by
Stacey L. Hanlon 1,* and R. Scott Hawley 1,2
and R. Scott Hawley 1,2
and R. Scott Hawley 1,2
1
Stowers Institute for Medical Research, Kansas City, MO 64110, USA
2
Department of Molecular and Integrative Physiology, University of Kansas Medical Center, Kansas City, KS 66160, USA
Genes 2018, 9(10), 470; https://doi.org/10.3390/genes9100470 - 27 Sep 2018
Cited by 6 | Viewed by 6876
Abstract
Our current knowledge of B chromosome biology has been augmented by an increase in the number and diversity of species observed to carry B chromosomes as well as the use of next-generation sequencing for B chromosome genomic analysis. Within the genus Drosophila,
[...] Read more.
Our current knowledge of B chromosome biology has been augmented by an increase in the number and diversity of species observed to carry B chromosomes as well as the use of next-generation sequencing for B chromosome genomic analysis. Within the genus Drosophila, B chromosomes have been observed in a handful of species, but recently they were discovered in a single laboratory stock of Drosophila melanogaster. In this paper, we review the B chromosomes that have been identified within the Drosophila genus and pay special attention to those recently found in D. melanogaster. These newly-discovered B chromosomes have centromeres, telomeres, and a number of simple satellite repeats. They also appear to be entirely heterochromatic since next-generation sequencing of isolated B chromosomes did not detect sequences associated with known genic regions. We also summarize what effects the B chromosomes have been found to have on the A chromosomes. Lastly, we highlight some of the outstanding questions regarding B chromosome biology and discuss how studying B chromosomes in Drosophila melanogaster, which is a versatile model system with a wealth of genetic and genomic tools, may advance our understanding of the B chromosome’s unique biology.
Full article
(This article belongs to the Special Issue Evolution, Composition and Regulation of Supernumerary B Chromosomes)
▼
Show Figures
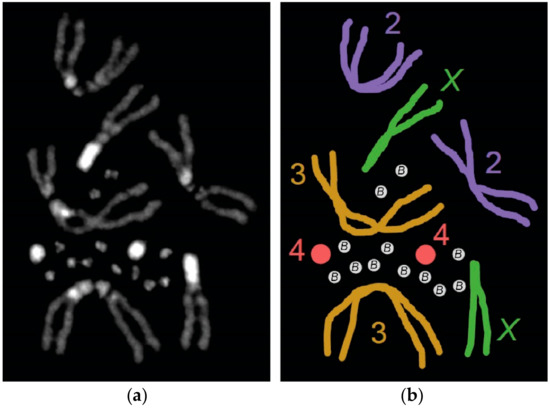
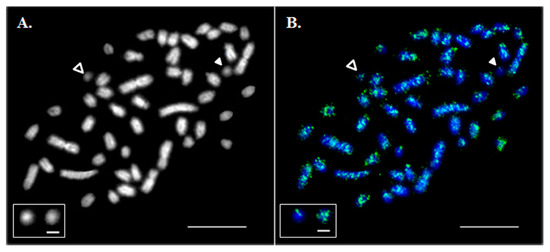
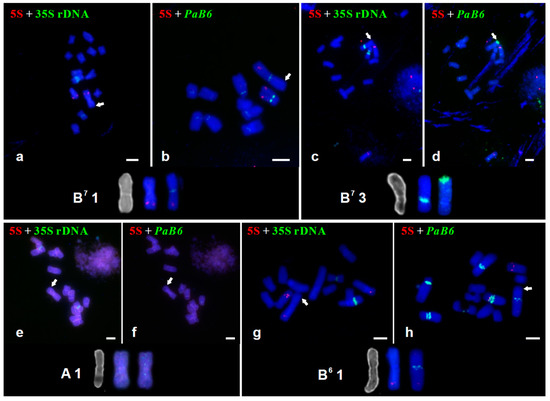
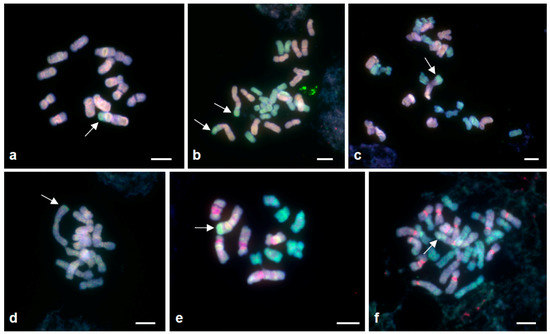
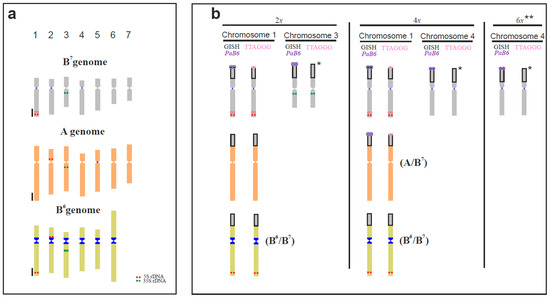
Figure 1


{kind=link}
{kind=link}
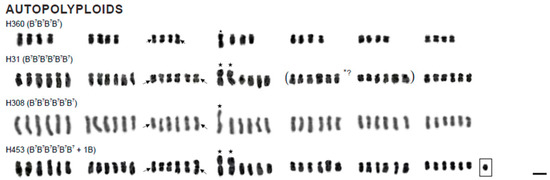{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}