Abstract
A novel consecutive three-component synthesis of 3-(hetero)aryl-1H-pyrazoles via room temperature Sonogashira arylation of propynal diethylacetal used as a propargyl aldehyde synthetic equivalent has been disclosed. The final acetal cleavage-cyclocondensation with hydrazine hydrochloride at 80 °C rapidly furnishes the title compounds in a one-pot fashion.
1. Introduction
Pyrazoles, i.e., diazoles with two adjacent nitrogen atoms, display a rich chemistry and are used in numerous applications [1,2,3]. Their broad spectrum of biological activities, such as anti-hyperglycemic, analgesic, anti-inflammatory, antipyretic, anti-bacterial, and sedative-hypnotic properties, has attracted considerable interest in medicinal chemistry [4,5,6]. In addition, several 3,5-diaryl substituted pyrazoles also reversibly inhibit monoamine oxidases A and B [7]. In crop protection, 1,4-dialkyl-3,5-diphenylpyrazoles are known as potent herbicides [8]. Furthermore, pyrazoles are pluripotent as ligands in coordination chemistry [9,10,11,12], building blocks in heterocyclic synthesis [13,14], and units in supramolecular entities [15,16,17]. In addition, they have received attention as optical brighteners [18] and UV stabilizers [19] as well as photoinduced electron transfer systems [20,21].
Since multi-component reactions (MCRs) are highly efficient and address the fundamental principles of chemo-, regio-, and stereoselectivity, they are receiving an increasing interest in academia and industry [22,23,24,25,26,27,28,29,30,31,32,33,34,35]. With respect to economical and ecological considerations conceptually novel synthetic approaches [36,37,38,39] are directed towards the improvement of processes [40,41], while simultaneously reducing the input of work and the generation of waste. Therefore, MCRs are not only elegant, but also beneficial for the environment [39].
In the past years, we have developed and elaborated the concept of MCR syntheses of multiple classes of heterocycles via Sonogashira coupling of alkynes and (hetero)aroyl chlorides with alkynyl ketones, followed by Michael addition-cyclocondensation, eventually in a one-pot fashion [42,43,44,45]. Alkynyl ketones and aldehydes are ideal three-carbon building blocks as synthetic equivalents of β-dicarbonyl compounds. However, their electrophilicity is enhanced and the inherent regioselectivity is imposed in the Michael-type nucleophilic additions. Within this conceptual framework we have devised regioselective three-component [46] and four-component syntheses [47] of highly luminescent tri- and tetrasubstituted pyrazoles. Here, we communicate a straightforward consecutive three-component synthesis of 3-(hetero)aryl-1H-pyrazoles, i.e., monosubstituted pyrazole derivatives, via the concatenation of a Sonogashira alkynylation of (hetero)aryl iodides with the commercially available propynal diethylacetal, an in situ acetal cleavage, and a final cyclocondensation with hydrazine hydrochloride.
2. Results and Discussion
Monosubstituted 3-(hetero)aryl pyrazoles can be retrosynthetically traced back either to (hetero)aroyl chlorides and TMS-acetylene via 3-trimethylsilylalk-2-yn-1-ones or to (hetero)aryl halides and propargyl aldehyde (Scheme 1).
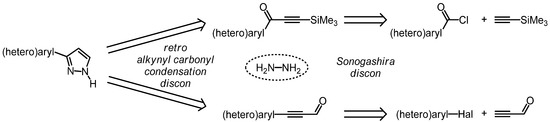
Scheme 1.
Retrosynthetic conception of three-component pyrazole syntheses by virtue of alkynyl carbonyl condensation.
Both disconnection approaches proceed via a retro alkynyl carbonyl condensation, thus leading to hydrazine and either to an alkynyl ketone [46,47,48] or a propargyl aldehyde [49,50,51,52,53,54]. While the former disconnection establishes the three-carbon skeleton by virtue of Sonogashira acylation [46,55,56], the latter analysis takes advantage of the ligation of the aryl substituent to a propargyl aldehyde synthon via Sonogashira arylation. Since the direct alkynylation of propargyl aldehyde has not been reported, presumably due to its pronounced electrophilicity, propynal diethylacetal apparently represents a suitable synthetic equivalent [57,58,59]. Although the most recent report on the coupling of aryl halides with propynal diethylacetal taking advantage of a tetradentate phosphane ligand based upon the cyclopentane scaffold affords high yields and requires low catalyst loadings, the requisite high reaction temperatures are less favorable for thermally sensitive functionalities [57].
Therefore, we first set out to optimize the reaction conditions for the Sonogashira coupling step. The transformation of p-iodoanisole (1a) and propynal diethylacetal (2) into 1-(p-anisyl)-3,3-diethoxyprop-1-yne (3a) under standard Sonogashira conditions at room temperature was chosen as a model reaction (Scheme 2). The amount of triethylamine, the solvent, and the reaction time were modified (Table 1).
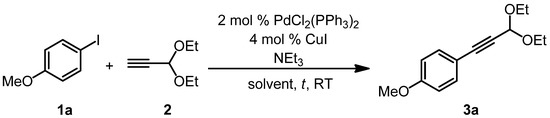
Scheme 2.
Optimization of the Sonogashira coupling of p-iodoanisole (1a) and propynal diethylacetal (2) to furnish 1-(p-anisyl)-3,3-diethoxyprop-1-yne (3a).

Table 1.
Optimization study for the synthesis of 1-(p-anisyl)-3,3-diethoxyprop-1-yne (3a).
| Entry 1 | Alkyne 2 | NEt3 | Solvent | Reaction time | Yield of 3a 2 |
|---|---|---|---|---|---|
| 1 | 1.0 equiv. | 2.0 equivs. | THF | 2 h | 64% |
| 2 | 1.0 equiv. | 2.0 equivs. | 1,4-dioxane | 2 h | 80% |
| 3 | 1.1 equivs. | 1.1 equivs. | 1,4-dioxane | 2 h | 64% |
| 4 3 | 1.1 equivs. | 2.0 equivs. | 1,4-dioxane | 2 h | 89% |
| 5 | 1.1 equivs. | 2.0 equivs. | 1,4-dioxane | 1 h | 87% |
| 6 | 1.1 equivs. | 2.0 equivs. | 1,4-dioxane | 3 h | 91% |
1 The reactions were carried out on a 2.0 mmol scale (c(1a) = 0.4 M); 2 All yields were determined after isolation and purification on silica gel; 3 This reaction was additionally carried out on a 5.0 mmol scale [c(1a) = 0.4 M] yielding 81% of 3a.
1,4-Dioxane is apparently a better solvent for the coupling than THF (Table 1, entries 1 and 2), a small excess of alkyne 2 furnishes a higher yield (Table 1, entry 3), and two equivalents of triethylamine are more favorable for complete conversion (Table 1, entries 4–6), which is reached after 2 h (Table 1, entry 4). Therefore, the parameters of entry 4 were considered to be optimal for the first step of the sequence (vide infra).
Due to the sensitivity of the deprotected 3-aryl propargyl aldehydes towards oligo- and polymerization in the presence of nucleophiles we then addressed the sequential acetal cleavage-cyclocondensation of 1-(p-anisyl)-3,3-diethoxyprop-1-yne (3a) in the presence of hydrazine hydrochloride [60,61,62] giving rise to the formation of 3-(p-anisyl)-1H-pyrazole (4a) (Scheme 3, Table 2).
The optimal temperature of the sequential acetal cleavage-cyclocondensation is 80 °C (Table 2, entries 1–4), as preliminary experiments showed that lower temperatures yield poorer results and higher temperatures lead to no further improvement. Conductive heating (Table 2, entries 2–4) turns out to be superior to dielectric heating in the microwave oven (Table 2, entry 1).
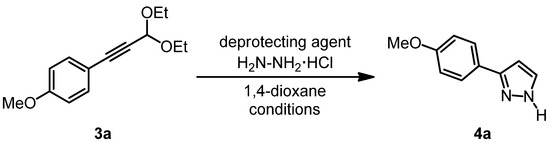
Scheme 3.
Optimization of the acetal cleavage-cyclocondensation of 1-(p-anisyl)-3,3-diethoxyprop-1-yne (3a) to furnish 3-(p-anisyl)-1H-pyrazole (4a).
Table 2.
Optimization study for the synthesis of 3-(p-anisyl)-1H-pyrazole (4a).
| Entry | Deprotecting agent 1 | H2N-NH2·HCl | Reaction temperature | Reaction time | Yield of 4a 2 |
|---|---|---|---|---|---|
| 1 3 | H2O | 2.0 equivs. | 80 °C (MW) | 15 min | 53% |
| 2 4 | H2O | 2.0 equivs. | 80 °C 5 | 15 min | 65% |
| 3 3 | H2O | 2.0 equivs. | 80 °C 5 | 1 h | 62% |
| 4 4 | H2O | 1.0 equiv. | 80 °C 5 | 15 min | 22% |
| 5 3 | HClaq (1 N) | 2.0 equivs. | 130 °C 5 | 10 min | 55% |
1 2.5 mL/mmol were added; 2 All yields were determined after isolation and purification on silica gel; 3 The reactions were carried out on a 0.5 mmol scale [c(3a) = 0.4 M]; 4 The reactions were carried out on a 1.0 mmol scale [c(3a) = 0.4 M]; 5 Preheated oil bath.
While the complete conversion is already achieved within 15 min (Table 2, entries 2 and 3), the amount of hydrazine hydrochloride is crucial for the success of the sequence (Table 2, entries 2 and 4). Strongly Brønsted acidic conditions (Table 2, entry 5) are not required and the combination of water and hydrazine hydrochloride is sufficient to trigger the acetal cleavage and the Michael addition followed by cyclocondensation. Thereafter, the combination of Sonogashira coupling and sequential acetal cleavage-cyclocondensation was performed with the established model system (Scheme 4, Table 3).
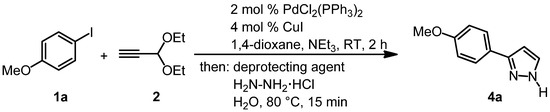
Scheme 4.
Optimization of the consecutive three-component Sonogashira coupling-acetal cleavage-cyclocondensation synthesis of 3-(p-anisyl)-1H-pyrazole (4a).
Table 3.
Optimization study for the one-pot synthesis of 3-(p-anisyl)-1H-pyrazole (4a).
| Entry | Deprotecting agent | Yield of 4a 1 |
|---|---|---|
| 1 | not isolated | |
| 2 | 1.0 equiv. of HClaq (1 N) | 50% |
| 3 | 1.0 equiv. of CH3COOH | 25% |
| 4 | 1.0 equiv. of PTSA·H2O | 56% |
| 5 | 2.0 equivs. of PTSA·H2O | 55% |
1 All yields were determined after isolation and purification on silica gel.
Although the acetal cleavage-cyclocondensation conditions only require a moderate acidity (vide supra), the excess triethylamine from the coupling step has to be buffered (Table 3, compare entry 1 with entries 2–5), which is most efficiently achieved by addition of a stoichiometric amount of p-toluenesulfonic acid (PTSA) (Table 3, entry 4).
With these optimized conditions for the whole sequence in hand, the stage was set for testing the range of applicable (hetero)aryl iodides 1 in this novel consecutive three-component synthesis of 3-(hetero)aryl-1H-pyrazoles 4. After coupling of (hetero)aryl iodides 1 with propynal diethylacetal (2) at room temperature for two hours the subsequent addition of PTSA·H2O, hydrazine hydrochloride, and water, heating to 80 °C for 15 min gave rise to the isolation of 3-(hetero)aryl-1H-pyrazoles 4 in moderate to good yields (Scheme 5).
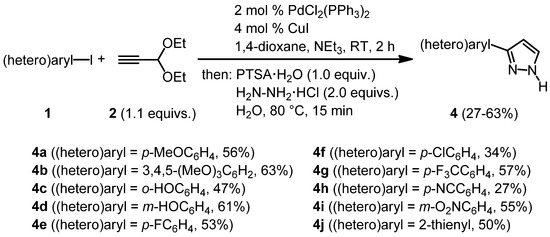
Scheme 5.
Consecutive three-component Sonogashira coupling-acetal cleavage-cyclocondensation synthesis of 3-(hetero)aryl-1H-pyrazoles 4.
This first study on the methodological scope of this novel three-component pyrazole synthesis shows that electron-rich as well as electron-poor aryl substituents can be readily introduced. Also, chloro and fluoro substituents and even unprotected phenol derivatives, which are incompatible with the coupling of acid chlorides, are well tolerated. Although a thienyl substituent can be carried through the sequence, all attempts to react other heteroaryl iodides such as 3- and 4-pyridyl iodides under the standard conditions met with failure.
3. Experimental
3.1. General
All cross coupling reactions were carried out in oven dried Schlenk or microwave tubes using septa and syringes under a nitrogen or argon atmosphere. Dry tetrahydrofuran and 1,4-dioxane were supplied by a MBraun system MB-SPS-800 solvent purification system. Chemicals were either commercially obtained from ABCR GmbH & Co KG, Acros Organics, Alfa Aesar GmbH & Co KG, Fluka, Merck KGaA, Riedel-de Haën, Sigma-Aldrich Co., and used as supplied or were already available in the research group.
All products were purified via column chromatography on silica gel 60 M (0.04–0.063 mm) from Macherey-Nagel using the flash technique under a pressure of 2 bar. The crude mixtures were absorbed on Celite® 545 (0.02–0.10 mm) from Merck KGaA, Darmstadt before chromatographic purification. The reaction progress was observed qualitatively using TLC Silica gel 60 F254 aluminium sheets. The spots were detected with UV light at 254 nm and with aqueous potassium permanganate solution.
1H-, 13C-, and 13C-135 DEPT NMR spectra were recorded on Bruker AVIII-300 spectrometer, using CDCl3 or DMSO-d6 as solvents. The resonances of CDCl3 or DMSO-d6 were locked as internal standards (CDCl3: 1H δ 7.26, 13C δ 77.0; DMSO-d6: 1H δ 2.49, 13C δ 39.7). The multiplicities of signals were abbreviated as follows: s: singlet; d: doublet; dd: doublet of doublets; dq: doublet of quartets; ddd: doublet of doublets of doublets; t: triplet; tdd: triplet of doublets of doublets; m: multiplet and br: broad signal. The type of carbon atom was determined on the basis of 13C-135 DEPT NMR spectra. For the description of the 13C-NMR spectra primary carbon atoms are abbreviated as CH3, secondary carbon atoms as CH2, tertiary carbon atoms as CH, and quaternary carbon atoms as Cquat. EI mass spectra were measured on Finnigan MAT 200 spectrometer. IR spectra were either obtained on Shimadzu IRAffinity or on Bruker Vector 22 FT-IR instruments. The intensity of the signals was abbreviated as follows: s (strong), m (medium), w (weak). The melting points (uncorrected) were measured on a Reichert Thermovar apparatus. Combustion analyses were carried out on a Perkin Elmer Series II Analyser 2400 in the microanalytical laboratory of the Institut für Pharmazeutische und Medizinische Chemie der Heinrich-Heine-Universität Düsseldorf.
3.2. Experimental Procedure for the Synthesis of 1-(3,3-Diethoxyprop-1-yn-1-yl)-4-methoxybenzene (3a)
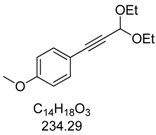
In a screw-cap Schlenk tube PdCl2(PPh3)2 (28 mg, 2 mol%, 0.04 mmol) and CuI (16 mg, 4 mol%, 0.08 mmol) were tempered. Then, 1-iodo-4-methoxybenzene (1a, 278 mg, 2.00 mmol), 3,3-diethoxyprop-1-yne (2, 0.32 mL, 2.20 mmol, 1.1 equiv.), degassed dry 1,4-dioxane (5.00 mL), and triethylamine (0.55 mL, 4.00 mmol, 2.0 equiv.) were successively added under an argon atmosphere. The mixture was stirred for 2 h until complete conversion of 1a (TLC monitoring). The crude mixture was absorbed onto Celite® and purified chromatographically on silica gel with hexanes/ethylacetate (30:1, Rf = 0.26) to give the alkyne 3a (415 mg, 1.80 mmol, 89%) as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.27 (t, J = 7.1 Hz, 6H), 3.65 (dq, J = 9.5 Hz, J = 7.1 Hz, 2H), 3.80 (s, 3H), 3.81 (dq, J = 9.5 Hz, J = 7.1 Hz, 2H), 5.48 (s, 1H), 6.80–6.85 (m, 2H), 7.38–7.43 (m, 2H). 13C-NMR (CDCl3, 75 MHz): δ 15.1 (CH3), 55.2 (CH3), 60.8 (CH2), 83.0 (Cquat), 85.2 (Cquat), 91.7 (CH), 113.8 (CH), 113.9 (Cquat), 133.4 (CH), 159.9 (Cquat). EI + MS (m/z (%)): 234 (M+, 7), 205 ((M-C2H5)+, 2), 190 (16), 189 ((M-C2H5O)+, 100), 175 (12), 162 (20), 161 (95), 160 (C10H8O2+, 14), 159 (22), 133 (28), 132 (18), 131 (C9H7O+, 10), 118 (12), 89 (15). IR (Oil; Shimadzu IRAffinity): 2974 (w) cm−1, 2932 (w), 2884 (w), 2839 (w), 2224 (w), 1604 (w), 1570 (w), 1508 (s), 1458 (w), 1443 (w), 1391 (w), 1356 (w), 1329 (w), 1290 (m), 1246 (s), 1172 (m), 1090 (s), 1049 (s), 1032 (s), 1005 (s), 978 (m), 895 (w), 831 (s), 806 (m), 739 (w), 665 (w), 642 (w). Anal. calcd. for: C14H18O3 (234.3): C 71.77, H 7.74; Found: C 71.75, H 7.71.
3.3. Typical Experimental Procedure (Synthesis of Pyrazole 4b)
In a screw-cap Schlenk tube PdCl2(PPh3)2 (28 mg, 0.04 mmol) and CuI (16 mg, 0.08 mmol) were tempered. Then, 1-iodo-3,4,5-trimethoxybenzene (1b, 600 mg, 2.00 mmol), 3,3-diethoxyprop-1-yne (2, 0.32 mL, 2.20 mmol), degassed dry 1,4-dioxane (5.00 mL), and triethylamine (0.55 mL, 4.00 mmol) were successively added under an argon atmosphere. The mixture was stirred for 2 h at room temperature (water bath) until complete conversion of 1b (according to TLC monitoring). Then, hydrazine hydrochloride (280 mg, 4.00 mmol), deionized water (5.00 mL), and PTSA·H2O (388 mg, 2.00 mmol) were added. The mixture was stirred at 80 °C (preheated oil bath) until complete conversion (according to TLC monitoring). After extraction with CH2Cl2 (10 × 10 mL) the combined organic phases were dried with MgSO4. The solvents were removed in vacuo, the crude mixture was purified by chromatography on silica gel (CH2Cl2/MeOH/NH3 (aq)) to give 3-(3,4,5-tri-methoxyphenyl)-1H-pyrazole (4b, 295 mg, 63%) as an orange solid, Mp 95 °C. 1H-NMR (CDCl3, 300 MHz): δ 3.88 (s, 3H), 3.90 (s, 6H), 6.58 (d, J = 1.7 Hz, 1H), 6.99 (s, 2H), 7.61 (d, J = 1.8 Hz, 1H). 13C-NMR (CDCl3, 75 MHz): δ 56.1 (CH3), 60.9 (CH3), 102.5 (CH), 103.0 (CH), 127.9 (Cquat), 132.6 (CH), 138.0 (Cquat), 149.5 (Cquat), 153.5 (Cquat). EI+MS (m/z (%)): 235 (13), 234 (M+, 100), 219 ((M−CH3)+, 68), 205 ((M−HN2)+, 2), 192 ((M−CH2N2)+, 2), 191 (30), 176 (30), 161 (20), 159 (15), 149 (13), 131 (15), 105 (22), 58 (23), 43 (67). IR (neat; Shimadzu IRAffinity): 3576 (w) cm−1, 3146 (w), 3041 (w), 2995 (w), 2963 (w), 2930 (m), 2860 (w), 2826 (w), 1730 (w), 1587 (m), 1516 (m), 1474 (m), 1449 (m), 1416 (m), 1325 (m), 1288 (w), 1261 (w), 1238 (m), 1121 (s), 1092 (m), 1057 (m), 1034 (w), 995 (s), 928 (w), 889 (w), 845 (s), 826 (m), 760 (s), 739 (m), 665 (w), 627 (m). Anal. calcd. for C12H14N2O3 (234.3): C 61.53, H 6.03, N 11.96; Found: C 61.63, H 6.29, N 11.68.
The pyrazoles 4a, 4c–j were similarly prepared following the general procedure described above. The corresponding reactions conditions and work-ups are summarized in Table 4.
Table 4.
Synthesized 3-substituted 1H-pyrazoles 4 via alkynylation-cyclocondensation sequence.
| Entry | (Hetero)Aryl iodide 1 | Reaction time t | Pyrazole 4 | Chromatographic |
|---|---|---|---|---|
| (2.00 mmol) | 2nd step | (isolated yield) | purification | |
| 1 | 1a, 1-Iodo-4-methoxybenzene | 15 min | 195 mg | CH2Cl2/MeOH/NH3 |
| Merck | (1.12 mmol, 56%) | |||
| 478 mg | 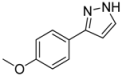 | |||
| 4a | 100:2:1 | |||
| 2 | 1b, 1-Iodo-3,4,5-trimethoxybenzene | 15 min | 295 mg | CH2Cl2/MeOH/NH3 |
| Alfa Aesar | (1.26 mmol, 63%) | |||
| 600 mg | 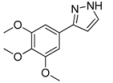 | |||
| 4b | 100:2:1 | |||
| 3 | 1c, 1-Iodo-2-hydroxybenzene | 15 min | 150 mg | CH2Cl2/MeOH/NH3 |
| ABCR | (0.94 mmol, 47%) | |||
| 449 mg | 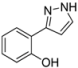 | |||
| 4c | 100:1:1 | |||
| 4 | 1d, 1-Iodo-3-hydroxybenzene | 15 min | 196 mg | CH2Cl2/MeOH/NH3 |
| Alfa Aesar | (1.23 mmol, 61%) | |||
| 449 mg | 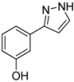 | |||
| 4d | 100:1:1 → 100:3:1 → 100:7:1 | |||
| 5 | 1e, 1-Iodo-4-fluorobenzene | 15 min | 170 mg | CH2Cl2/MeOH/NH3 |
| ABCR | (1.05 mmol, 53%) | |||
| 0.32 mL | 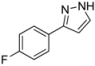 | |||
| 4e | 100:1:1 → 100:2:1 | |||
| 6 | 1f, 1-Iodo-4-chlorobenzene | 30 min | 121 mg | CH2Cl2/MeOH/NH3 |
| ABCR | (0.68 mmol, 34%) | |||
| 482 mg | 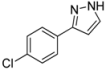 | |||
| 4f | 100:2:1 | |||
| 7 | 1g, 1-Iodo-4-trifluoromethylbenzene | 15 min | 244 mg | CH2Cl2/MeOH/NH3 |
| Alfa Aesar | (1.15 mmol, 57%) | |||
| 0.30 mL | 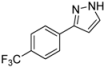 | |||
| 4g | 100:1:1 → 100:2:1 | |||
| 8 | 1h, 1-Iodo-4-cyanobenzene | 15 min | 90 mg | CH2Cl2/NH3 100:1 → CH2Cl2/MeOH/NH3 |
| ABCR | (0.53 mmol, 27%) | |||
| 467 mg | 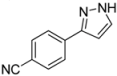 | |||
| 4h | 100:1:1 | |||
| 9 | 1i, 1-Iodo-3-nitrobenzene | 15 min | 209 mg | CH2Cl2/MeOH/NH3 |
| ABCR | (1.10 mmol, 55%) | |||
| 508 mg | 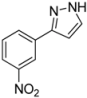 | |||
| 4i | 100:2:1 | |||
| 10 | 1j, 2-Iodothiophene | 1 h | 151 mg | CH2Cl2/MeOH/NH3 |
| ABCR | (1.00 mmol, 50%) | |||
| 429 mg | 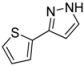 | |||
| 4j | 100:1:1 |
3.4. Spectroscopic Data of 1H-Pyrazoles 4
3.4.1. 3-(4-Methoxyphenyl)-1H-pyrazole (4a)
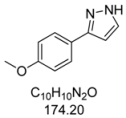
195 mg (1.12 mmol, 56%) as a pale yellow solid. Mp 128 °C. 1H-NMR (CDCl3, 300 MHz): δ 3.83 (s, 3H), 6.52 (d, J = 2.0 Hz, 1H), 6.89–6.94 (m, 2H), 7.59 (d, J = 2.1 Hz, 1H), 7.65–7.69 (m, 2H), 10.9 (br, 1H). 13C-NMR (CDCl3, 75 MHz): δ 55.3 (CH3), 102.0 (CH), 114.1 (CH), 124.7 (Cquat), 127.1 (CH), 133.6 (CH), 148.5 (Cquat), 159.5 (Cquat). EI + MS (m/z (%)): 175 (12), 174 (M+, 100), 159 ((M−CH3)+, 52), 145 ((M−HN2)+, 2), 132 ((M−CH2N2)+, 5), 131 (37), 77 (11). IR (Neat; Shimadzu IRAffinity): 3048 (w) cm−1, 2965 (w), 2914 (w), 2904 (w), 2824 (w), 2783 (w), 2725 (w), 1611 (w), 1526 (w), 1508 (m), 1454 (m), 1439 (m), 1417 (w), 1275 (m), 1248 (s), 1182 (s), 1113 (w), 1098 (m), 1055 (w), 1026 (s), 952 (m), 934 (m), 897 (m), 853 (m), 831 (s), 795 (m), 773 (s), 729 (w), 611 (m). Anal. calcd. for C10H10N2O (174.2): C 68.95, H 5.79, N 16.08; Found: C 68.73, H 5.48, N 15.88.
3.4.2. 3-(3,4,5-Trimethoxyphenyl)-1H-pyrazole (4b)
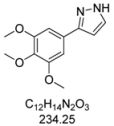
295 mg (1.26 mmol, 63%) as an orange solid. Mp 60 °C. 1H-NMR (CDCl3, 300 MHz): δ 3.88 (s, 3H), 3.90 (s, 6H), 6.58 (d, J = 1.7 Hz, 1H), 6.99 (s, 2H), 7.61 (d, J = 1.8 Hz, 1H). 13C-NMR (CDCl3, 75 MHz): δ 56.1 (CH3), 60.9 (CH3), 102.5 (CH), 103.0 (CH), 127.9 (Cquat), 132.6 (CH), 138.0 (Cquat), 149.5 (Cquat), 153.5 (Cquat). EI + MS (m/z (%)): 235 (13), 234 (M+, 100), 219 ((M-CH3)+, 68), 205 ((M-HN2)+, 2), 192 ((M-CH2N2)+, 2), 191 (30), 176 (30), 161 (20), 159 (15), 149 (13), 131 (15), 105 (22), 58 (23), 43 (67). IR (Neat; Shimadzu IRAffinity): 3576 (w) cm−1, 3146 (w), 3041 (w), 2995 (w), 2963 (w), 2930 (m), 2860 (w), 2826 (w), 1730 (w), 1587 (m), 1516 (m), 1474 (m), 1449 (m), 1416 (m), 1325 (m), 1288 (w), 1261 (w), 1238 (m), 1121 (s), 1092 (m), 1057 (m), 1034 (w), 995 (s), 928 (w), 889 (w), 845 (s), 826 (m), 760 (s), 739 (m), 665 (w), 627 (m). Anal. calcd. for C12H14N2O3 (234.3): C 61.53, H 6.03, N 11.96; Found: C 61.63, H 6.29, N 11.68.
3.4.3. 2-(1H-Pyrazol-3-yl)phenol (4c)
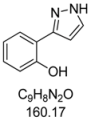
150 mg (0.94 mmol, 47%) as a yellow solid. Mp 92 °C. 1H-NMR (CDCl3, 300 MHz): δ 6.73 (d, J = 2.6 Hz, 1H), 6.93 (ddd, J = 7.7 Hz, J = 7.3 Hz, J = 2.0 Hz, 1H), 7.05 (dd, J = 8.2 Hz, J = 1.0 Hz, 1H), 7.24 (ddd, J = 8.2 Hz, J = 7.3 Hz, J = 2.0 Hz, 1H), 7.61 (dd, J = 7.7 Hz, J = 1.6 Hz, 1H), 7.64 (d, J = 2.6 Hz, 1H), 9.9–11.2 (br, 2 H). 13C-NMR (CDCl3, 75 MHz): δ 102.1 (CH), 116.5 (Cquat), 117.0 (CH), 119.3 (CH), 126.6 (CH), 129.1 (CH), 129.3 (CH), 152.0 (Cquat), 155.8 (Cquat). EI + MS (m/z (%)): 161 (11), 160 (M+, 100), 132 (12), 131 ((M−HN2)+, 53), 104 (12). IR (KBr; Bruker Vector 22 FT-IR): 3277 (s) cm−1, 1736 (w), 1719 (w), 1686 (w), 1655 (w), 1625 (m), 1589 (s), 1560 (w), 1518 (m), 1451 (s), 1399 (m), 1286 (m), 1256 (s), 1200 (m), 1126 (m), 1110 (m), 1080 (m), 1047 (m), 952 (m), 826 (m), 779 (s), 746 (s), 600 (m), 561 (m), 517 (m). Anal. calcd. for C9H8N2O (160.2): C 67.49, H 5.03, N 17.49; Found: C 67.41, H 4.90, N 17.26.
3.4.4. 3-(1H-Pyrazol-3-yl)phenol (4d)
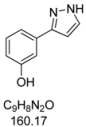
196 mg (1.23 mmol, 61%) as a pale brown solid. Mp 154 °C. 1H-NMR (DMSO-d6, 300 MHz): δ 6.60 (d, J = 2.0 Hz, 1H), 6.68-6.70 (m, 1H), 7.15-7.31 (m, 3H), 7.7 (br, 1H), 9.4 (br, 1H), 12.4–13.5 (2xbr, 1H).13C-NMR (DMSO-d6, 75 MHz): δ 102.1 (CH), 112.2 (CH), 114.7 (CH), 116.3 (CH), 129.9 (CH), 135.3 (Cquat), 150.3 (Cquat), 157.8 (Cquat). EI + MS (m/z (%)): 161 (11), 160 (M+, 100), 131 ((M-HN2)+, 25), 93 (C6H5O+, 3). IR (KBr, Bruker Vector 22 FT-IR): 3245 (s) cm−1, 1610 (m), 1591 (s), 1541 (w), 1494 (m), 1478 (s), 1439 (s), 1382 (m), 1292 (m), 1258 (s), 1239 (m), 1218 (m), 1161 (w), 1117 (m), 1097 (m), 1082 (m), 1053 (m), 987 (w), 940 (m), 862 (s), 758 (s), 681 (s), 605 (w), 540 (w). Anal. calcd. for C9H8N2O (160.2): C 67.49, H 5.03, N 17.49; Found: C 67.50, H 5.04, N 17.31.
3.4.5. 3-(4-Fluorophenyl)-1H-pyrazole (4e)
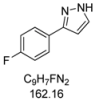
170 mg (1.05 mmol, 53%) as a pale brown solid. Mp 104 °C. 1H-NMR (CDCl3, 300 MHz): δ 6.56 (m, 1H), 7.07 (t, J = 8.7 Hz, 2H), 7.59 (m, 1H), 7.69-7.74 (m, 2H), 11.0-12.2 (br, 1H). 13C-NMR (CDCl3, 75 MHz): δ 102.5 (CH), 115.7 (d, J = 21.7 Hz, CH), 127.5 (d, J = 8.1 Hz, CH), 128.6 (Cquat), 132.5 (CH), 148.9 (Cquat), 162.6 (d, J = 247.1 Hz, Cquat). EI + MS (m/z (%)): 163 (11), 162 (M+, 100), 161 (11), 135 (16), 133 ((M-HN2)+, 27), 108 (11), 95 (C6H4F+, 10). IR (Neat; Shimadzu IRAffinity): 3121 (w) cm−1, 3051 (w), 3034 (w), 2959 (w), 2918 (w), 2901 (w), 2882 (w), 2860 (w), 2841 (w), 2814 (w), 2776 (w), 2745 (w), 2725 (w), 2675 (w), 2627 (w), 2581 (w), 1651 (w), 1607 (w), 1524 (m), 1506 (m), 1452 (m), 1408 (w), 1352 (w), 1337 (w), 1233 (s), 1223 (s), 1204 (w), 1157 (m), 1111 (w), 1094 (m), 1082 (w), 1051 (m), 1015 (w), 953 (m), 934 (m), 885 (w), 839 (s), 812 (s), 770 (s), 752 (w), 692 (w), 673 (w), 602 (m). Anal. calcd. for C9H7FN2 (162.2): C 66.66, H 4.35, N 17.27; Found: C 66.92, H 4.41, N 17.00.
3.4.6. 3-(4-Chlorophenyl)-1H-pyrazole (4f)
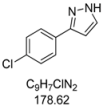
121 mg (0.68 mmol, 34 %) as an orange-brown solid. Mp 95 °C. 1H-NMR (CDCl3, 300 MHz): δ 6.60 (s, 1H), 7.36 (d, J = 8.4 Hz, 2H), 7.60 (s, 1H), 7.69 (d, J = 8.5 Hz, 2H), 8.5–12.1 (br, 1H). 13C-NMR (CDCl3, 75 MHz): δ 102.8 (CH), 127.1 (CH), 128.9 (CH), 131.0 (Cquat), 132.2 (CH), 133.8 (Cquat), 149.0 (Cquat). EI + MS (m/z (%)): 180 (M(37Cl)+, 30), 179 (14), 178 (M(35Cl)+, 100), 151 ((M−HN2)(37Cl)+, 10), 149 ((M−HN2)(35Cl)+, 4), 116 (10), 115 (19), 113 (C6H437Cl+, 5), 111 (C6H435Cl+, 7), 89 (12). IR (Neat; Shimadzu IRAffinity): 3165 (w) cm−1, 2955 (w), 2918 (m), 2876 (w), 2847 (w), 1510 (m), 1447 (m), 1406 (w), 1115 (w), 1090 (s), 1080 (m), 1047 (m), 1013 (m), 953 (m), 924 (m), 878 (w), 833 (s), 761 (s), 723 (m), 692 (m), 613 (m). Anal. calcd. for C9H7ClN2 (178.6): C 60.52, H 3.95, N 15.68; Found: C 60.78, H 4.15, N 15.55.
3.4.7. 3-[4-(Trifluoromethyl)phenyl]-1H-pyrazole (4g)
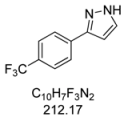
244 mg (1.15 mmol, 57%) as a pale brown solid. Mp 162 °C. 1H-NMR (CDCl3, 300 MHz): δ 6.68 (m, 1H), 7.62 (m, 1H), 7.63 (d, J = 8.1 Hz, 2H), 7.87 (d, J = 8.1 Hz, 2H), 10.9–12.7 (br, 1H). 13C-NMR (CDCl3, 75 MHz): δ 103.0 (CH), 124.1 (q, J = 271.9 Hz, Cquat), 125.7 (q, J = 3.8 Hz, CH), 125.9 (CH), 129.8 (q, J = 32.5 Hz, Cquat), 132.0 (CH), 135.9 (Cquat), 149.1 (Cquat). EI + MS (m/z (%)): 213 (11), 212 (M+, 100), 211 (12), 193 ((M−F)+, 7), 185 (11), 183 ((M−HN2)+, 5), 145 (14). IR (Neat; Shimadzu IRAffinity): 3122 (w) cm−1, 3030 (w), 3013 (w), 2959 (w), 2918 (w), 2900 (w), 2868 (w), 2779 (w), 2727 (w), 2679 (w), 1620 (m), 1468 (w), 1416 (w), 1321 (s), 1275 (w), 1182 (s), 1159 (m), 1109 (s), 1097 (s), 1064 (s), 1053 (m), 1013 (m), 957 (m), 930 (w), 881 (w), 841 (s), 783 (m), 770 (s), 741 (m), 692 (m), 673 (w), 617 (w). Anal. calcd. for C10H7F3N2 (212.2): C 56.61, H 3.33, N 13.20; Found: C 56.83, H 3.08, N 13.04.
3.4.8. 4-(1H-Pyrazol-3-yl)benzonitrile (4h)
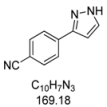
90 mg (0.53 mmol, 27%) as a pale yellow solid. Mp 143 °C. 1H-NMR (CDCl3, 300 MHz): δ 6.70 (m, 1H), 7.66 (m, 1H), 7.68 (d, J = 8.5 Hz, 2H), 7.89 (d, J = 8.5 Hz, 2H), 10.4–12.5 (br, 1H). 13C-NMR (CDCl3, 75 MHz): δ 103.5 (CH), 111.1 (Cquat), 118.9 (Cquat), 126.1 (CH), 131.3 (CH), 132.6 (CH), 137.2 (Cquat), 149.1 (Cquat). EI + MS (m/z (%)): 170 (12), 169 (M+, 100), 142 (15), 140 ((M−HN2)+, 12), 102 (C7H4N+, 7). IR (Neat; Shimadzu IRAffinity): 3248 (w) cm−1, 3237 (w), 3129 (w), 3057 (w), 2980 (m), 2972 (m), 2889 (w), 2814 (w), 2220 (w), 1929 (w), 1609 (m), 1508 (w), 1464 (w), 1449 (w), 1393 (w), 1350 (w), 1233 (w), 1221 (w), 1184 (m), 1157 (w), 1074 (w), 1051 (m), 968 (w), 951 (s), 934 (w), 841 (s), 768 (s), 731 (m), 685 (w), 608 (s). Anal. calcd. for C10H7N3 (169.2): C 70.99, H 4.17, N 24.84; Found: C 70.78, H 4.10, N 24.59.
3.4.9. 3-(3-Nitrophenyl)-1H-pyrazole (4i)
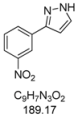
209 mg (1.10 mmol, 55%) as a pale brown solid. Mp 121 °C. 1H-NMR (CDCl3, 300 MHz): δ 6.74 (d, J = 2.3 Hz, 1H), 7.58 (t, J = 8.0 Hz, 1H), 7.70 (d, J = 2.4 Hz, 1H), 8.15 (tdd, J = 8.1 Hz, J = 2.2 Hz, J = 1.1 Hz, 2H), 8.64 (t, J = 1.9 Hz, 1H), 9.8–12.3 (br, 1H). 13C-NMR (CDCl3, 75 MHz): δ 103.2 (CH), 120.6 (CH), 122.5 (CH), 129.7 (CH), 131.1 (CH), 131.6 (CH), 134.7 (Cquat), 148.7 (Cquat), 149.1 (Cquat). EI + MS (m/z (%)): 190 (12), 189 (M+, 100), 143 ((M−NO2)+, 33), 142 (10), 116 (39), 89 (28). IR (Neat; Shimadzu IRAffinity): 3173 (w) cm−1, 3067 (w), 2961 (w), 2926 (w), 2851 (w), 1557 (w), 1530 (m), 1518 (m), 1504 (m), 1487 (m), 1398 (w), 1375 (m), 1344 (m), 1315 (w), 1275 (m), 1265 (w), 1221 (w), 1202 (w), 1072 (w), 993 (m), 972 (w), 897 (w), 851 (w), 829 (w), 775 (m), 735 (m), 698 (s), 683 (m), 671 (m), 615 (m). Anal. calcd. for C9H7N3O2 (189.2): C 57.14, H 3.73, N 22.21; Found: C 57.19, H 4.02, N 22.18.
3.4.10. 3-(Thiophen-2-yl)-1H-pyrazole (4j)
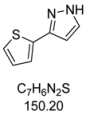
151 mg (1.00 mmol, 50%) as an orange solid. Mp 89 °C. 1H-NMR (CDCl3, 300 MHz): δ 6.54 (d, J = 2.1 Hz, 1H), 7.07 (dd, J = 5.1 Hz, J = 3.6 Hz, 1H), 7.27 (dd, J = 5.1 Hz, J = 1.0 Hz, 1H), 7.35 (dd, J = 3.6 Hz, J = 1.1 Hz, 1H), 7.63 (d, J = 2.3 Hz, 1H), 11.55 (s, 1H). 13C-NMR (CDCl3, 75 MHz): δ 102.7 (CH), 124.2 (CH), 124.6 (CH), 127.6 (CH), 131.5 (CH), 135.7 (Cquat), 145.6 (Cquat). EI + MS (m/z (%)): 152 (13), 151 (26), 150 (M+, 100), 123 (20), 122 (16), 121 ((M−HN2)+, 35), 105 ((M−CHS)+, 7), 96 (22), 78 (12). IR (Neat; Shimadzu IRAffinity): 3146 (w) cm−1, 3129 (w), 3117 (w), 3102 (w), 3022 (w), 2957 (w), 2913 (w), 2872 (w), 2855 (w), 2808 (w), 2361 (w), 1732 (w), 1560 (w), 1522 (w), 1464 (w), 1410 (w), 1371 (w), 1273 (w), 1179 (m), 1047 (m), 1030 (m), 910 (m), 845 (m), 824 (m), 756 (s), 723 (w), 692 (s), 644 (w), 602 (m). Anal. calcd. for C7H6N2S (150.2): C 55.97, H 4.03, N 18.65; Found: C 56.12, H 4.12, N 18.46.
4. Conclusions
In summary, we have disclosed a novel consecutive three-component synthesis of 3-(hetero)aryl-1H-pyrazoles 4 in moderate to good yields starting with the Sonogashira coupling of (hetero)aryl iodides 1 and the commercially available propynal diethylacetal (2) as a propargyl aldehyde synthetic equivalent, followed by a rapid sequential acetal cleavage-cyclocondensation with hydrazine hydrochloride. Further studies directed to the synthesis of luminescent pyrazole derivatives as ligands for metal organic frameworks (MOFs) are currently underway.
Acknowledgments
The work was supported by the Fonds der Chemischen Industrie and the authors gratefully thank Merck Serono, Darmstadt, for the donation of chemicals.
References
- Kost, A.N.; Grandberg, I.I. Progress in pyrazole chemistry. Adv. Heterocycl. Chem. 1966, 6, 347–429. [Google Scholar] [CrossRef]
- Elguero, J. Pyrazoles and their benzo derivatives. In Comprehensive Heterocyclic Chemistry; Katritzky, A.R., Rees, C.W., Eds.; Pergamon: Oxford, UK, 1984; Volume 5, pp. 167–303. [Google Scholar]
- Elguero, J. Pyrazoles. In Comprehensive Heterocyclic Chemistry II; Katritzky, A.R., Rees, C.W., Scriven, E.F.V., Eds.; Elsevier: Oxford, UK, 1996; Volume 3, pp. 1–75. [Google Scholar]
- Wustrow, D.J.; Capiris, T.; Rubin, R.; Knobelsdorf, J.A.; Akunne, H.; Davis, M.D.; MacKenzie, R.; Pugsley, T.A.; Zoski, K.T.; Heffner, T.G.; et al. Pyrazolo[1,5-a]pyrimidine CRF-1 receptor antagonists. Bioorg. Med. Chem. Lett. 1998, 8, 2067–2070. [Google Scholar]
- Menozzi, G.; Mosti, L.; Fossa, P.; Mattioli, F.; Ghia, M. ω-Dialkylaminoalkyl ethers of phenyl-(5-substituted 1-phenyl-1H-pyrazol-4-yl)methanols with analgesic and anti-inflammatory activity. J. Heterocycl. Chem. 1997, 34, 963–968. [Google Scholar] [CrossRef]
- Penning, T.D.; Talley, J.J.; Bertenshaw, S.R.; Carter, J.S.; Collins, P.W.; Docter, S.; Graneto, M.J.; Lee, L.F.; Malecha, J.W.; Miyashiro, J.M.; et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: Identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, Celecoxib). J. Med. Chem. 1997, 40, 1347–1365. [Google Scholar] [CrossRef]
- Chimenti, F.; Fioravanti, R.; Bolasco, A.; Manna, F.; Chimenti, P.; Secci, D.; Befani, O.; Turini, P.; Ortuso, F.; Alcaro, S. Monoamine oxidase isoform-dependent tautomeric influence in the recognition of 3,5-diaryl pyrazole inhibitors. J. Med. Chem. 2007, 50, 425–428. [Google Scholar] [CrossRef]
- Walworth, B.L.; Klingsberg, E. Neue herbicide Zusammensetzungen. DE 2260485 19730628, 1973. [Google Scholar]
- Trofimenko, S. Coordination chemistry of pyrazole-derived ligands. Chem. Rev. 1972, 72, 497–509. [Google Scholar] [CrossRef]
- Mukherjee, R. Coordination chemistry with pyrazole-based chelating ligands: Molecular structural aspects. Coord. Chem. Rev. 2000, 203, 151–218. [Google Scholar]
- Trofimenko, S. Scorpionates: Genesis, milestones, prognosis. Polyhedron 2004, 23, 197–203. [Google Scholar] [CrossRef]
- Ward, M.D.; McCleverty, J.A.; Jeffery, J.C. Coordination and supramolecular chemistry of multinucleating ligands containing two or more pyrazolyl-pyridine ‘arms’. Coord. Chem. Rev. 2001, 222, 251–272. [Google Scholar]
- Harb, A.-F.A.; Abbas, H.H.; Mostafa, F.H. Pyrazoles as building blocks in heterocyclic synthesis: Synthesis of some new substituted 1-triazinylpyrazolo[3,4-d]pyrimidine and 1-triazinylpyrazolo[3,4-b]pyridine derivatives. Chem. Pap. 2005, 59, 187–195. [Google Scholar]
- Harb, A.-F.A.; Abbas, H.H.; Mostafa, F.H. Pyrazoles as building blocks in heterocyclic synthesis: Synthesis of pyrazolo[3,4-d]pyrimidine, pyrazolo[3,4-e][1,4]diazepine, pyrazolo[3,4-d][1,2,3]-triazine and pyrrolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine derivatives. J. Iranian Chem. Soc. 2005, 2, 115–123. [Google Scholar] [CrossRef]
- Sachse, A.; Penkova, L.; Noël, G.; Dechert, S.; Varzatskii, O.A.; Fritsky, I.O.; Meyer, F. Efficient syntheses of some versatile 3,5-bifunctional pyrazole building blocks. Synthesis 2008, 800–806. [Google Scholar]
- Maeda, H.; Ito, Y.; Kusunose, Y.; Nakanishi, T. Dipyrrolylpyrazoles: Anion receptors in protonated form and efficient building blocks for organized structures. Chem. Commun. 2007, 1136–1138. [Google Scholar]
- Gemming, S.; Schreiber, M.; Thiel, W.; Heine, T.; Seifert, G.; Avelino de Abreu, H.; Anderson Duarte, H. Tunable discotic building blocks for liquid crystalline displays. J. Luminescence 2004, 108, 143–147. [Google Scholar] [CrossRef]
- Dolars, A.; Schellhammer, C.-W.; Schroeder, J. Heterocycles as structural units in new optical brighteners. Angew. Chem. Int. Ed. Engl. 1975, 14, 665–679. [Google Scholar] [CrossRef]
- Catalan, J.; Fabero, F.; Claramunt, R.M.; Santa Maria, M.D.; Foces-Foces, M.C.; Hernandez Cano, F.; Martinez-Ripoll, M.; Elguero, J.; Sastre, R. New ultraviolet stabilizers: 3- and 5-(2'-Hydroxyphenyl)pyrazoles. J. Am. Chem. Soc. 1992, 114, 5039–5048. [Google Scholar]
- Karatsu, T.; Shiochi, N.; Aono, T.; Miyagawa, N.; Kitamura, A. Photoinduced electron transfer reactions of 3H-pyrazole derivatives. Formation of solvent adduct by specific sensitizer. Bull. Chem. Soc. Jpn. 2003, 76, 1227–1231. [Google Scholar] [CrossRef]
- Yen, Y.-P.; Huang, T.-M.; Tseng, Y.-P.; Lin, H.-Y.; Lai, C.-C. Photoinduced electron transfer reactions of 3,3-dialkylated 4,5-diphenyl-3H-pyrazoles: A new route to the formation of the solvent adducts. J. Chin. Chem. Soc. 2004, 51, 393–398. [Google Scholar]
- Zhu, J.; Bienaymé, H. Multicomponent Reactions; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Touré, B.B.; Hall, D.G. Natural product synthesis using multicomponent reaction strategies. Chem. Rev. 2009, 109, 4439–4486. [Google Scholar] [CrossRef]
- Sunderhaus, J.D.; Martin, S.F. Applications of multicomponent reactions to the synthesis of diverse heterocyclic scaffolds. Chem. Eur. J. 2009, 15, 1300–1308. [Google Scholar] [CrossRef]
- Isambert, N.; Lavilla, R. Heterocycles as key substrates in multicomponent reactions: The fast lane towards molecular complexity. Chem. Eur. J. 2008, 14, 8444–8454. [Google Scholar] [CrossRef]
- D’Souza, D.M.; Müller, T.J.J. Multi-component syntheses of heterocycles by transition-metal catalysis. Chem. Soc. Rev. 2007, 36, 1095–1108. [Google Scholar]
- Dömling, A. Recent developments in isocyanide based multicomponent reactions in applied chemistry. Chem. Rev. 2006, 106, 17–89. [Google Scholar]
- Orru, R.V.A.; de Greef, M. Recent advances in solution-phase multicomponent methodology for the synthesis of heterocyclic compounds. Synthesis 2003, 1471–1499. [Google Scholar]
- Bienaymé, H.; Hulme, C.; Oddon, G.; Schmitt, P. Maximizing synthetic efficiency: Multi-component transformations lead the way. Chem. Eur. J. 2000, 6, 3321–3329. [Google Scholar] [CrossRef]
- Dömling, A.; Ugi, I. Multicomponent reactions with isocyanides. Angew. Chem. Int. Ed. Engl. 2000, 39, 3168–3210. [Google Scholar] [CrossRef]
- Ugi, I.; Dömling, A.; Werner, B. Since 1995 the new chemistry of multicomponent reactions and their libraries, including their heterocyclic chemistry. J. Heterocycl. Chem. 2000, 37, 647–658. [Google Scholar] [CrossRef]
- Weber, L.; Illgen, K.; Almstetter, M. Discovery of new multi-component reactions with combinatorial methods. Synlett 1999, 366–374. [Google Scholar]
- Armstrong, R.W.; Combs, A.P.; Tempest, P.A.; Brown, S.D.; Keating, T.A. Multiple-component condensation strategies for combinatorial library synthesis. Acc. Chem. Res. 1996, 29, 123–131. [Google Scholar] [CrossRef]
- Ugi, I.; Dömling, A.; Hörl, W. Multicomponent reactions in organic chemistry. Endeavour 1994, 18, 115–122. [Google Scholar] [CrossRef]
- Posner, G.H. Multicomponent one-pot annulations forming 3 to 6 bonds. Chem. Rev. 1986, 86, 831–844. [Google Scholar]
- Tietze, L.F. Domino reactions in organic synthesis. Chem. Rev. 1996, 96, 115–136. [Google Scholar]
- Tietze, L.F.; Beifuss, U. Sequential transformations in organic chemistry: A synthetic strategy with a future. Angew. Chem. Int. Ed. Engl. 1993, 32, 131–163. [Google Scholar] [CrossRef]
- Tietze, L.F. Domino-reactions: The tandem-Knoevenagel-hetero-Diels-Alder reaction and its application in natural product synthesis. J. Heterocycl. Chem. 1990, 27, 47–69. [Google Scholar] [CrossRef]
- Coquerel, Y.; Boddaert, T.; Presset, M.; Mailhol, D.; Rodriguez, J. Multiple bond-forming transformations: The key concept toward eco-compatible synthetic organic chemistry. In Ideas in Chemistry and Molecular Sciences: Advances in Synthetic Chemistry; Pignataro, B., Ed.; Wiley-VCH: Weinheim, Germany, 2010; pp. 187–202. [Google Scholar]
- Vries, J.G. The Heck reaction in the production of fine chemicals. Can. J. Chem. 2001, 79, 1086–1092. [Google Scholar] [CrossRef]
- Pommer, H. The Wittig reaction in industrial practice. Angew. Chem. Int. Ed. Engl. 1977, 16, 423–429. [Google Scholar] [CrossRef]
- Müller, T.J.J. Palladium-copper catalyzed alkyne activation as an entry to multicomponent syntheses of heterocycles. Top. Heterocycl. Chem. 2010, 25, 25–94. [Google Scholar] [CrossRef]
- Willy, B.; Müller, T.J.J. Multi-component heterocycle syntheses via catalytic generation of alkynones. Curr. Org. Chem. 2009, 13, 1777–1790. [Google Scholar] [CrossRef]
- Willy, B.; Müller, T.J.J. Consecutive multi-component syntheses of heterocycles via palladium-copper catalyzed generation of alkynones. ARKIVOC 2008, 195–208. [Google Scholar]
- Müller, T.J.J. Multi-component syntheses of heterocycles by virtue of palladium catalyzed generation of alkynones and chalcones. Targets Heterocycl. Syst. 2006, 10, 54–65. [Google Scholar]
- Willy, B.; Müller, T.J.J. Regioselective three-component synthesis of highly fluorescent 1,3,5-trisubstituted pyrazoles. Eur. J. Org. Chem. 2008, 4157–4168. [Google Scholar]
- Willy, B.; Müller, T.J.J. Rapid one-pot, four-step synthesis of highly fluorescent 1,3,4,5-tetrasubstituted pyrazoles. Org. Lett. 2011, 13, 2082–2085. [Google Scholar]
- Bagley, M.C.; Lubinu, M.C.; Mason, C. Regioselective microwave-assisted synthesis of substituted pyrazoles from ethynyl ketones. Synlett 2007, 704–708. [Google Scholar]
- Evans, G.B.; Furneaux, R.H.; Gainsford, G.J.; Hanson, J.C.; Kicska, G.A.; Sauve, A.A.; Schramm, V.L.; Tyler, P.C. 8-Aza-immucillins as transition-state analogue inhibitors of purine nucleoside phosphorylase and nucleoside hydrolases. J. Med. Chem. 2003, 46, 155–160. [Google Scholar]
- Zhou, J.; Yang, M.; Akdag, A.; Wang, H.; Schneller, S.W. Carbocyclic 4′-epi-formycin. Tetrahedron 2008, 64, 433–438. [Google Scholar]
- Zhou, J.; Yang, M.; Schneller, S.W. A model study to carbocyclic formycin A and B analogues. Tetrahedron Lett. 2004, 45, 8233–8234. [Google Scholar]
- Buchanan, J.G.; Edgar, A.R.; Hutchison, R.J.; Stobie, A.; Wightman, R.H. C-nucleoside studies. Part 10. A new synthesis of 3-(2,3,5-tri-O-benzyl-β-D-ribofuranosyl)pyrazole and its conversion into 4-nitro-3(5)-β-D-ribofuranosylpyrazole. J. Chem. Soc. Perkin Trans. 1 1980, 2567–2571. [Google Scholar]
- Buchanan, J.G.; Dunn, A.D.; Edgar, A.R.; Hutchison, R.J.; Power, M.J.; Williams, G.C. C-nucleoside studies. Part 6. Synthesis of 3-[2,3,5-tri-O-benzyl-β-(and α)-D-ribofuranosyl]prop-2-yn-1-ol and related compounds; a new synthesis of 3(5)-(2,3,5-tri-O-benzyl-β-D-ribofuranosyl)pyrazole. J. Chem. Soc. Perkin Trans. 1 1977, 1786–1791. [Google Scholar]
- Hüttel, R.; Büchele, F.; Jochum, P. Über Nitro-, Nitroso- und Azopyrazole. Chem. Ber. 1955, 88, 1577–1585. [Google Scholar] [CrossRef]
- Karpov, A.S.; Müller, T.J.J. New entry to a three-component pyrimidine synthesis by TMS-ynones via Sonogashira coupling. Org. Lett. 2003, 5, 3451–3454. [Google Scholar] [CrossRef]
- Bagley, M.C.; Hughes, D.D.; Sabo, H.M.; Taylor, P.H.; Xiong, X. One-pot synthesis of pyridines or pyrimidines by tandem oxidation-heteroannulation of propargylic alcohols. Synlett 2003, 1443–1446. [Google Scholar]
- Lemhadri, M.; Doucet, H.; Santelli, M. Sonogashira reaction of aryl halides with propiolaldehydediethyl acetal catalyzed by a tetraphosphine/palladium complex. Tetrahedron 2005, 61, 9839–9847. [Google Scholar]
- Cacchi, S.; Fabrizi, G.; Marinelli, F.; Moro, L.; Pace, P. Palladium-catalysed hydroarylation and hydrovinylation of 3,3-dialkoxy-1-aryl-1-propynes. An approach to 3-aryl- and 3-vinylquinolines. Tetrahedron 1996, 52, 10225–10240. [Google Scholar] [CrossRef]
- Sakamoto, T.; Shiga, F.; Yasuhara, A.; Uchiyama, D.; Kondo, Y.; Yamanaka, H. Preparation of ethyl arylpropiolates from aryl iodides by palladium-catalyzed cross-coupling reaction. Synthesis 1992, 746–748. [Google Scholar]
- Stoit, A.; Coolen, H.K.A.C.; Kruse, C.G.; Terwel, L.J.H. Heterocyclic compounds with affinity to muscarinic receptors. PCT Int. Appl. WO 2008129054 A2, 30 October 2008..
- Buchanan, J.G.; Quijano, M.L.; Wightman, R.H. C-nucleoside studies. Part 23. New and more direct synthesis of 3-(β-D-xylofuranosyl)pyrazole. J. Chem. Soc. Perkin Trans. 1 1992, 1573–1576. [Google Scholar]
- Jones, R.G.; Mann, M.J. New methods of synthesis of p-aminoethylpyrazoles. J. Am. Chem. Soc. 1953, 75, 4048–4052. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 4 are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).