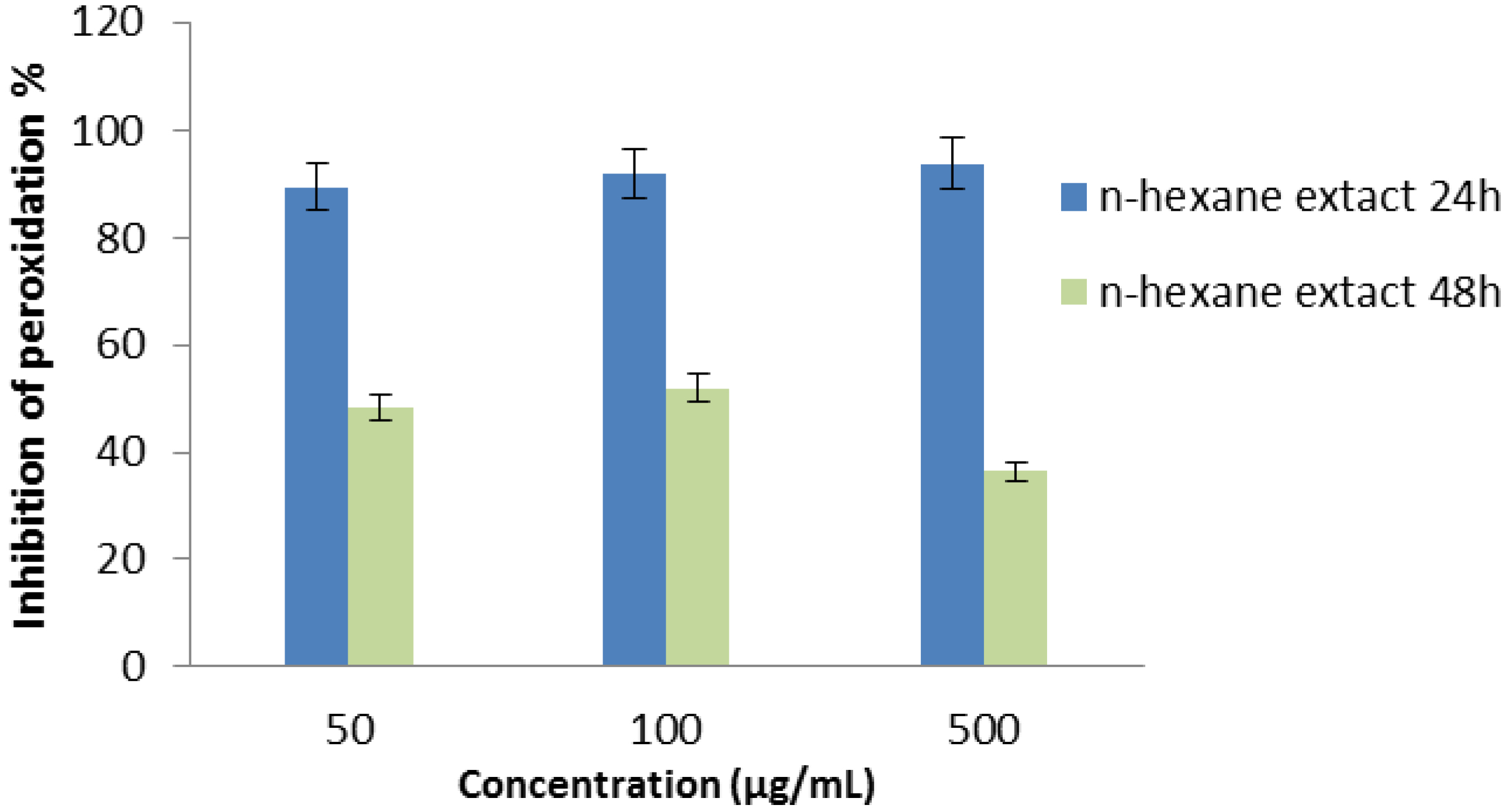
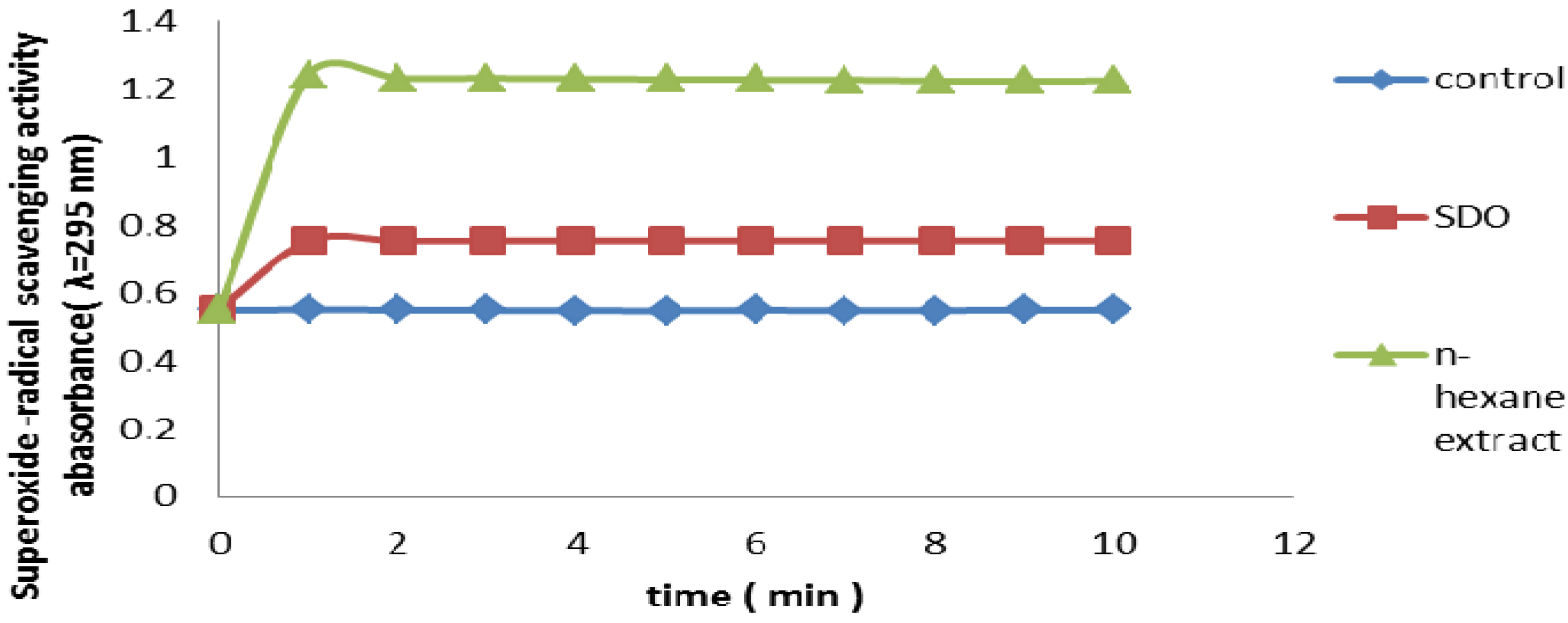
2.2.8. Superoxide Radical Scavenging Assay

2.3. Antioxidant Activity in Vivo
2.3.1. Measurements of GSH, SOD, CAT and GPx

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Normal Control | Diabetic control | Diabetic+HS (200 mg/kg) | Diabetic+HS (400 mg/kg) | Diabetic+ GB (5 mg/kg) |
|---|---|---|---|---|---|
| SOD-Liver | 7.65 ± 2.54 | 3.29 ± 1.57 a | 5.87 ± 1.29 c | 6.41 ± 1.79 b | 6.82 ± 2.18 c |
| SOD-Kidney | 14.03 ± 0.36 | 8.04 ± 2.49 a | 10.47 ± 1.37 b | 12.19 ± 2.58 b | 12.97 ± 1.26 b |
| SOD-Pancreas | 54.1 ± 3.54 | 35.76 ± 5.15 | 39.36 ± 3.42 b | 47.59 ± 3.52 c | 51.78 ± 4.77 b |
| CAT-Liver | 82.10 ± 1.79 | 43.35 ± 4.94 a | 61.25 ± 3.47 c | 70.63 ± 2.80 b | 74.67 ± 5.49 b |
| CAT-Kidney | 34.85 ± 2.65 | 21.49 ± 1.58 a | 30.27 ± 4.43 b | 33.90 ± 2.60 c | 34.32 ± 1.76 b |
| CAT-Pancreas | 59.6 ± 3.17 | 25.41 ± 3.12 a | 35.47 ± 2.80 c | 49.67 ± 4.21 c | 51.29 ± 4.48 c |
| GSH-Liver | 46.48 ± 2.34 | 23.72 ± 1.80 a | 37.17 ± 4.54 b | 43.26 ± 1.73 b | 42.87 ± 3.31 b |
| GSH-Kidney | 24.11 ± 0.73 | 5.78 ± 0.84 a | 16.73 ± 2.41 b | 20.21 ± 2.68 b | 19.86 ± 1.13 b |
| GSH-Pancreas | 11.9 ± 1.23 | 6.58 ± 0.91 a | 8.09 ± 2.39 c | 10.26 ± 1.87 c | 10.98 ± 1.54 c |
| GPx-Liver | 7.43 ± 2.17 | 4.56 ± 0.24 a | 5.69 ± 1.52 c | 6.23 ± 1.27 b | 5.90 ± 0.75 b |
| GPx-Kidney | 5.89 ± 0.78 | 3.49 ± 0.18 a | 4.11 ± 1.36 b | 4.67 ± 0.17 b | 4.53 ± 0.90 b |
| GPx-Pancreas | 4.12 ± 1.09 | 2.18 ± 0.63 a | 2.98 ± 0.53 c | 3.67 ± 0.82 b | 3.89 ± 0.68 c |
2.3.2. TBARS Levels in the Liver and Pancreas
| Group | Liver | Pancreas | |
|---|---|---|---|
| Normal control | 1.15 ± 0.35 | 0.484 ± 0.001 | |
| Diabetic control | 2.18 ± 0.61 a | 2.87 ± 0.054 a | |
| Diabetic + HS (200 mg/kg) | 1.73 ± 0. 12 b | 1.62 ± 0.061 b | |
| Diabetic+ HS (400 mg/kg) | 1.19 ± 0.23 b | 1.49 ± 0.036 b | |
| Diabetic+ GB (0.5 mg/kg) | 1.03 ± 0. 40 b | 1.22 ± 0.029 b | |
2.4. In Vitro Glycation of Proteins
2.4.1. BSA-Glucose and BSA-Methylglyoxal Assays
| Inducer | Treatment | AGEs IC50 (mg/mL) |
|---|---|---|
| Glucose | Hexane extract (HS) | 0.420 ± 0.062 |
| Aminoguanidine | 0.323 ± 0.081 | |
| Phloroglucinol | 0.070 ± 0.0049 | |
| Methylglyoxal | Hexane extract (HS) | 0.286 ± 0.039 |
| Aminoguanidine | 0.195 ± 0.021 | |
| Phloroglucinol | 0.060 ± 0.0072 |
2.4.2. Glycation of Hemoglobin
2.4.3. In Vitro Glycation of LDL
| Groups | GHb | HbA1c | Glycosylated protein |
|---|---|---|---|
| Negative Control | 8.9 ± 0.06 | 7.9 ± 0.98 | 15.3 ± 1.47 |
| Positive control | 27.6 ± 1.34 | 17.5 ± 1.56 | 23.7 ± 2.19 |
| Methanol extract | 18.6 ± 1.53 a | 14.9 ± 1.25 a | 19.1 ± 2.04 a |
| Glutathione | 8.1 ± 0.08 a | 9.0 ± 0.67 a | - |
| Aminoguanidine | - | - | 20.2 ± 1.87 a |
| Groups | MDA formation (nmol/mg LDL protein) |
|---|---|
| Control | 13.40 ± 1.58 |
| AB 5 µM | 22.25 ± 4.39 a |
| AB 10 µM | 34.27 ± 2.91 a |
| AB 10 µM + HS (5 µM) | 28.11 ± 3.89 a |
| Treatment | With EDTA | Without EDTA | ||
|---|---|---|---|---|
| Glycation | Oxidation | Glycation | Oxidation | |
| LDL | 2.9 ± 0.076 | 3.4 ± 0.51 | 3.7 ± 1.02 | 21.49 ± 2.62 |
| LDL+glucose | 17.6 ± 2.94 a | 4.8 ± 1.64 a | 22.83 ± 2.35 a | 58.761 ± 3.29 a |
| HS (5 µM) | 6.39 ± 1.39 a,b | 3.2 ± 0.46 b | 15.27 ± 0.95 a,b | 46.50 ± 4.30 a,b |
2.5. Anti-AGES Activity Assay in Vivo
Effect of HS on Renal Glucose, Mitochondrial TBA-reactive Substance, Renal Weight, and AGE Levels
| Groups | TBA-reactive substance (mmol/mg protein) | Renal Weight (g) | AGE (AU) | Renal glucose (mg/g wet tissue) |
|---|---|---|---|---|
| Normoglucemic | 1.85 ± 0.043 a | 0.75 ± 0.074 a | 16.03 ± 2.19 a | 0.81 ± 0.004 a |
| Diabetic | 2.75 ± 0.012 | 1.09 ± 0.065 | 24.25 ± 3.28 | 6.49 ± 1.67 |
| HS | 1.92 ± 0.082 a | 0.96 ± 0.012 a | 14.82 ± 2.38 a | 4.43 ± 1.36 a |
| Aminoguanidine | 1.87 ± 0.065 a | 0.94 ± 0.048 a | 12.87 ± 1.74 a | 4.10 ± 1.07 a |
3. Experimental
3.1. Plant Material and Preparation of Extracts
3.2. Estimation of Total Phenolic Content
3.3. Antioxidant Activity in Vitro
3.3.1. 1,1-Diphenyl-2-picrylhydrazyl (DPPH) Assay
3.3.2. Trolox Equivalent Antioxidant Capacity (TEAC) Assay
3.3.3. Oxygen Radical Absorbance Capacity (ORAC) Assay
3.3.4. Ferrous Ion Chelating Ability
3.3.5. Nitric Oxide Radical Scavenging Assay
3.3.6. β-Carotene Bleaching (BCB) Assay
3.3.7. Thiocyanate Method
3.3.8. Superoxide Radical Scavenging Assay
3.4. Antioxidant Activity Assay in Vivo
3.4.1. Animals
3.4.2. Streptozotocin (STZ)-Induced Diabetic Model
3.4.3. Experimental Design
3.4.4. Measurements of GSH, SOD, CAT and GPx
3.4.5. Statistical Analysis
3.5. Anti-AGES Activity Assay in Vitro
3.5.1. Bovine Serum Albumin (BSA)-Glucose Assay
3.5.2. BSA-Methylglyoxal Assay
3.5.3. Glycation of Hemoglobin
3.5.4. LDL Oxidation Measurement
3.5.5. In Vitro Glycation of LDL
3.6. Anti-AGES Activity Assay in Vivo
Mitochondrial TBA-Reactive Substance Level and AGE Level in Kidney
4. Conclusions
Acknowledgements
- Sample Availability: Samples of the hexane, chloroform and methanol extracts are available from the authors.
References
- Aruoma, O.I.; Halliwell, B.; Hoey, B.M.; Butler, J. The antioxidant action of N-acetylcysteine: Its reaction with hydrogen peroxide, Hydroxyl radical, Superoxide and hypochlorous acid. Free Radic. Biol. Med. 1989, 6, 593–597. [Google Scholar] [CrossRef]
- Ames, B.N. Dietary carcinogens and anticarcinogens: Oxygen radicals and degenerative diseases. Science 1983, 221, 1256–1264. [Google Scholar]
- Sumino, M.; Sekine, T.; Ruangrungsi, N.; Igarashi, K.; Ikegami, F. Ardisiphenols and other antioxidant principles from the fruits of Ardisia colorata. Chem. Pharm. Bull. 2002, 50, 1484–1487. [Google Scholar] [CrossRef]
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [CrossRef]
- Garcia, A.; Leyva, M.; Martinez, J.; Stashenko, E. Determinación de la composición química y actividad antioxidante in vitro del aceite esencial de Piper auritum Kunth (Piperaceae). Scientia Et Technica 2007, 33, 439–442. [Google Scholar]
- Navarro, M.C.; Montilla, M.P.; Cabo, M.M.; Galisteu, M.; Caceres, A.; Morales, C.; Berger, I. Antibacterial, Antiprotozoal and antioxidant activity of five plants seed in Izabal for infectious diseases. Phytother. Res. 2003, 17, 325–329. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improvement ABTS radical cation decolourization assay. Free Radic. Biol. Med. 1999, 26, 1232–1237. [Google Scholar]
- Sanchez-Moreno, C.; Larrauri, J.A.; Saura-Calixto, F. A procedure to measure the antiradical efficiency of polyphenols. J. Sci. Food Agric. 1998, 76, 270–276. [Google Scholar] [CrossRef]
- Frankel, E.N.; Meyer, A.S. The problems of using one-dimensional methods to evaluate multifunctional food and biological antioxidants. J. Sci. Food Agric. 2000, 80, 1925–1940. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 1999; pp. 325–327. [Google Scholar]
- Reichenbach, G.; Momi, S.; Gresele, P. Nitric oxide and its antithrombotic action in the cardiovascular system. Curr. Drug Targets Cardiovasc. Haematol. Disord. 2005, 5, 65–74. [Google Scholar] [CrossRef]
- Koleva, I.I.; van Beek, T.A.; Linssen, J.P.H.; de Groot, A.; Evstatieva, L.N. Screening of plant extracts for antioxidant activity: A comparative study of three testing methods. Phytochem. Anal. 2002, 13, 8–17. [Google Scholar] [CrossRef]
- Haber, F.; Weiss, J. The catalytic decomposition of hydrogen peroxide by iron salts. Proce. Royal Soc. Lon. 1934, 147, 332–351. [Google Scholar] [CrossRef]
- Goh, S.; Cooper, M.E. The role of advanced glycation end products in progression and complications of diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 1143–1152. [Google Scholar] [CrossRef]
- Nakagawa, S. Immunochemical detection of advanced glycation end products in lens crystallins from streptozotozin-induced diabetic rats. Diabetes 1993, 42, 345–350. [Google Scholar]
- Gohil, T.; Pathak, N.; Jivani, N.; Devmurari, Y.; Atel, J. Treatment with extract of Eugenia jambolana seed and Aegle marmelos leaf extracts prevents hyperglycemia and hyperlipidemia in alloxan-induced diabetic. Afr. J. Pharm. Pharmacol. 2010, 4, 270–275. [Google Scholar]
- Liu, P.; Hu, Y.; Guo, D.; Lu, B.; Rahman, K.; Mu, Li.; Wang, D. Antioxidant activity of oligosaccharide ester extracted from Polygala tenuifolia roots in senescense-accelerated mice. Pharm. Biol. 2010, 48, 828–833. [Google Scholar] [CrossRef]
- Monnier, V.M. Intervention against the Maillard reaction in vivo. Arch. Biochem. Biophys. 2003, 419, 1–15. [Google Scholar] [CrossRef]
- Dorman, H.; Peltoketo, A.; Hiltunen, R.; Tikkanen, M.J. Characterisation of the antioxidant properties of de-odorised aqueous extracts from selected Lamiaceae herbs. Food Chem. 2003, 83, 255–262. [Google Scholar] [CrossRef]
- Handl, S.; Hellweg, P.; Khol-Parisini, A.; Rossmann, B.; Thurner, K.; Luf, W.; Novak, J.; Zentek, J. Effect of oregano (O. majorana X O. vulgare) on performance and antioxidative capacity of quails fed a diet rich in Ω-3 fatty acids. J. Anim. Physiol. Anim. Nutr. 2008, 92, 242–245. [Google Scholar] [CrossRef]
- Pushparaj, P.N.; Low, H.K.; Manikandan, J.; Tan, B.K.; Tan, C.H. Antidiabetic effects of Cichorium intybus in streptozotocin-induced diabetic rats. J. Ethnopharmacol. 2007, 111, 430–434. [Google Scholar] [CrossRef]
- Wolff, S.P.; Jang, Z.Y.; Hunt, L.V. Protein glycation and oxidative stress in diabetes mellitus and ageing. Free Radic. Biol. Med. 1991, 10, 339–352. [Google Scholar] [CrossRef]
- Giardino, I.; Edelstein, D.; Brownlee, M. Nonenzymatic glycosylation in vitro and in bovine endothelial cells alters basic fibroblast growth factor activity. A model for intracellular glycosylation in diabetes. J. Clin. Invest. 1994, 94, 110–117. [Google Scholar] [CrossRef]
- Andrea, M.V.; Jarnes, W.R.; Philip, L.; Eva, L.F. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr. Rev. 2004, 25, 612–623. [Google Scholar] [CrossRef]
- Gutteridge, J.M. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin. Chem. 1995, 41, 1819–1828. [Google Scholar]
- Davies, K.J. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 2000, 50, 279–289. [Google Scholar] [CrossRef]
- Strain, J.J. Disturbances of micronutrient and antioxidant status in diabetes. Proc. Nutr. Soc. 1991, 50, 591–604. [Google Scholar] [CrossRef]
- Ceriello, A.M.D. New insights on oxidative stress and diabetic complications may lead to a ‘causal’ antioxidant therapy. Diabetes Care 2003, 26, 1589–1596. [Google Scholar] [CrossRef]
- Singleton, V.L.; Orthofer, R.; Lamuela-Raventós, R.M. Analysis of total phenols and other oxidation substrates and antioxidants by means of folin-ciocalteu reagent. Meth. Enzymol. 1999, 299, 152–178. [Google Scholar]
- Szabo, M.R.; Iditoiu, C.; Chambre, D.; Lupea, A.X. Improved DPPH determination for antioxidant activity spectrophotometric assay. Chem. Mater. Sci. 2007, 61, 214–216. [Google Scholar]
- Van den Berg, R.; Haenen, G.R.M.N.; Berg, H.; Bast, A. Applicability of an improved Trolox equivalent antioxidant capacity (TEAC) assay for evaluation of antioxidant capacity measurements of mixtures. Food Chem. 1999, 66, 511–517. [Google Scholar] [CrossRef]
- Cao, G.; Alessio, H.M.; Cutler, R.G. Oxygen-radical absorbance capacity assay for antioxidants. Free Rad. Biol. Med. 1993, 14, 303–311. [Google Scholar] [CrossRef]
- Jain, S.K.; Palmer, M. The effect of oxygen radicals metabolites and vitamin E on glycosylation of proteins. Free Radic. Biol. Med. 1997, 22, 593–596. [Google Scholar]
- Esterbauer, H.; Puhl, H.; Dieber-Rotheneder, M.; Waeg, G.; Rabl, H. Effect of antioxidants on oxidative modification of LDL. Ann. Med. 1991, 23, 573–581. [Google Scholar] [CrossRef]
- Li, D.; Devaraj, S.; Fuller, C.; Bucala, R.; Jialal, I. Effect of alpha-tocopherol on LDL oxidation and glycation: In vitro and in vivo studies. J. Lipid Res. 1996, 1978–1986. [Google Scholar]
- Duell, P.B.; Oram, J.F.; Biermann, E.L. Nonenzymatic glycation of HDL resulting in inhibition of high-affinity binding to cultured human fibroblasts. Diabetes 1990, 39, 1257–1263. [Google Scholar]
- Jung, K.; Pergande, M. Influence of cyclosporin A on the respiration of isolated rat kidney mitochondria. Fed. Eur. Biochem. Soc. Lett. 1985, 183, 167–169. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Perez Gutierrez, R.M.; Flores Cotera, L.B.; Gonzalez, A.M.N. Evaluation of the Antioxidant and Anti-glication Effects of the Hexane Extract from Piper auritum Leaves in Vitro and Beneficial Activity on Oxidative Stress and Advanced Glycation End-Product-Mediated Renal Injury in Streptozotocin-Treated Diabetic Rats. Molecules 2012, 17, 11897-11919. https://doi.org/10.3390/molecules171011897
Perez Gutierrez RM, Flores Cotera LB, Gonzalez AMN. Evaluation of the Antioxidant and Anti-glication Effects of the Hexane Extract from Piper auritum Leaves in Vitro and Beneficial Activity on Oxidative Stress and Advanced Glycation End-Product-Mediated Renal Injury in Streptozotocin-Treated Diabetic Rats. Molecules. 2012; 17(10):11897-11919. https://doi.org/10.3390/molecules171011897
Chicago/Turabian StylePerez Gutierrez, Rosa Martha, Luis B. Flores Cotera, and Adriana Maria Neira Gonzalez. 2012. "Evaluation of the Antioxidant and Anti-glication Effects of the Hexane Extract from Piper auritum Leaves in Vitro and Beneficial Activity on Oxidative Stress and Advanced Glycation End-Product-Mediated Renal Injury in Streptozotocin-Treated Diabetic Rats" Molecules 17, no. 10: 11897-11919. https://doi.org/10.3390/molecules171011897
APA StylePerez Gutierrez, R. M., Flores Cotera, L. B., & Gonzalez, A. M. N. (2012). Evaluation of the Antioxidant and Anti-glication Effects of the Hexane Extract from Piper auritum Leaves in Vitro and Beneficial Activity on Oxidative Stress and Advanced Glycation End-Product-Mediated Renal Injury in Streptozotocin-Treated Diabetic Rats. Molecules, 17(10), 11897-11919. https://doi.org/10.3390/molecules171011897