A Derivate of Benzimidazole-Isoquinolinone Induces SKP2 Transcriptional Inhibition to Exert Anti-Tumor Activity in Glioblastoma Cells

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
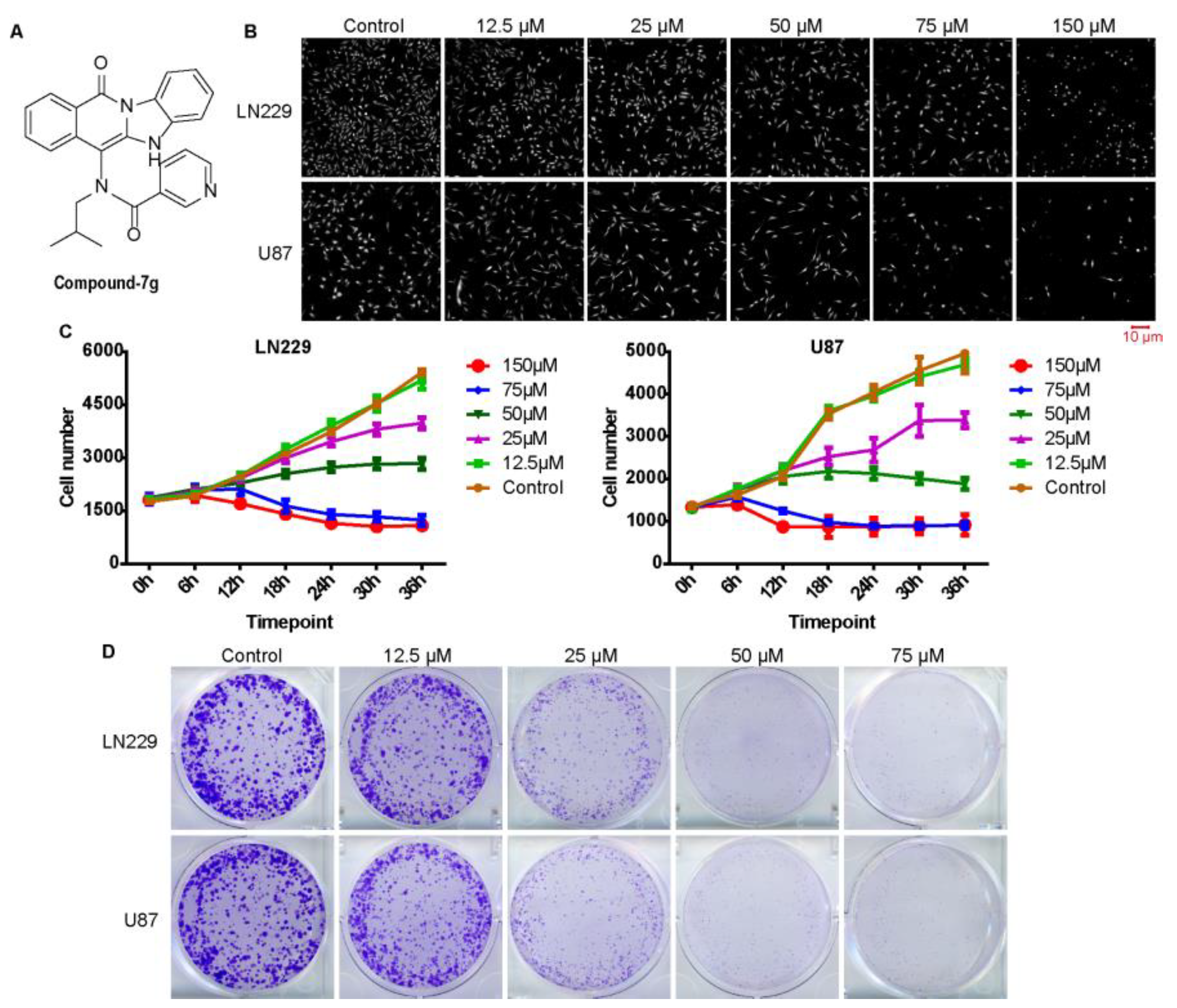
2.1. Compound-7g, as An Effective Anticancer Inhibitor, Can Significantly Inhibit Cell Proliferation and Growth in Glioblastoma Cells
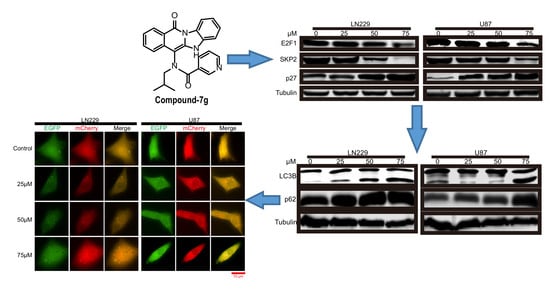
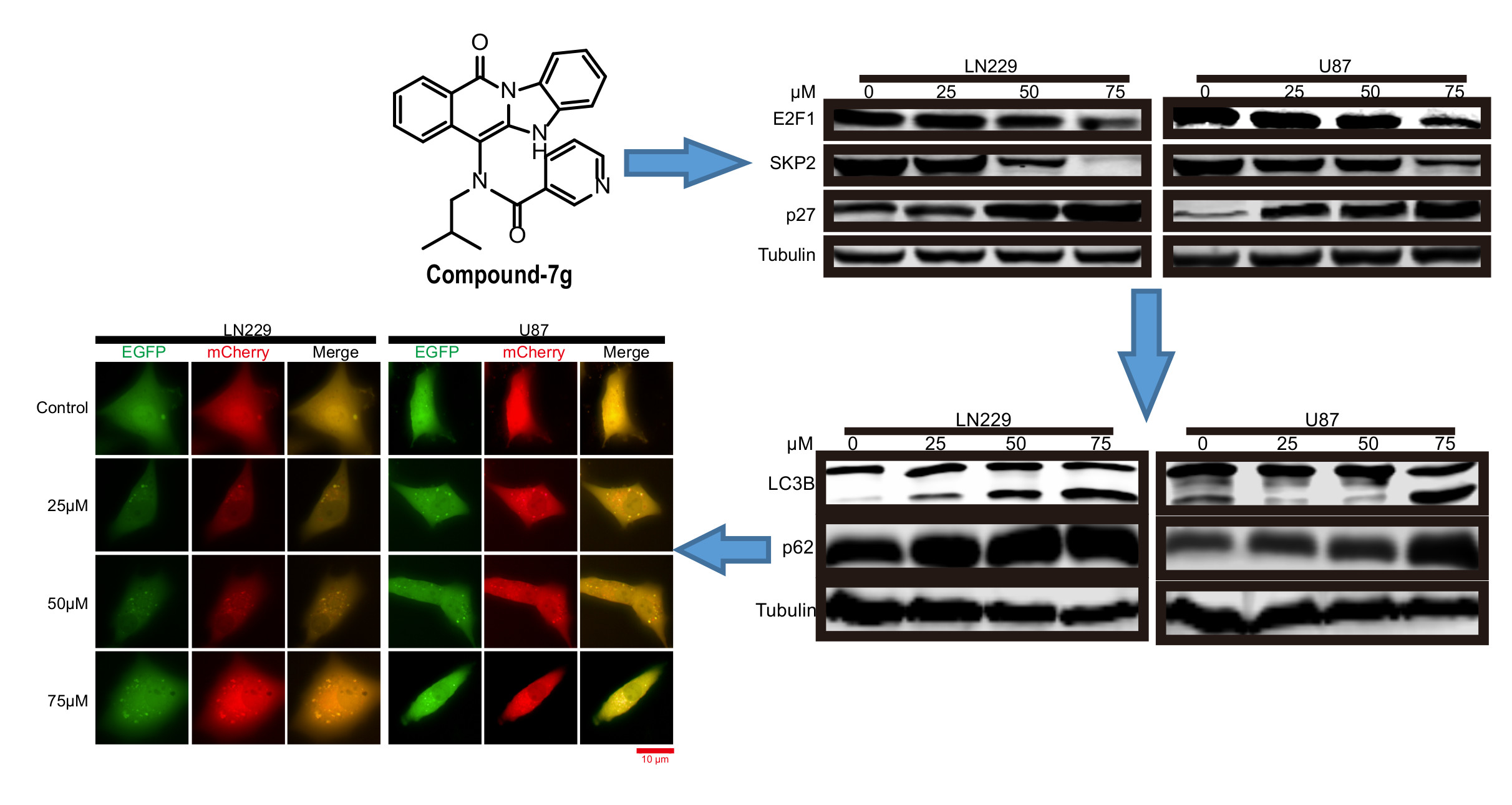
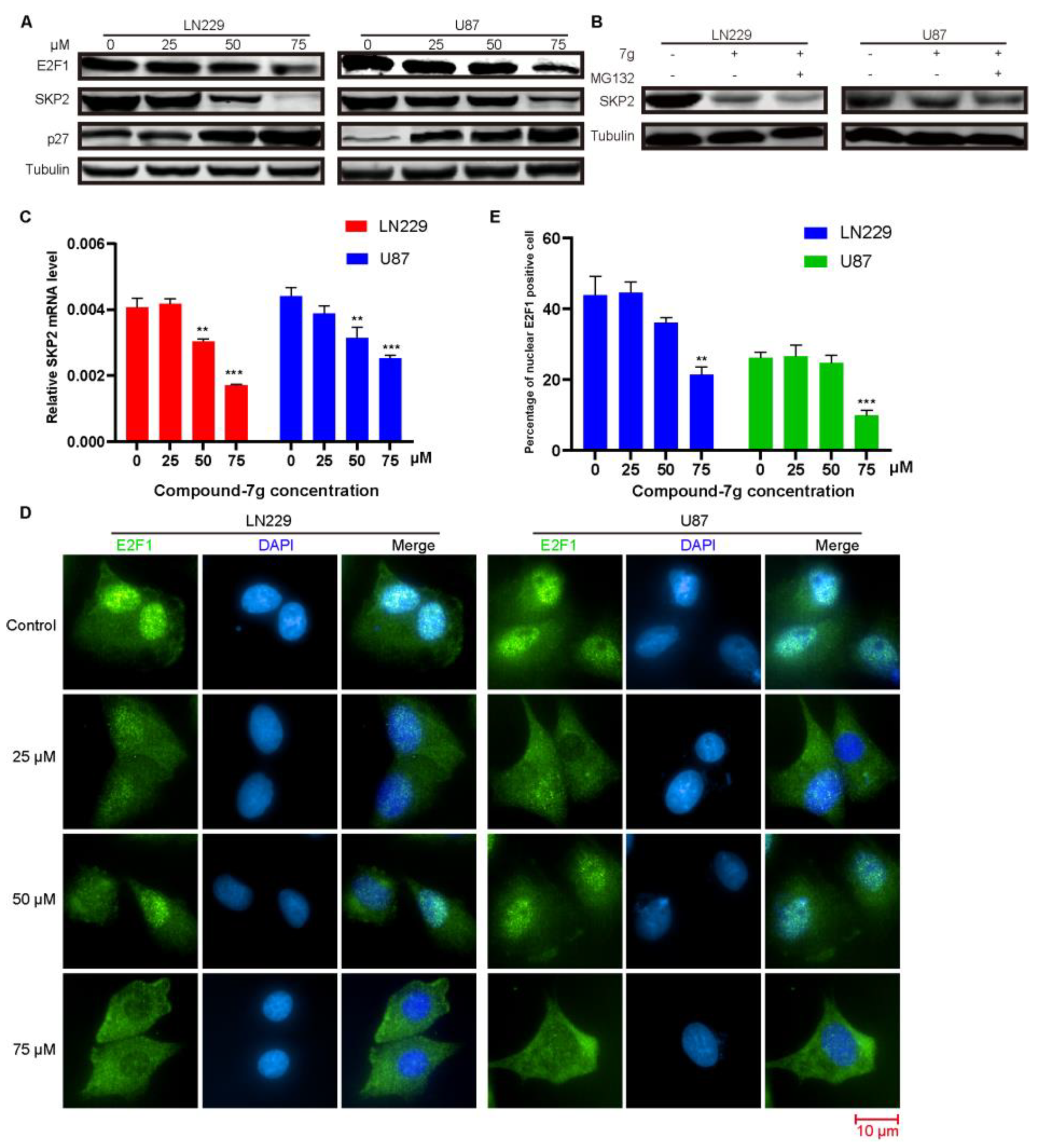
2.2. Compound-7g Induces Transcriptional Repression of Skp2 By Promoting E2F-1 Degradation
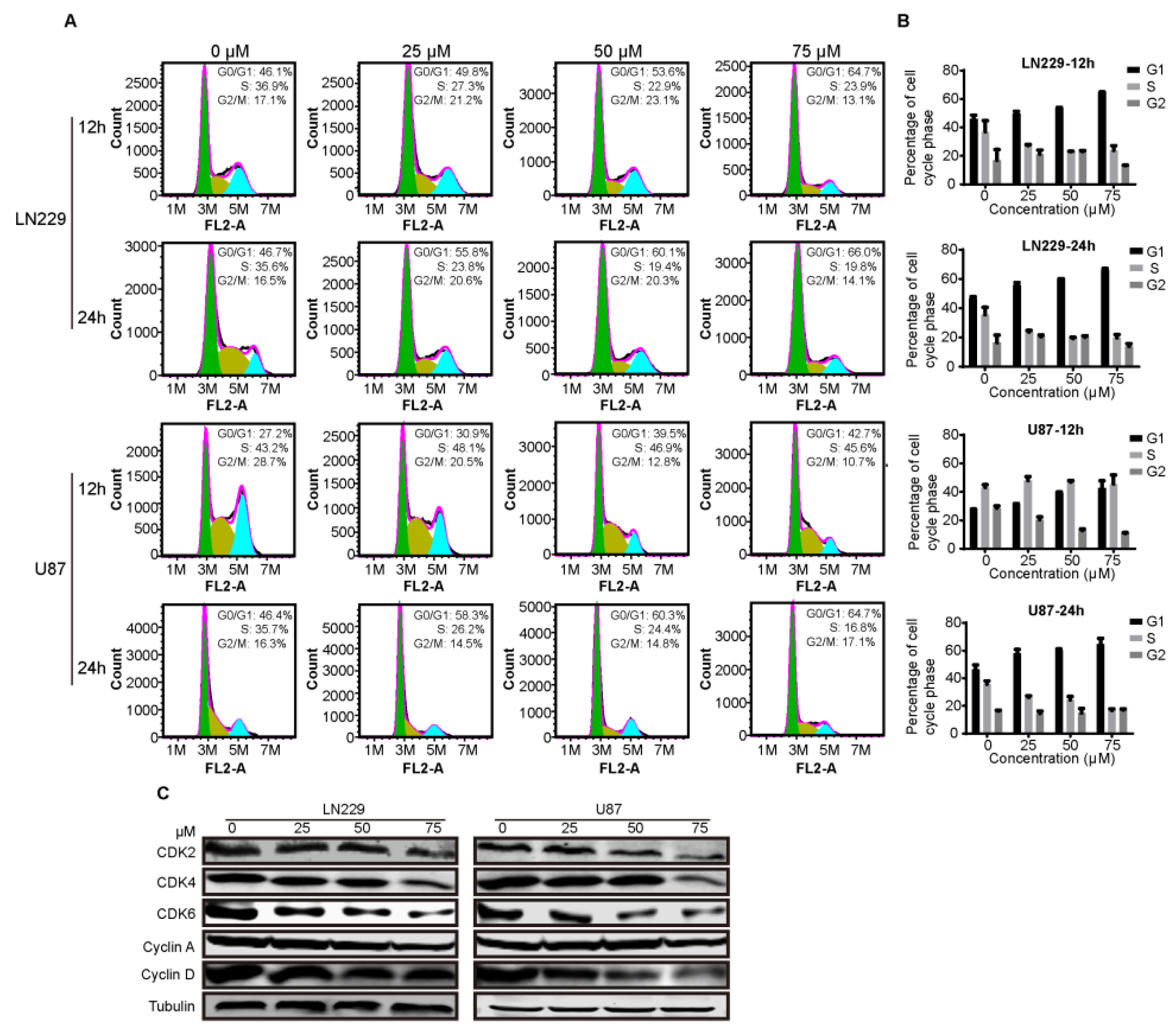
2.3. Compound-7g Induces G1-Phase Arrest of Glioblastoma Cells
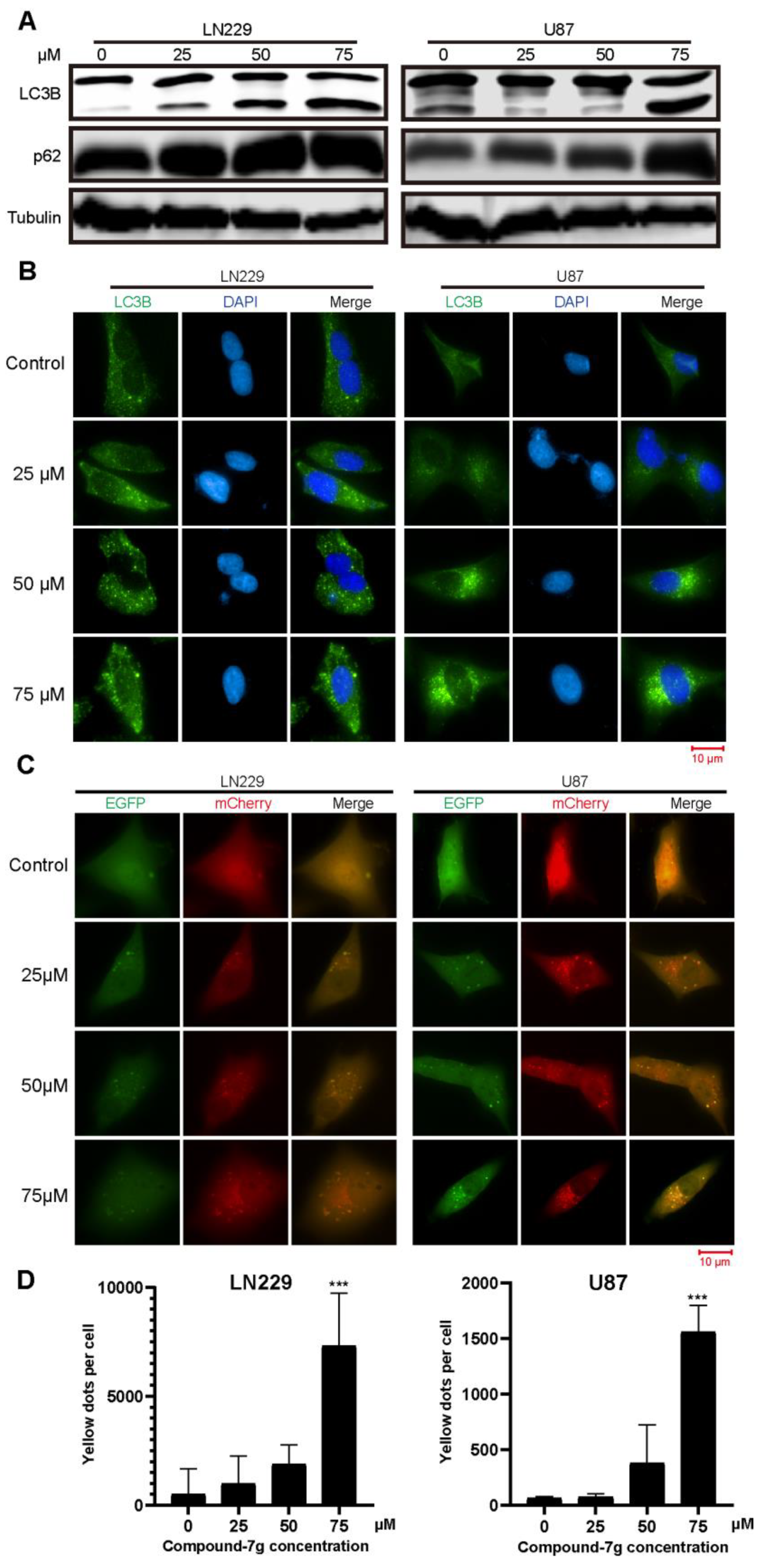
2.4. Compound-7g Inhibits the Fusion Between Autophagosome And Lysosome in Glioblastoma Cells
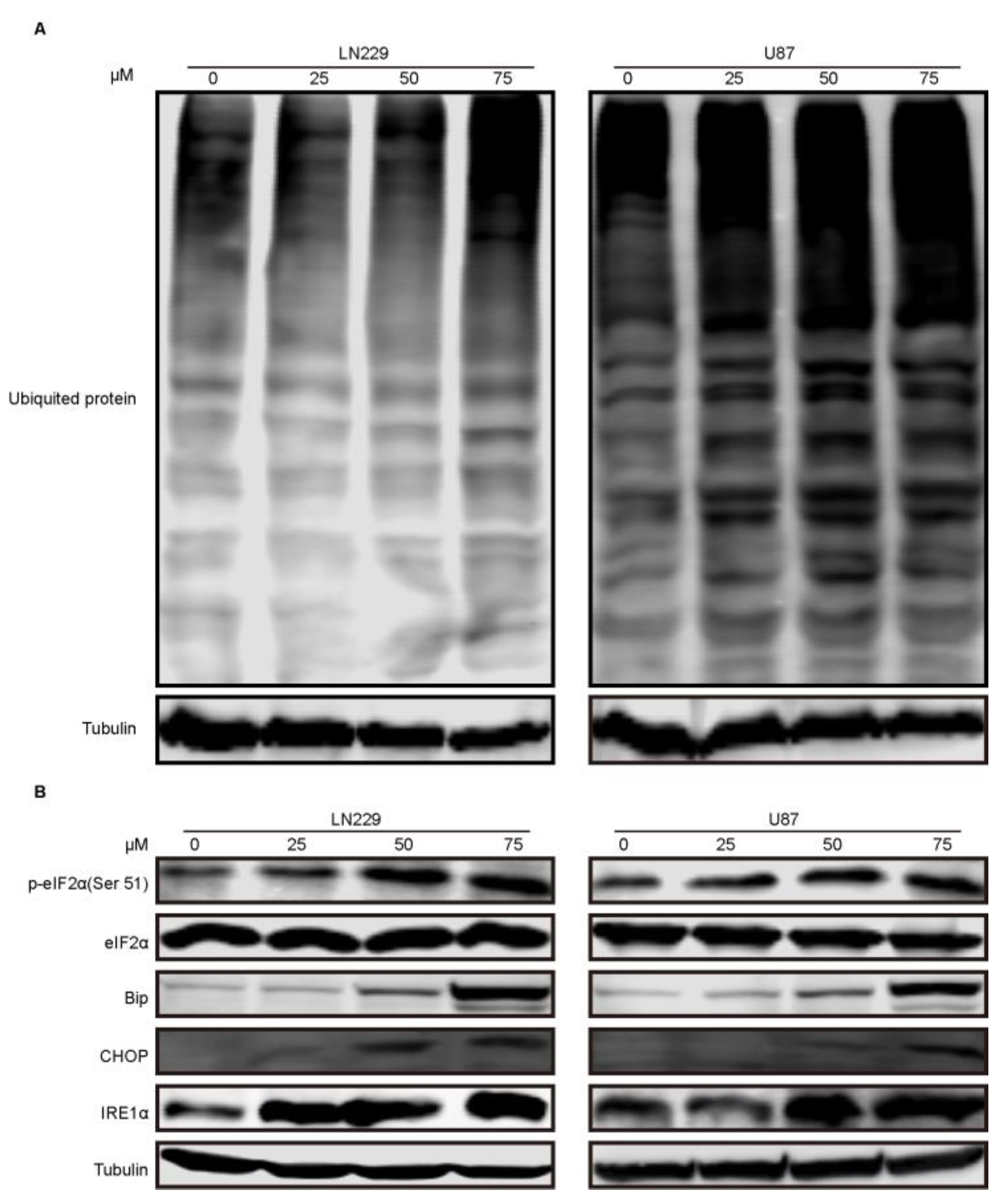
2.5. Compound-7g Induced Autophagic Flux Blockage Results in Accumulation of Ubiquitinated Proteins and Subsequent ER Stress
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Lines and Culture
4.3. Cell Viability Analysis
4.4. Colony Formation Assay
4.5. Immunoblotting
4.6. Cell Cycle Measurement
4.7. RNA Extraction and Real-Time Fluorescent Quantitative PCR
4.8. Immunofluorescence Staining
4.9. Fluorescence Observation of Mcherry-GFP-LC3B
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Sathornsumetee, S.; Reardon, D.A.; Desjardins, A.; Quinn, J.A.; Vredenburgh, J.J.; Rich, J.N. Molecularly targeted therapy for malignant glioma. Cancer 2007, 110, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Hueng, D.-Y.; Hsieh, C.-H.; Cheng, Y.-C.; Tsai, W.-C.; Chen, Y. Cordycepin inhibits migration of human glioblastoma cells by affecting lysosomal degradation and protein phosphatase activation. J. Nutr. Biochem. 2017, 41, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fan, C.; Pu, L.; Wei, C.; Jin, H.; Teng, Y.; Zhao, M.; Yu, A.C.H.; Jiang, F.; Shu, J. Phloretin induces cell cycle arrest and apoptosis of human glioblastoma cells through the generation of reactive oxygen species. J. Neuro-Oncol. 2016, 128, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lim, H.-S. Skp2 inhibitors: Novel anticancer strategies. Curr. Med. Chem. 2016, 23, 2363–2379. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, P.; Inuzuka, H.; Wei, W. Roles of F-box proteins in cancer. Nat. Rev. Cancer 2014, 14, 233. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.-H.; Morrow, J.K.; Zhang, S.; Lin, H.-K. Skp2: A dream target in the coming age of cancer therapy. Cell Cycle 2014, 13, 679–680. [Google Scholar] [CrossRef] [PubMed]
- Chu, I.M.; Hengst, L.; Slingerland, J.M. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 2008, 8, 253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, C. F-box protein Skp2: A novel transcriptional target of E2F. Oncogene 2006, 25, 2615. [Google Scholar] [CrossRef]
- Reichert, M.; Saur, D.; Hamacher, R.; Schmid, R.M.; Schneider, G. Phosphoinositide-3-kinase signaling controls S-phase kinase–associated protein 2 transcription via E2F1 in pancreatic ductal adenocarcinoma cells. Cancer Res. 2007, 67, 4149–4156. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Bauzon, F.; Fu, H.; Cui, J.; Zhao, H.; Nakayama, K.; Nakayama, K.I.; Zhu, L. Skp2 suppresses apoptosis in Rb1-deficient tumours by limiting E2F1 activity. Nat. Commun. 2014, 5, 3463. [Google Scholar] [CrossRef] [PubMed]
- Salon, C.; Merdzhanova, G.; Brambilla, C.; Brambilla, E.; Gazzeri, S.; Eymin, B. E2F-1, Skp2 and cyclin E oncoproteins are upregulated and directly correlated in high-grade neuroendocrine lung tumors. Oncogene 2007, 26, 6927. [Google Scholar] [CrossRef] [PubMed]
- He, L.-J.; Yang, D.-L.; Li, S.-Q.; Zhang, Y.-J.; Tang, Y.; Lei, J.; Frett, B.; Lin, H.-K.; Li, H.-Y.; Chen, Z.-Z. Facile construction of fused benzimidazole-isoquinolinones that induce cell-cycle arrest and apoptosis in colorectal cancer cells. Bioorgan Med. Chem. 2018, 26, 3899–3908. [Google Scholar] [CrossRef] [PubMed]
- Kurland, J.F.; Tansey, W.P. Crashing waves of destruction: The cell cycle and APCCdh1 regulation of SCFSkp2. Cancer Cell 2004, 5, 305–306. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Wang, Y.; Wang, X.; Li, R.; Yin, D. MicroRNA-365 accelerates cardiac hypertrophy by inhibiting autophagy via the modulation of Skp2 expression. Biochem. Biophys. Res. Commun. 2017, 484, 304–310. [Google Scholar] [CrossRef]
- Jung, D.; Khurana, A.; Roy, D.; Kalogera, E.; Bakkum-Gamez, J.; Chien, J.; Shridhar, V. Quinacrine upregulates p21/p27 independent of p53 through autophagy-mediated downregulation of p62-Skp2 axis in ovarian cancer. Sci. Rep. 2018, 8, 2487. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Zhang, L.; Zhao, Q.; Zhao, Z.; Zhi, F.; Qin, Y.; Cui, J. SKP2 attenuates NF-κ B signaling by mediating IKK β degradation through autophagy. J. Mol. Cell Boil. 2018, 10, 205–215. [Google Scholar] [CrossRef]
- Hao, Z.; Huang, S. E3 ubiquitin ligase Skp2 as an attractive target in cancer therapy. Front. Biosci. (Landmark Ed.) 2015, 20, 474–490. [Google Scholar] [CrossRef]
- Lu, W.; Liu, S.; Li, B.; Xie, Y.; Adhiambo, C.; Yang, Q.; Ballard, B.R.; Nakayama, K.I.; Matusik, R.J.; Chen, Z. SKP2 inactivation suppresses prostate tumorigenesis by mediating JARID1B ubiquitination. Oncotarget 2015, 6, 771. [Google Scholar] [CrossRef]
- Wei, Z.; Jiang, X.; Qiao, H.; Zhai, B.; Zhang, L.; Zhang, Q.; Wu, Y.; Jiang, H.; Sun, X. STAT3 interacts with Skp2/p27/p21 pathway to regulate the motility and invasion of gastric cancer cells. Cell. Signal. 2013, 25, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Zhou, Y.; Wang, B.; Li, Q.; Chen, Y.; Lan, H. Immunohistochemically detected expression of Skp2, p27 kip1, and p-p27 (Thr187) in patients with cholangiocarcinoma. Tumor. Biol. 2015, 36, 5119–5125. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Nan, H.; Ma, J.; Jiang, L.; Guo, Q.; Han, L.; Zhang, Y.; Nan, K.; Guo, H. High Skp2/low p57Kip2 expression is associated with poor prognosis in human breast carcinoma. Breast Cancer Basic Clin. Res. 2015, 9, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.T.; So, M.; Ng, J.; Yang, H.W.; Chang, M.L.; Lai, M.W.; Chen, T.C.; Lin, C.Y.; Yeh, T.S.; Lee, W.C. Hepatitis B virus–DNA level and basal core promoter A1762T/G1764A mutation in liver tissue independently predict postoperative survival in hepatocellular carcinoma. Hepatology 2010, 52, 1922–1933. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cui, J.; Bauzon, F.; Zhu, L. A comparison between Skp2 and FOXO1 for their cytoplasmic localization by Akt1. Cell Cycle 2010, 9, 1021–1022. [Google Scholar] [CrossRef] [PubMed]
- Beth, L.; Guido, K. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.-P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Shin, H.-J.R.; Kim, H.; Oh, S.; Lee, J.-G.; Kee, M.; Ko, H.-J.; Kweon, M.-N.; Won, K.-J.; Baek, S.H. AMPK–SKP2–CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 2016, 534, 553–557. [Google Scholar] [CrossRef]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [Green Version]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef]
Sample Availability: Sample of the compound-7g is available from the authors. |





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.-y.; He, L.-j.; Li, S.-q.; Zhang, Y.-j.; Huang, J.-h.; Qin, H.-x.; Wang, J.-l.; Li, Q.-y.; Yang, D.-l. A Derivate of Benzimidazole-Isoquinolinone Induces SKP2 Transcriptional Inhibition to Exert Anti-Tumor Activity in Glioblastoma Cells. Molecules 2019, 24, 2722. https://doi.org/10.3390/molecules24152722
Chen H-y, He L-j, Li S-q, Zhang Y-j, Huang J-h, Qin H-x, Wang J-l, Li Q-y, Yang D-l. A Derivate of Benzimidazole-Isoquinolinone Induces SKP2 Transcriptional Inhibition to Exert Anti-Tumor Activity in Glioblastoma Cells. Molecules. 2019; 24(15):2722. https://doi.org/10.3390/molecules24152722
Chicago/Turabian StyleChen, He-ying, Liu-jun He, Shi-qiang Li, Ya-jun Zhang, Jiu-hong Huang, Hong-xia Qin, Juan-li Wang, Qian-yin Li, and Dong-lin Yang. 2019. "A Derivate of Benzimidazole-Isoquinolinone Induces SKP2 Transcriptional Inhibition to Exert Anti-Tumor Activity in Glioblastoma Cells" Molecules 24, no. 15: 2722. https://doi.org/10.3390/molecules24152722