Synthesis of Flavone Derivatives via N-Amination and Evaluation of Their Anticancer Activities

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
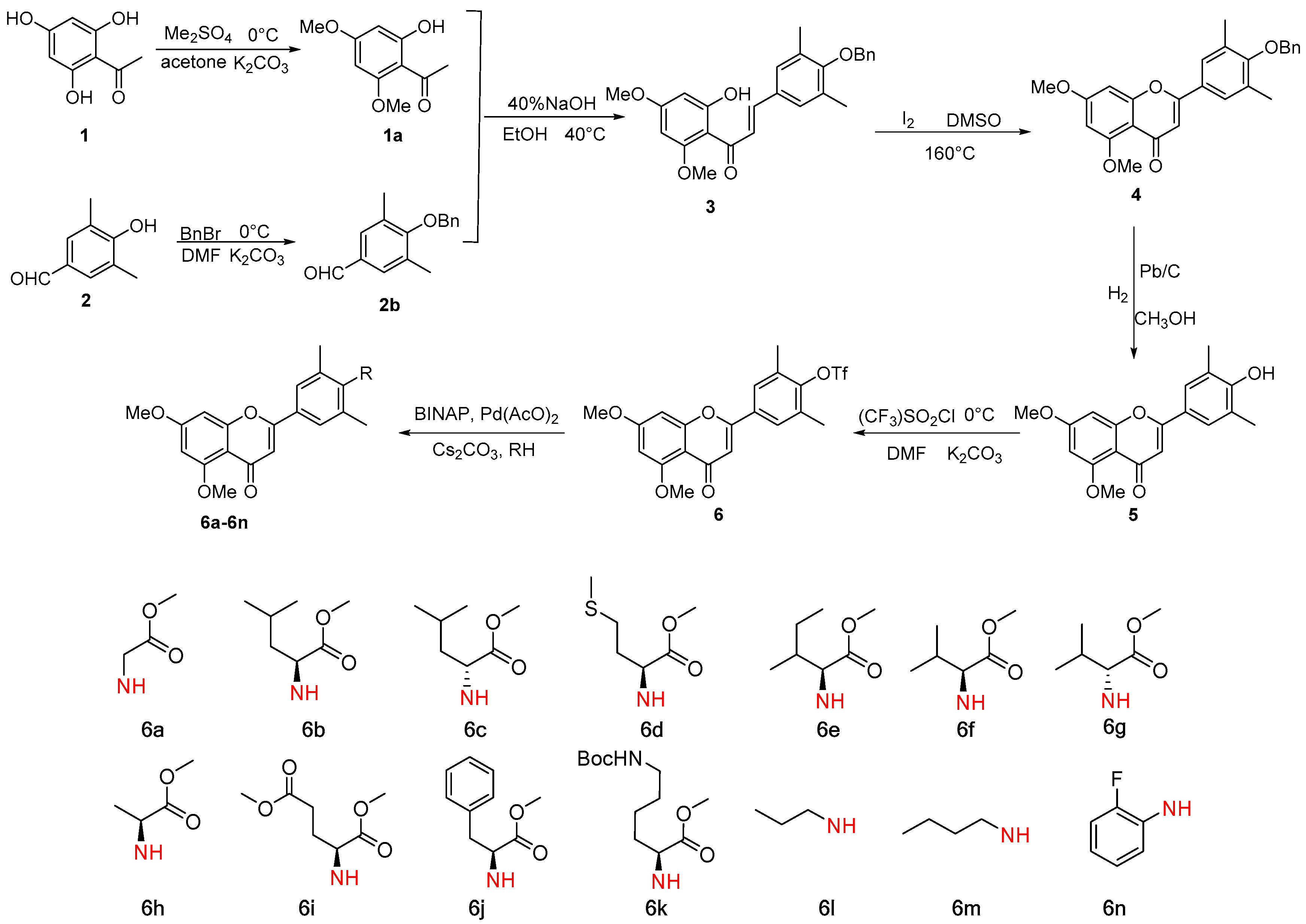
2.1. Chemistry
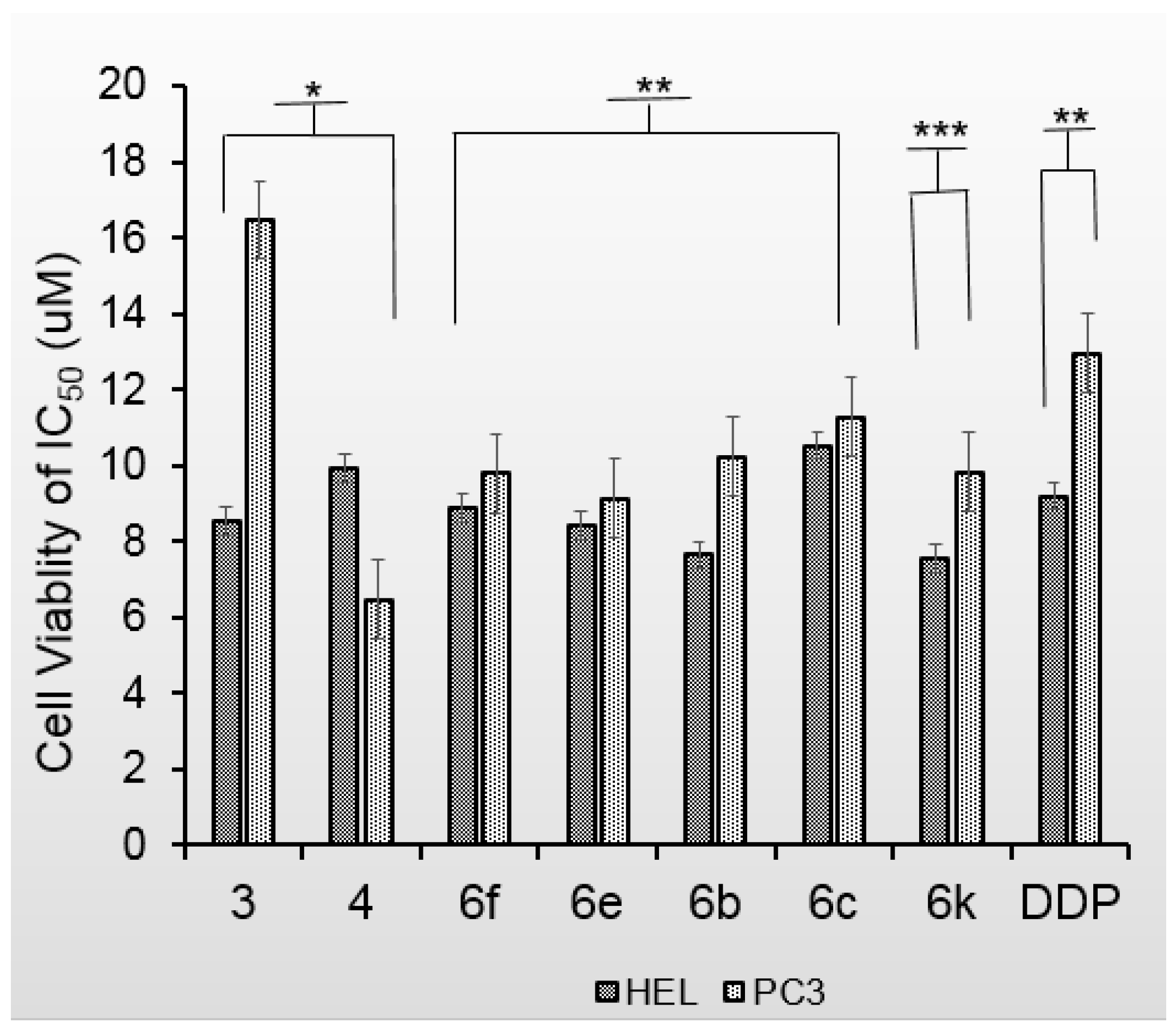
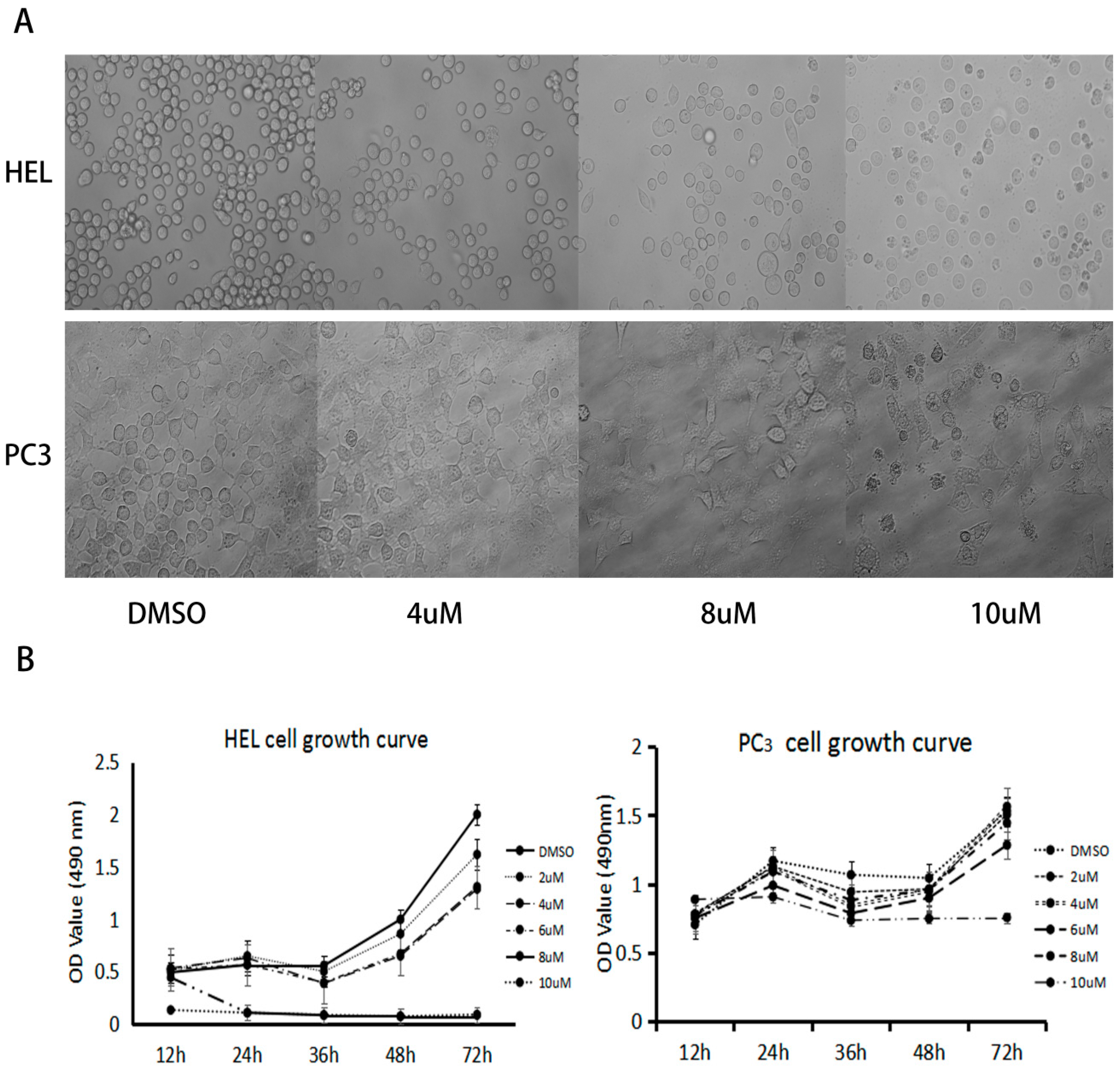
2.2. Anticancer Bioactivity
3. Materials and Methods
3.1. Instruments and Materials
3.2. Methods of Synthesis
3.2.1. Synthesis of 2,4-Dimethoxy-3-hydroxyacetophenone 1a
3.2.2. Synthesis of 3,5-Dimethyl-4-benzyloxybenzaldehyde 2b
3.2.3. Synthesis of 2′,4′-Dimethoxyl-5′-hydroxy-3,5-dimethyl-4-benzyloxychalcone 3
3.2.4. Synthesis of 5,7-Dimethoxy- 3′,5′-dimethyl-4′-benzyloxyflavone 4
3.2.5. Synthesis of 5,7-Dimethoxy- 3′,5′-dimethyl-4′-hydroxyflavone 5
3.2.6. Synthesis of 5,7-Dimethoxy-3′,5′-dimethyl-4′-trifluoromethanesulfonyloxyflavone 6
3.2.7. Synthesis of 6a–6n
3.3. Method of Bioactivity Study
3.3.1. Cell Lines and Cell Culture
3.3.2. Cell Viability Assay
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 686, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Weerink, L.B.; Gant, C.M.; Leeuwen, B.L.V.; De Bock, G.H.; Kouwenhoven, E.A.; Faneyte, I.F. Long-term survival in octogenarians after surgical treatment for colorectal cancer: Prevention of postoperative complications is key. Ann. Surg. Oncol. 2018, 25, 3874–3882. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the Last 25 years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.L.; Yuan, X.Y.; Wang, J.B.; Feng, Y.F.; Fei, J.; Li, Z.Y.; Bian, J.L. A review on flavones targeting serine/threonine protein kinases for potential anticancer drugs. Bioorg. Med. Chem. 2019, 27, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Khan, F. 3D-QSAR, Docking, ADME/Tox studies on Flavone analogs reveal anticancer activity through tankyrase inhibition. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sedlacek, H.H.; Czech, J.; Naik, R.; Kaur, G.; Worland, P.; Losiewicz, M.; Parker, B.; Carlson, B.; Smith, A.; Sendeerowicz, A.; et al. Flavopiridol (L86 8275; NSC 649890), a new kinase inhibitor for tumor therapy. Int. J. Oncol. 1996, 9, 1143–1168. [Google Scholar]
- Mark, C.; Dhanapalan, N.; Robert, L.G. Synthesis and evaluation of hydroxylated flavones and related compounds as potential inhibitors of the protein-tyrosine kinase P56lck. J. Nat. Prod. 1991, 54, 1345–1352. [Google Scholar]
- Ferriola, P.C.; Cody, V.; Middleton, E. Protein kinase C inhibition by plant flavoneoids kinetic mechanisms and structure-activity relationships. Biochem. Pharmacol. 1989, 38, 1617–1624. [Google Scholar] [CrossRef]
- Golub, A.G.; Bdzhola, V.G.; Ostrynska, O.V.; Kyshenia, I.V.; Sapelkin, V.M.; Prykhod’ko, A.O.; Kukharenko, O.P.; Yarmoluk, S.M. Discovery and characterization of synthetic 4′-hydroxyflavones-New CK2 inhibitors from flavone family. Bioorg. Med. Chem. 2013, 21, 6681–6689. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Brasier, A.R.; Tian, B.; Liu, Z.Q.; Chen, H.Y.; Rytting, E. Inhibitors of Bromodomain-Containing Protein 4(BRD4) Priority Paragraph. U.S. Patent WO2018/112037, 2018. [Google Scholar]
- Obreque-Balboa, J.E.; Sun, Q.; Bernhardt, G.; Konig, B.; Buschauer, A. Flavonoid derivatives as selective ABCC1 modulators: Synthesis and functional characterization. Eur. J. Med. Chem. 2016, 109, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, J.N.; Lee, H.K.; Yoon, K.S.; Shin, K.D.; Kwon, B.M.; Han, D.C. Biological evaluation of KRIBB3 analogs as a microtubule polymerization inhibitor. Bioorg. Med. Chem. Lett. 2011, 21, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Stacko, P.; Solomek, T.; Klan, P. Electronic-state switching strategy in the photochemical synthesis of indanones from o-methyl phenacyl epoxides. Org. Lett. 2011, 13, 6556–6559. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, X.; Hu, X.F.; Li, Y.; Yin, S.F. Synthesis and Calm Activity of Quinoline and Flavone Derivatives of Helicid. Chin. J. Org. Chem. 2011, 31, 1878–1883. [Google Scholar]
- Cabrera, M.; Simoens, M.; Falchi, G.; Lavaggi, M.L.; Piro, O.E.; Castellano, E.E.; Vidal, A.; Azqueta, A.; Monge, A.; Cerain, A.L.; et al. Synthetic chalcones, flavanones, and flavones as antitumoral agents: Biological evaluation and structure-activity relationships. Bioorg. Med. Chem. 2007, 15, 3356–3367. [Google Scholar] [CrossRef] [PubMed]
- Zaveri, N.T. Synthesis of a 3,4,5-Trimethoxybenzoyl Ester Analogue of Epigallocatechin-3-gallate (EGCG): A potential route to the natural product green tea catechin, EGCG. Org. Lett. 2001, 3, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.F.; Wang, C.; Chen, L.J.; Liang, R.X.; Yu, Y.F.; Jiang, H.F. Modular approach for synthesis of vicinal diamines containing axial chiral 1,10-Binaphthyl from 1,2-Diaminoethane byPd-Catalyzed N-Arylation Reactions. Org. Lett. 2011, 13, 1146–1149. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.J.; Qin, L.N.; Ren, X.F.; Lu, Y.P.; Li, Y.X.; Zhou, J.R. Selective arylation and vinylation at the a position of vinylarenes. Chem. Eur. J. 2013, 19, 3504–3511. [Google Scholar] [CrossRef] [PubMed]
- Gerlier, D.; Thomasset, N. Use of MTT colorimetric assay to measure cell activation. J. Immunol. Methods 1986, 94, 57–63. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
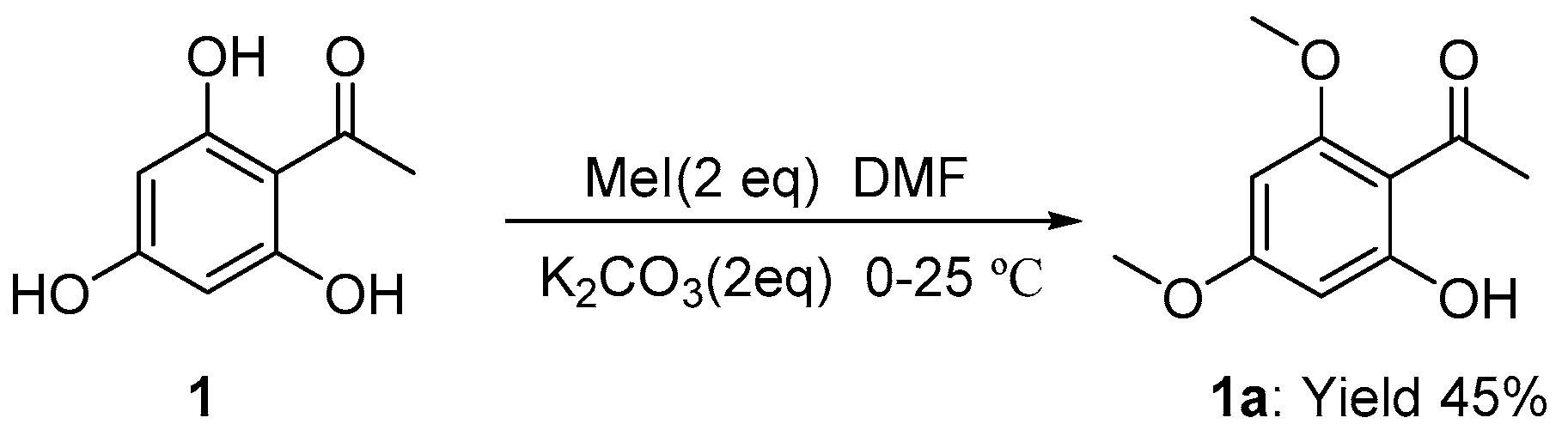{kind=link}
| Compd | IC50 (μM) | Compd | IC50 (μM) | ||
|---|---|---|---|---|---|
| HEL | PC3 | HEL | PC3 | ||
| 5 | >20 | >20 | 6n | >20 | >20 |
| 6 | >20 | >20 | 6k | 7.563 ± 0.844 | 9.836 ± 0.939 |
| 6a | >20 | >20 | 6b | 7.649 ± 0.837 | 10.242 ± 0.952 |
| 6d | >20 | >20 | 6e | 8.416 ± 0.888 | 9.140 ± 0.904 |
| 6g | >20 | >20 | 3 | 8.547 ± 0.932 | 16.471 ± 0.872 |
| 6h | >20 | >20 | 6f | 8.886 ± 0.872 | 9.795 ± 0.991 |
| 6i | >20 | >20 | 4 | 9.945 ± 0.930 | 6.473 ± 0.811 |
| 6j | >20 | >20 | 6c | 10.526 ± 0.992 | 11.266 ± 0.971 |
| 6l | >20 | >20 | Apigenin | >20 | >20 |
| 6m | >20 | >20 | cisplatin | 8.783 ± 0.818 | 11.873 ± 1.075 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, N.; Yang, J.; Li, K.; Luo, J.; Yang, S.; Song, J.-R.; Chen, C.; Pan, W.-D. Synthesis of Flavone Derivatives via N-Amination and Evaluation of Their Anticancer Activities. Molecules 2019, 24, 2723. https://doi.org/10.3390/molecules24152723
Zhang N, Yang J, Li K, Luo J, Yang S, Song J-R, Chen C, Pan W-D. Synthesis of Flavone Derivatives via N-Amination and Evaluation of Their Anticancer Activities. Molecules. 2019; 24(15):2723. https://doi.org/10.3390/molecules24152723
Chicago/Turabian StyleZhang, Ni, Jin Yang, Ke Li, Jun Luo, Su Yang, Jun-Rong Song, Chao Chen, and Wei-Dong Pan. 2019. "Synthesis of Flavone Derivatives via N-Amination and Evaluation of Their Anticancer Activities" Molecules 24, no. 15: 2723. https://doi.org/10.3390/molecules24152723
APA StyleZhang, N., Yang, J., Li, K., Luo, J., Yang, S., Song, J.-R., Chen, C., & Pan, W.-D. (2019). Synthesis of Flavone Derivatives via N-Amination and Evaluation of Their Anticancer Activities. Molecules, 24(15), 2723. https://doi.org/10.3390/molecules24152723