by
Pravil Pokharel 1,2,† , Sabin Dhakal 1,2,† and Charles M. Dozois 1,2,3,*
, Sabin Dhakal 1,2,† and Charles M. Dozois 1,2,3,*
, Sabin Dhakal 1,2,† and Charles M. Dozois 1,2,3,*
1
Centre Armand-Frappier Santé Biotechnologie, Institut National de la Recherche Scientifique (INRS), 531 Boul des Prairies, Laval, QC H7V 1B7, Canada
2
Centre de Recherche en Infectiologie Porcine et Avicole (CRIPA), Faculté de Médecine Vétérinaire, Université de Montréal Saint-Hyacinthe, Saint-Hyacinthe, QC J2S 2M2, Canada
3
Pasteur Network, Laval, QC H7V 1B7, Canada
†
These authors equally contributed to this work.
Microorganisms 2023, 11(2), 344; https://doi.org/10.3390/microorganisms11020344 - 30 Jan 2023
Cited by 122 | Viewed by 37753
Abstract
Escherichia coli (E. coli) is a gram-negative bacillus and resident of the normal intestinal microbiota. However, some E. coli strains can cause diseases in humans, other mammals and birds ranging from intestinal infections, for example, diarrhea and dysentery, to extraintestinal infections,
[...] Read more.
Escherichia coli (E. coli) is a gram-negative bacillus and resident of the normal intestinal microbiota. However, some E. coli strains can cause diseases in humans, other mammals and birds ranging from intestinal infections, for example, diarrhea and dysentery, to extraintestinal infections, such as urinary tract infections, respiratory tract infections, meningitis, and sepsis. In terms of morbidity and mortality, pathogenic E. coli has a great impact on public health, with an economic cost of several billion dollars annually worldwide. Antibiotics are not usually used as first-line treatment for diarrheal illness caused by E. coli and in the case of bloody diarrhea, antibiotics are avoided due to the increased risk of hemolytic uremic syndrome. On the other hand, extraintestinal infections are treated with various antibiotics depending on the site of infection and susceptibility testing. Several alarming papers concerning the rising antibiotic resistance rates in E. coli strains have been published. The silent pandemic of multidrug-resistant bacteria including pathogenic E. coli that have become more difficult to treat favored prophylactic approaches such as E. coli vaccines. This review provides an overview of the pathogenesis of different pathotypes of E. coli, the virulence factors involved and updates on the major aspects of vaccine development against different E. coli pathotypes.
Full article
(This article belongs to the Special Issue Virulence Genes, Antimicrobial Resistance Profiles and Genetic Diversity of Escherichia coli)
▼
Show Figures

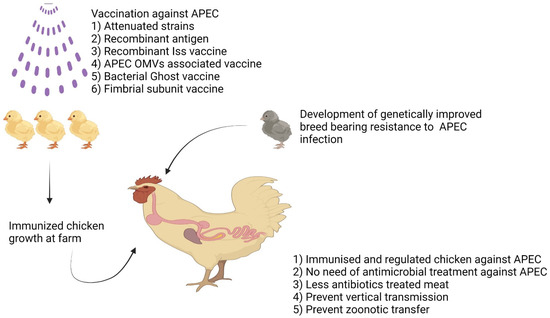
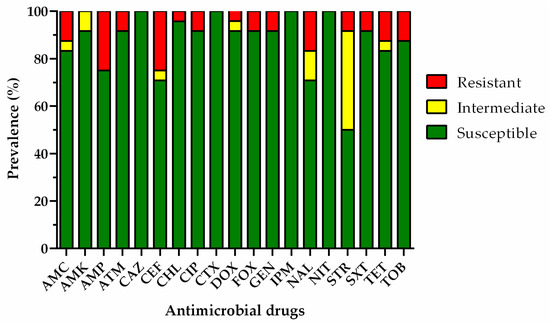
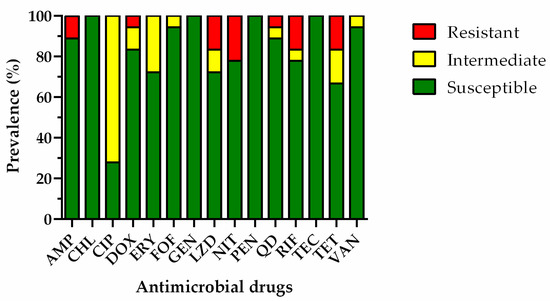
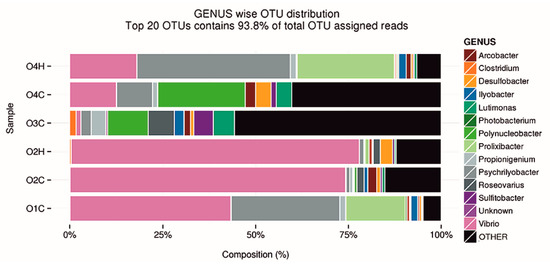
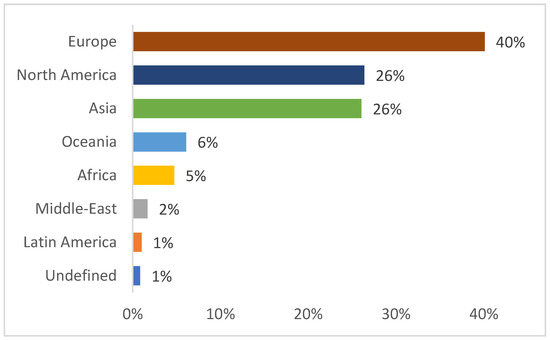
Figure 1




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}