Synthesis and Evaluation of Non-peptidic Cysteine Protease Inhibitors of P. falciparum Derived from Etacrynic Acid

Abstract
:
Introduction
Results and Discussion
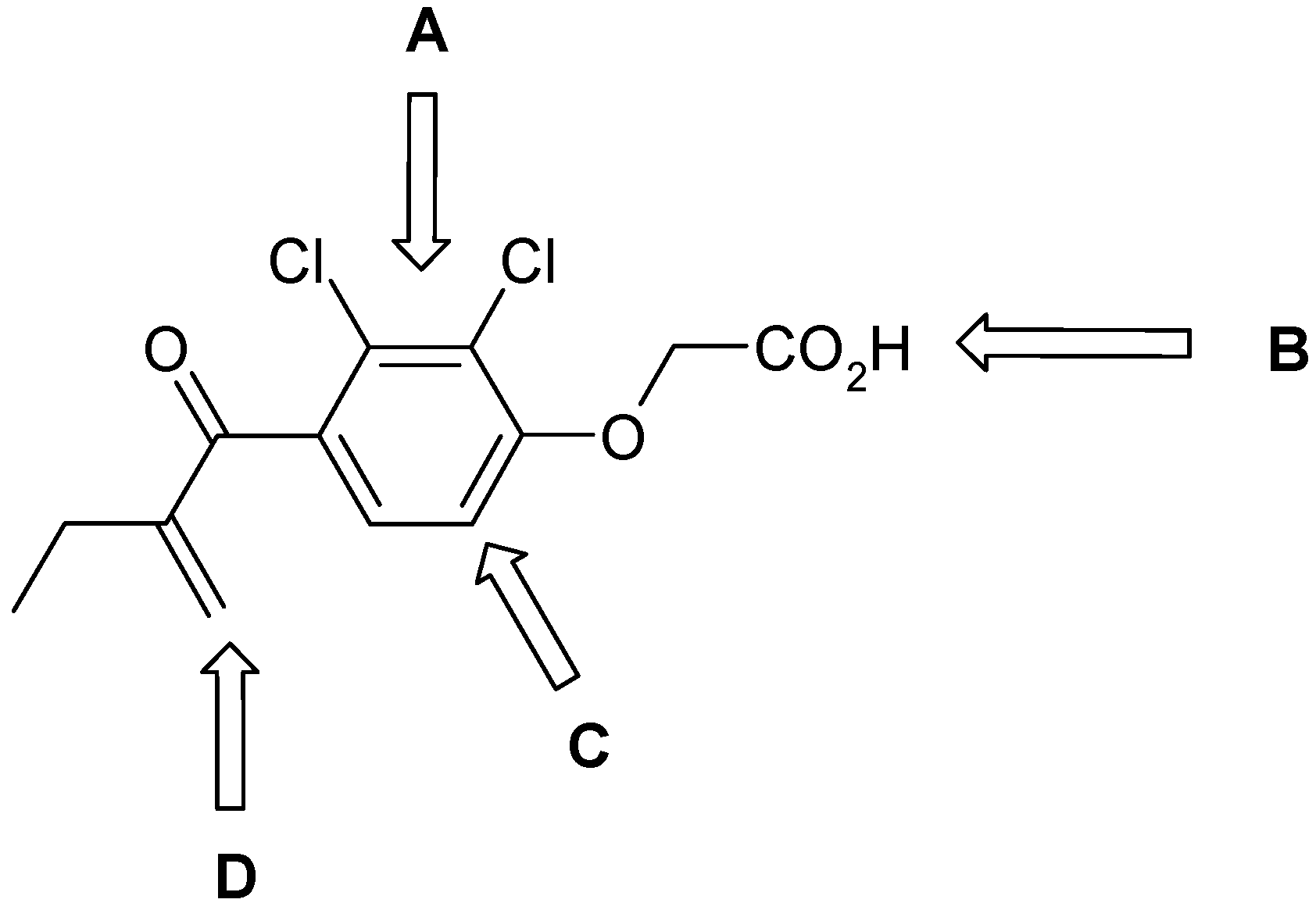
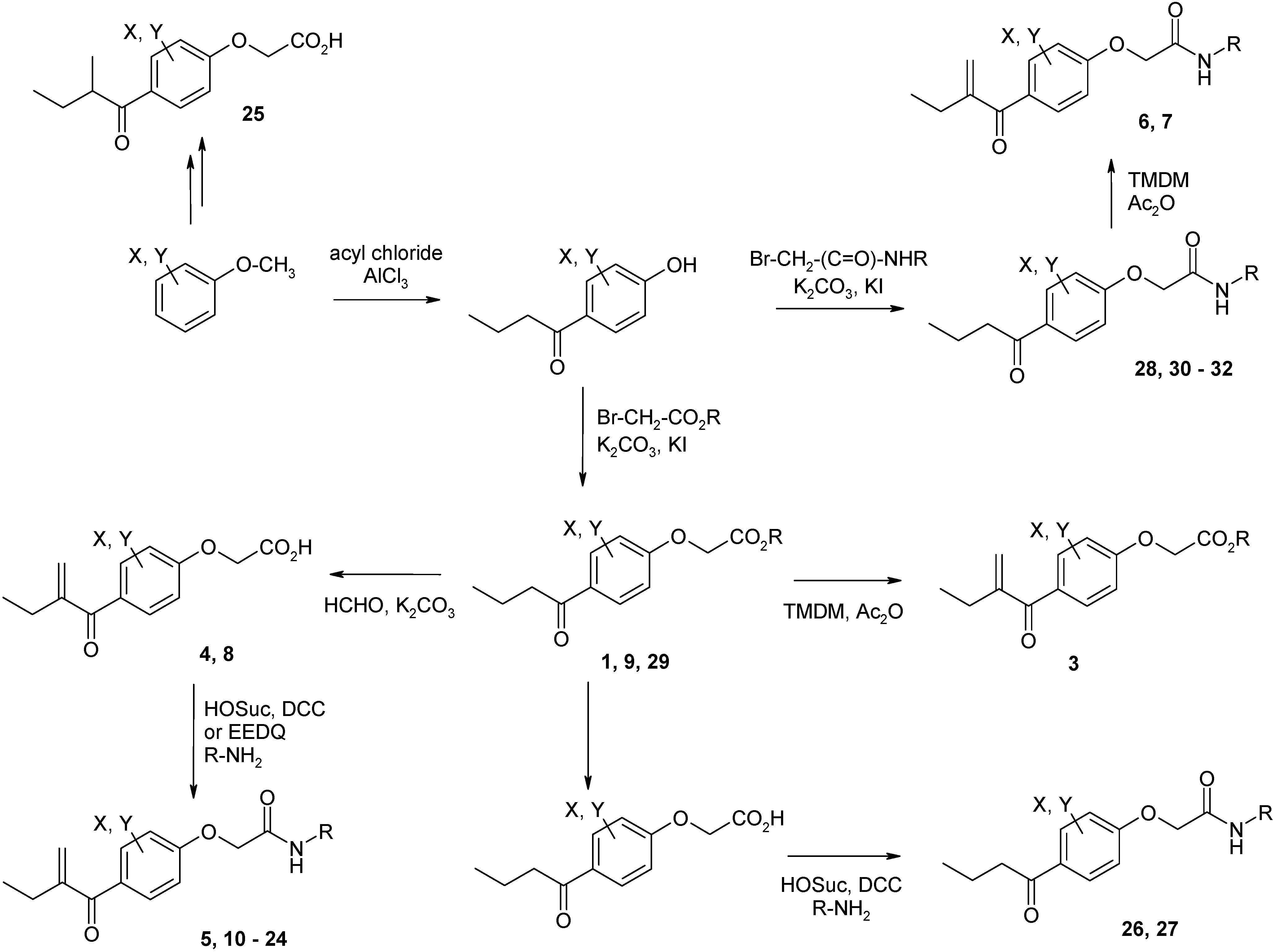

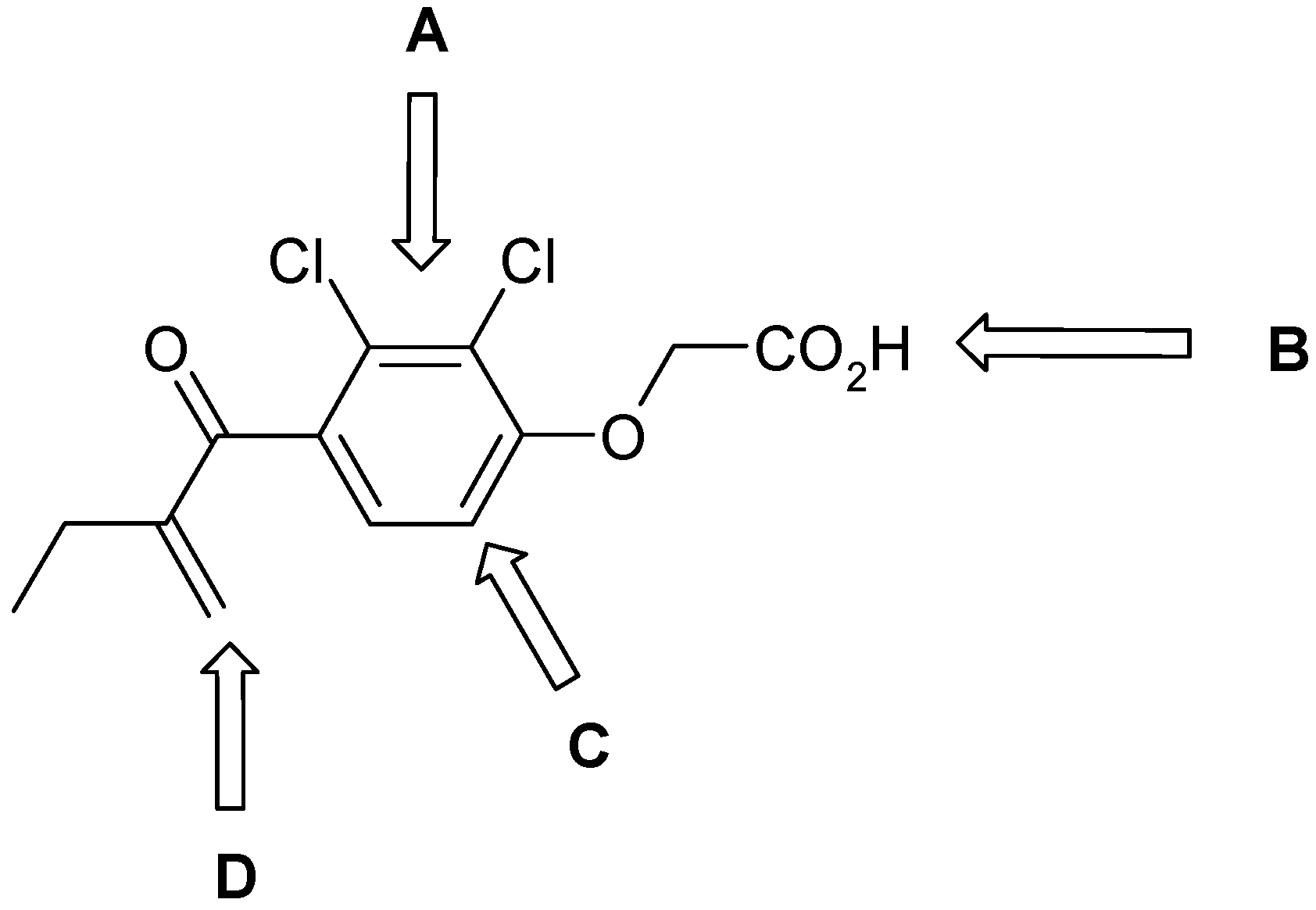
{kind=link}
{kind=link}
{kind=link}

| Cpd. | R1 | X | Y | R2 | FP-2 IC50, (mM) | FP-3 IC50, (mM) | P. f. 3D7/W2, IC50 (µM) |
|---|---|---|---|---|---|---|---|
| 1 | H5C2 | H | Cl | O-CH2-CO2Et | 498±8 | 346±14 | nic |
| 2 | H5C2 | O-CH2-CO2Et | H | Cl | 110±5 | 381±21 | nic |
| 3 | H5C2 | H | F | 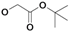 | ni | nd | 142±5b |
| 4 | H5C2 | H | F | O-CH2CO2H | nd | nd | 79.8±6b |
| 5 | H5C2 | H | F | 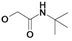 | 80±5 | nd | 205±9b |
| 6 | H5C2 | H | F |  | ni | nd | 141±4b |
| 7 | H5C2 | H | F | 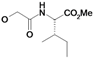 | ni | nd | nd |
| 8 | H5C2 | Cl | Cl | O-CH2-CO2H | 443±17 | ni | nic |
| 9 | H5C2 | Cl | Cl | O-CH2-CO2Et | 60.6±4.2 | 163±5.6 | nic |
| 10 | H5C2 | Cl | Cl |  | 178±14 | 56.7±5.7 | nic |
| 11 | H5C2 | Cl | Cl | 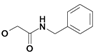 | 333±5 | 158±11 | nic |
| 12 | H5C2 | Cl | Cl | 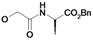 | 165±12 | nd | nd |
| 13 | H5C2 | Cl | Cl | 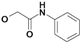 | 269±21 | 87.2±6 | nic |
| 14 | H5C2 | Cl | Cl | 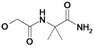 | 318±14 | ni | 29.3±3.4c |
| 15 | H5C2 | Cl | Cl | 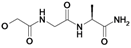 | 212±17 | ni | nic |
| 16 | H5C2 | Cl | Cl | 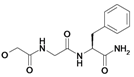 | 242±19 | ni | nic |
| 17 | H5C2 | Cl | Cl | 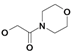 | 305±30 | 479±23 | nic |
| 18 | H5C2 | Cl | Cl | 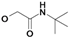 | 255±4 | 153±16 | nic |
| 19 | H5C2 | Cl | Cl | 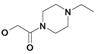 | 144±11 | 557±23 | 27.4±4.1c |
| 20 | H5C2 | Cl | Cl | 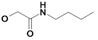 | 184±17 | 158±17 | nic |
| 21 | H5C2 | Cl | Cl |  | 182±9 | 123±8 | nic |
| 22 | H5C2 | Cl | Cl | 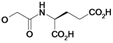 | ni | ni | nic |
| 23 | H5C2 | Cl | Cl | 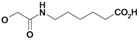 | 57.1±13 | 96.5±0.6 | 18.8±0.9c |
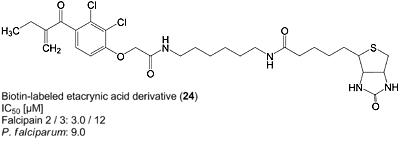
| 24 | H5C2 | Cl | Cl | 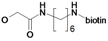 | 3.0±1.1 | 11.9±1.1 | 9.0±0.4c |
| 25 | 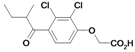 | 531a | ni | nic | |||
| 26 | 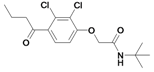 | 484a | ni | nic | |||
| 27 |  | 713a | ni | nic | |||
| 28 | 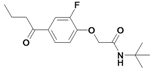 | 80±6 | nd | 66.4±2.2c | |||
| 29 | 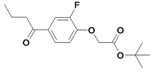 | 80±6 | nd | nd | |||
| 30 | 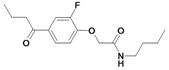 | ni | nd | nd | |||
| 31 | 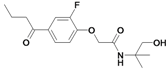 | ni | nd | nd | |||
| 32 | 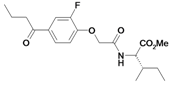 | ni | nd | nd | |||
| E-64 | 0.015±0.008 | 0.075±0.02 | 5.3±1.05c | ||||
| CQ (W2) | nd | nd | 0.24[19] | ||||
| CQ (3D7) | nd | nd | 0.01±0.0048 | ||||
Conclusions
Experimental
General
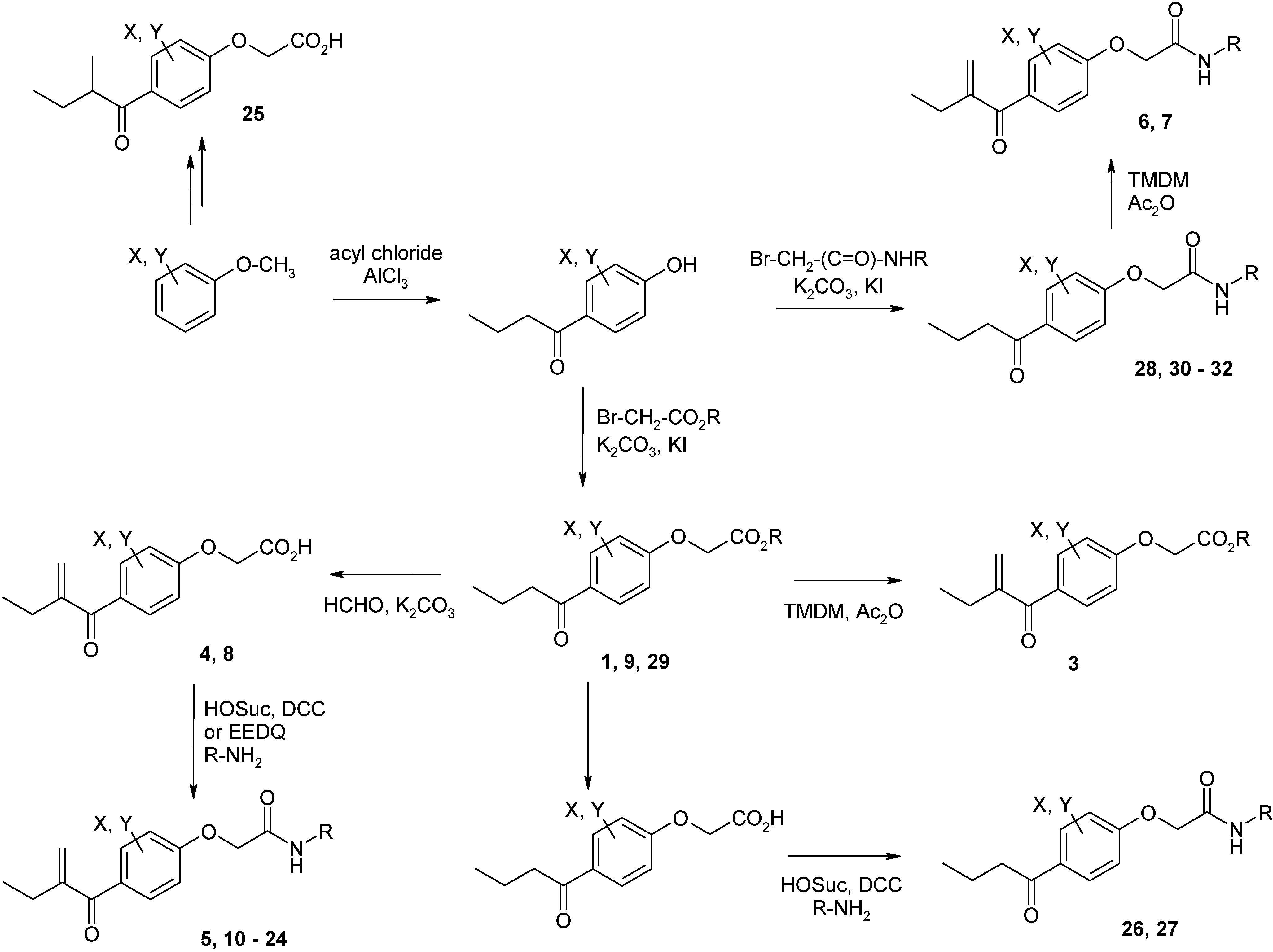
Syntheses of inhibitors
 : 61.1 (CHCl3, c = 0.1). 1H-NMR: δ 0.90–0.94 (m, 6H, Ile-CH2CH3 and Ile-CHCH3), 1.11 (t, 3H, 3J = 7.5 Hz, CqCH2CH3), 1.14–1.22 (m, 2H, Ile-CH2CH3), 1.40–1.48 (m, 1H, Ile-C=O), 1.93–1.99 (m, 1H, Ile-CHCH3), 2.47 (t, 2H, 3J = 7.3 Hz, CqCH2CH3), 3.74 (s, 3H, OCH3), 4.62 (s, 2H, OCH2C=O), 5.51 (s, Cq=CH2), 5.78 (s, Cq=CH2), 6.98 (dd, 1H, 3J = 8.2 Hz, CHarom.), 7.08 (d, 1H, J = 8.5 Hz, NH), 7.57–7.63 (m, 2H, CHarom.); 13C-NMR: δ 11.51 (Ile-CH2CH3 or Ile-CHCH3), 12.31 (CqCH2CH3), 15.50 (Ile-CH2CH3 or Ile-CHCH3), 25.08 (CqCH2CH3 or Ile-CHC=O), 25.43 (CqCH2CH3 or Ile-NCHC=O), 37.79 (Ile-CHCH3), 52.23 (OCH3), 68.32 (OCH2C=O), 114.25 (CHarom.), 117.86 (CHarom.), 123.26 (Cq=CH2), 126.81 (CHarom.), 132.46 (CqC=O), 149.37 (CqOCH2C=O), 150.66 (Cq=CH2), 153.14 (CqF), 166.83 (OCH2C=O), 171.68 (CqOCH3), 195.98 (CqC=O).
: 61.1 (CHCl3, c = 0.1). 1H-NMR: δ 0.90–0.94 (m, 6H, Ile-CH2CH3 and Ile-CHCH3), 1.11 (t, 3H, 3J = 7.5 Hz, CqCH2CH3), 1.14–1.22 (m, 2H, Ile-CH2CH3), 1.40–1.48 (m, 1H, Ile-C=O), 1.93–1.99 (m, 1H, Ile-CHCH3), 2.47 (t, 2H, 3J = 7.3 Hz, CqCH2CH3), 3.74 (s, 3H, OCH3), 4.62 (s, 2H, OCH2C=O), 5.51 (s, Cq=CH2), 5.78 (s, Cq=CH2), 6.98 (dd, 1H, 3J = 8.2 Hz, CHarom.), 7.08 (d, 1H, J = 8.5 Hz, NH), 7.57–7.63 (m, 2H, CHarom.); 13C-NMR: δ 11.51 (Ile-CH2CH3 or Ile-CHCH3), 12.31 (CqCH2CH3), 15.50 (Ile-CH2CH3 or Ile-CHCH3), 25.08 (CqCH2CH3 or Ile-CHC=O), 25.43 (CqCH2CH3 or Ile-NCHC=O), 37.79 (Ile-CHCH3), 52.23 (OCH3), 68.32 (OCH2C=O), 114.25 (CHarom.), 117.86 (CHarom.), 123.26 (Cq=CH2), 126.81 (CHarom.), 132.46 (CqC=O), 149.37 (CqOCH2C=O), 150.66 (Cq=CH2), 153.14 (CqF), 166.83 (OCH2C=O), 171.68 (CqOCH3), 195.98 (CqC=O).5-(2-Oxo-hexahydro-thieno[3,4-d]imidazol-4-yl)pentanoic acid (6-{2-[2,3-dichloro-4-(2-methylene-butyryl)-phenoxy]acetylamino}-hexyl)-amide (24)
= 61.1 (CHCl3, c = 0.1); 1H-NMR: δ 0.90–0.94 (m, 6H, Ile-CH2CH3 and Ile-CHCH3), 0.99 (t, 3H, 3J = 7.5 Hz, CH2CH2CH3), 1.12–1.23 (m, 2H, Ile-CH2CH3), 1.40–1.49 (m, 1H, Ile-CHC=O), 1.75 (sext, 3J = 7.3 Hz, CH2CH2CH3), 1.92–1.99 (m, 1H, Ile-CHCH3), 2.88 (t, 2H, 3J = 7.3 Hz, CH2CH2CH3), 3.74 (s, 3H, OCH3), 4.62 (s, 2H, OCH2C=O), 6.99 (dd, 1H, 3J = 8.5 Hz, CHarom.), 7.08 (d, 1H, J = 8.5 Hz, NH), 7.72–7.77 (m, 2H, CHarom.).Protein expression and purification
Enzyme assays, in-vitro assays
In vitro cytotoxicity assay
Acknowledgements
References and Notes
- WHO. World Malaria Report; World Health Organization: Geneva, Switzerland, 2005.
- Talisuna, A. O.; Okello, P. E.; Erhart, A.; Coosemans, M.; D'Alessandro, U. Intensity of malaria transmission and the spread of Plasmodium falciparum resistant malaria: a review of epidemiologic field evidence. Am. J. Trop. Med. Hyg. 2007, 77, 170–180. [Google Scholar]
- White, N. J. Antimalarial drug resistance. J. Clin. Invest. 2004, 113, 1084–1092. [Google Scholar]
- Otto, H.-H.; Schirmeister, T. Cysteine proteases and their inhibitors. Chem. Rev. 1997, 97, 133–172. [Google Scholar] [CrossRef]
- Rosenthal, P. J. Cysteine proteases of malaria parasites. Int. J. Parasitol. 2004, 34, 1489–1499. [Google Scholar] [CrossRef]
- Rosenthal, P. J.; Sijwali, P. S.; Singh, A.; Shenai, B. R. Cysteine proteases of malaria parasites: targets for chemotherapy. Curr. Pharm. Des. 2002, 8, 1659–1672. [Google Scholar] [CrossRef]
- Dahl, E. L.; Rosenthal, P. J. Biosynthesis, localization, and processing of falcipain cysteine proteases of Plasmodium falciparum. Mol. Biochem. Parasitol. 2005, 139, 205–212. [Google Scholar] [CrossRef]
- Rupp, I.; Bosse, R.; Schirmeister, T.; Pradel, G. Effect of protease inhibitors on exflagellation in Plasmodium falciparum. Mol. Biochem. Parasitol. 2008, 158, 208–212. [Google Scholar] [CrossRef]
- Lavrado, J.; Paulo, A.; Gut, J.; Rosenthal, P. J; Moreira, R. Cryptolepine analogues containing basic aminoalkyl side-chains at C-11: synthesis, antiplasmodial activity, and cytotoxicity. Bioorg. Med. Chem. Lett. 2008, 18, 1378–1381. [Google Scholar] [CrossRef]
- Vale, N.; Matos, J.; Gut, J.; Nogueira, F.; do Rosário, V.; Rosenthal, P. J.; Moreira, R.; Gomes, P. Imidazolidin-4-one peptidomimetic derivatives of primaquine: synthesis and antimalarial activity. Bioorg. Med. Chem. Lett. 2008, 18, 4150–4153. [Google Scholar] [CrossRef]
- Verissimo, E.; Berry, N.; Gibbons, P.; Cristiano, M. L.; Rosenthal, P. J.; Gut, J.; Ward, S. A.; O'Neill, P. M. Design and synthesis of novel 2-pyridone peptidomimetic falcipain 2/3 inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4210–4214. [Google Scholar] [CrossRef]
- Ettari, R.; Nizi, E.; Di Francesco, M. E.; Dude, M. A.; Pradel, G.; Vicík, R.; Schirmeister, T.; Micale, N.; Grasso, S.; Zappalà, M. Development of peptidomimetics with a vinyl sulfone warhead as irreversible falcipain-2 inhibitors. J. Med. Chem. 2008, 51, 988–996. [Google Scholar] [CrossRef]
- Sijwali, P. S.; Shenai, B. R..; Gut, J.; Singh, A.; Rosenthal, P. J. Expression and characterization of the Plasmodium falciparum haemoglobinase falcipain-3. Biochem. J. 2001, 360, 481–489. [Google Scholar] [CrossRef]
- Desai, P. V.; Patny, A.; Gut, J.; Rosenthal, P. J.; Tekwani, B.; Srivastava, A.; Avery, M. Identification of novel parasitic cysteine protease inhibitors by use of virtual screening. 2. The available chemical directory. J. Med. Chem. 2006, 49, 1576–1584. [Google Scholar] [CrossRef]
- Semenov, A.; Olson, P. J.; Rosenthal, P. J. Antimalarial synergy of cysteine and aspartic protease inhibitors. Antimicrob. Agents Chemother. 1998, 42, 2254–2258. [Google Scholar]
- Kaeppler, U.; Schirmeister, T. New non-peptidic inhibitors of papain derived from etacrynic acid. Med. Chem. 2005, 1, 361–370. [Google Scholar]
- Kaeppler, U.; Stiefl, N.; Schiller, M.; Vicik, R.; Breuning, A.; Schmitz, W.; Rupprecht, D.; Schmuck, C.; Baumann, K.; Ziebuhr, J.; Schirmeister, T. J. A new lead for nonpeptidic active-site-directed inhibitors of the severe acute respiratory syndrome coronavirus main protease discovered by a combination of screening and docking methods. J. Med. Chem. 2005, 48, 6832–6842. [Google Scholar] [CrossRef]
- Shenai, B. R.; Sijwali, P. S.; Singh, A.; Rosenthal, P. J. Stage-specific antimalarial activity of cysteine protease inhibitors. J. Biol. Chem. 2000, 275, 29000–29010. [Google Scholar]
- Schulz, F.; Gelhaus, C.; Degel, B.; Vicik, R.; Heppner, S.; Breuning, A.; Leippe, M.; Rosenthal, P. J.; Schirmeister, T. Screening of protease inhibitors as antiplasmodial agents. Part I: Aziridines and epoxides. ChemMedChem. 2007, 2, 1214–1224. [Google Scholar] [CrossRef]
- Sijwali, P. S.; Koo, J.; Singh, N.; Rosenthal, P. J. Gene disruptions demonstrate independent roles for the four falcipain cysteine proteases of Plasmodium falciparum. Mol. Biochem. Parasitol. 2006, 150, 96–106. [Google Scholar] [CrossRef]
- Mikus, J.; Steverding, D. A simple colorimetric method to screen drug cytotoxicity against Leishmania using the dye Alamar Blue. Parasitol. Int. 2000, 48, 265–269. [Google Scholar] [CrossRef]
- Ponte-Sucre, A.; Faber, J. H.; Gulder, T.; Kajahn, I.; Pedersen, S. E.; Schultheis, M.; Bringmann, G.; Moll, H. Activities of naphthylisoquinoline alkaloids and synthetic analogs against Leishmania major. Antimicrob. Agents Chemother. 2007, 51, 188–194. [Google Scholar] [CrossRef]
- Gelhaus, C.; Vicik, R.; Hilgenfeld, R.; Schmidt, C. L.; Leippe, M.; Schirmeister, T. Synthesis and antiplasmodial activity of a cysteine protease-inhibiting biotinylated aziridine-2,3-dicarboxylate. Biol. Chem. 2004, 385, 435–438. [Google Scholar]
- Pandey, K. C.; Wang, S. X.; Sijwali, P. S.; Lau, A. L.; McKerrow, J. H.; Rosenthal, P. J. The Plasmodium falciparum cysteine protease falcipain-2 captures its substrate, hemoglobin, via a unique motif. Proc. Natl. Acad. Sci. USA 2005, 102, 9138–9143. [Google Scholar] [CrossRef]
- Ramjee, M. K.; Flinn, N. S.; Pemberton, T. P.; Quibell, M.; Wang, Y.; Watts, J. P. Substrate mapping and inhibitor profiling of falcipain-2, falcipain-3 and berghepain-2: implications for peptidase anti-malarial drug discovery. Biochem. J. 2006, 399, 47–57. [Google Scholar] [CrossRef]
- Sijwali, P. S.; Rosenthal, P. J. Gene disruption confirms a critical role for the cysteine protease falcipain-2 in hemoglobin hydrolysis by Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 2004, 101, 4384–4389. [Google Scholar] [CrossRef]
- Musonda, C. C.; Gut, J.; Rosenthal, P. J.; Yardley, V.; Carvalho de Souza, R. C.; Chibale, K. Application of multicomponent reactions to antimalarial drug discovery. Part 2: New antiplasmodial and antitrypanosomal 4-aminoquinoline gamma- and delta-lactams via a 'catch and release' protocol. Bioorg. Med. Chem. 2006, 14, 5605–5615. [Google Scholar] [CrossRef]
- Smeijsters, L. J.; Zijlstra, N. M.; Franssen, F. F.; Overdulve, J. P. Simple, fast, and accurate fluorometric method to determine drug susceptibility of Plasmodium falciparum in 24-well suspension culture. Antimicrob. Agents Chemother. 1996, 40, 835–838. [Google Scholar]
- Makler, M. T.; Hinrichs, D. J. Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. Am. J. Trop. Med. Hyg. 1993, 48, 205–210. [Google Scholar]
- Ifediba, T.; Vanderberg, J. P. Complete in vitro maturation of Plasmodium falciparum gametocytes. Nature 1981, 294, 364–366. [Google Scholar] [CrossRef]
- GraphPad® Prism 4, GraphPad Software, Inc.: La Jolla, CA, USA, 2003.
- GraFit® 5.0.13, Erithacus Software Ltd.: Horley, Surrey, UK, 2006.
- Ahmed, S. A.; Gogal, R. M.; Walsh, J. E. A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes: an alternative to [3H]thymidine incorporation assay. J. Immunol. Methods 1994, 170, 211–224. [Google Scholar] [CrossRef]
- Sample Availability: Samples of compound 24 are available from the authors.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dude, M.-A.; Kaeppler, U.; Herb, M.; Schiller, M.; Schulz, F.; Vedder, B.; Heppner, S.; Pradel, G.; Gut, J.; Rosenthal, P.J.; et al. Synthesis and Evaluation of Non-peptidic Cysteine Protease Inhibitors of P. falciparum Derived from Etacrynic Acid. Molecules 2009, 14, 19-35. https://doi.org/10.3390/molecules14010019
Dude M-A, Kaeppler U, Herb M, Schiller M, Schulz F, Vedder B, Heppner S, Pradel G, Gut J, Rosenthal PJ, et al. Synthesis and Evaluation of Non-peptidic Cysteine Protease Inhibitors of P. falciparum Derived from Etacrynic Acid. Molecules. 2009; 14(1):19-35. https://doi.org/10.3390/molecules14010019
Chicago/Turabian StyleDude, Marie-Adrienne, Ulrich Kaeppler, Monika Herb, Markus Schiller, Franziska Schulz, Birgit Vedder, Saskia Heppner, Gabriele Pradel, Jiri Gut, Philip J. Rosenthal, and et al. 2009. "Synthesis and Evaluation of Non-peptidic Cysteine Protease Inhibitors of P. falciparum Derived from Etacrynic Acid" Molecules 14, no. 1: 19-35. https://doi.org/10.3390/molecules14010019