

Regulatory Mechanisms of Pollen Development: Transcriptomic and Bioinformatic Insights into the Role of β-1,3 Glucanase Gene (LbGlu1) in Lycium barbarum
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
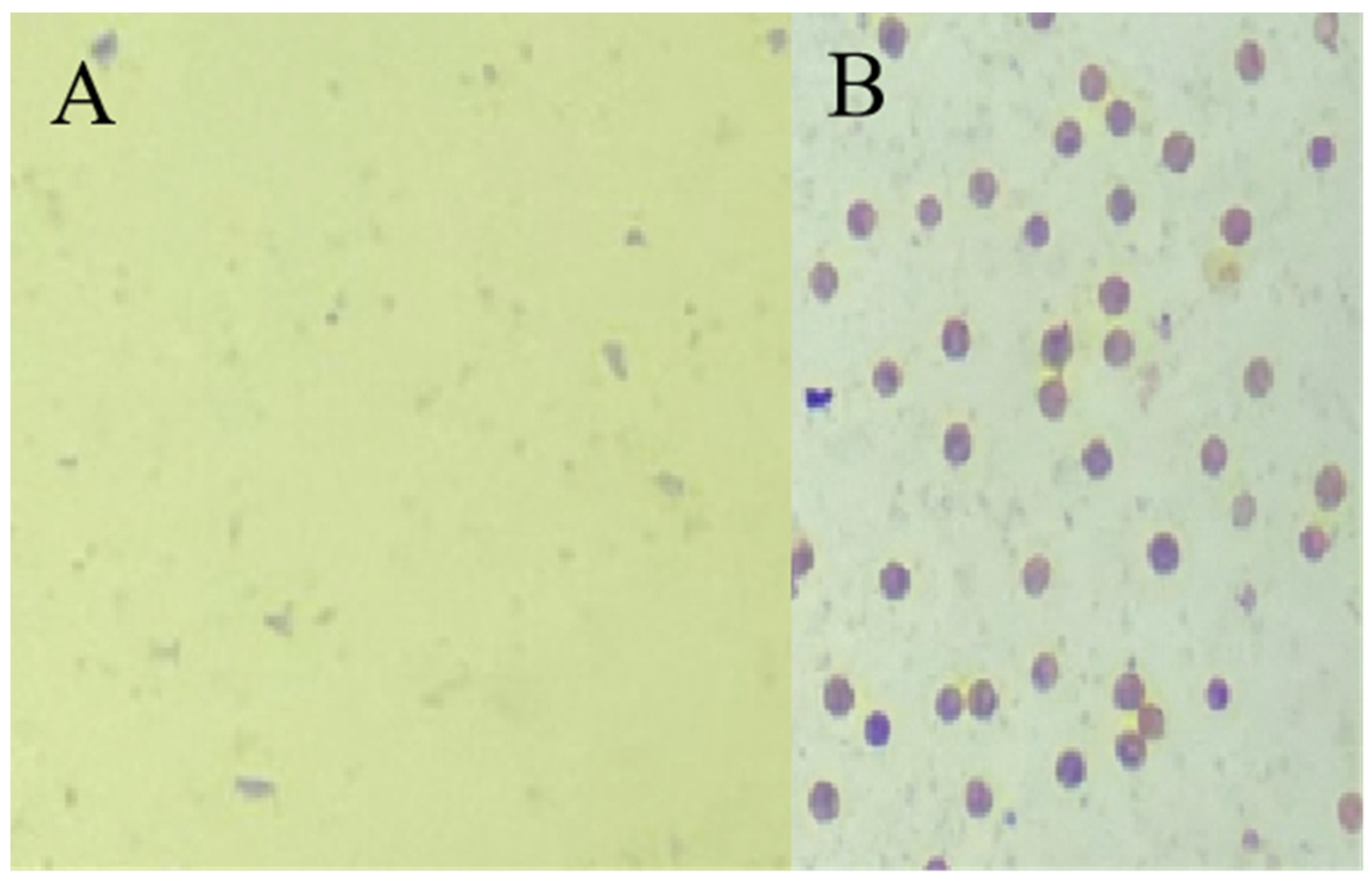
2.1. Plant Materials
2.2. RNA Sequencing
2.3. Sequential Data Processing, Mapping, and Annotation
2.4. Quantitative Reverse Transcription (qRT)-PCR Verification
2.5. Expression Annotation and Enrichment Analysis of DEGs
2.6. WGCNA
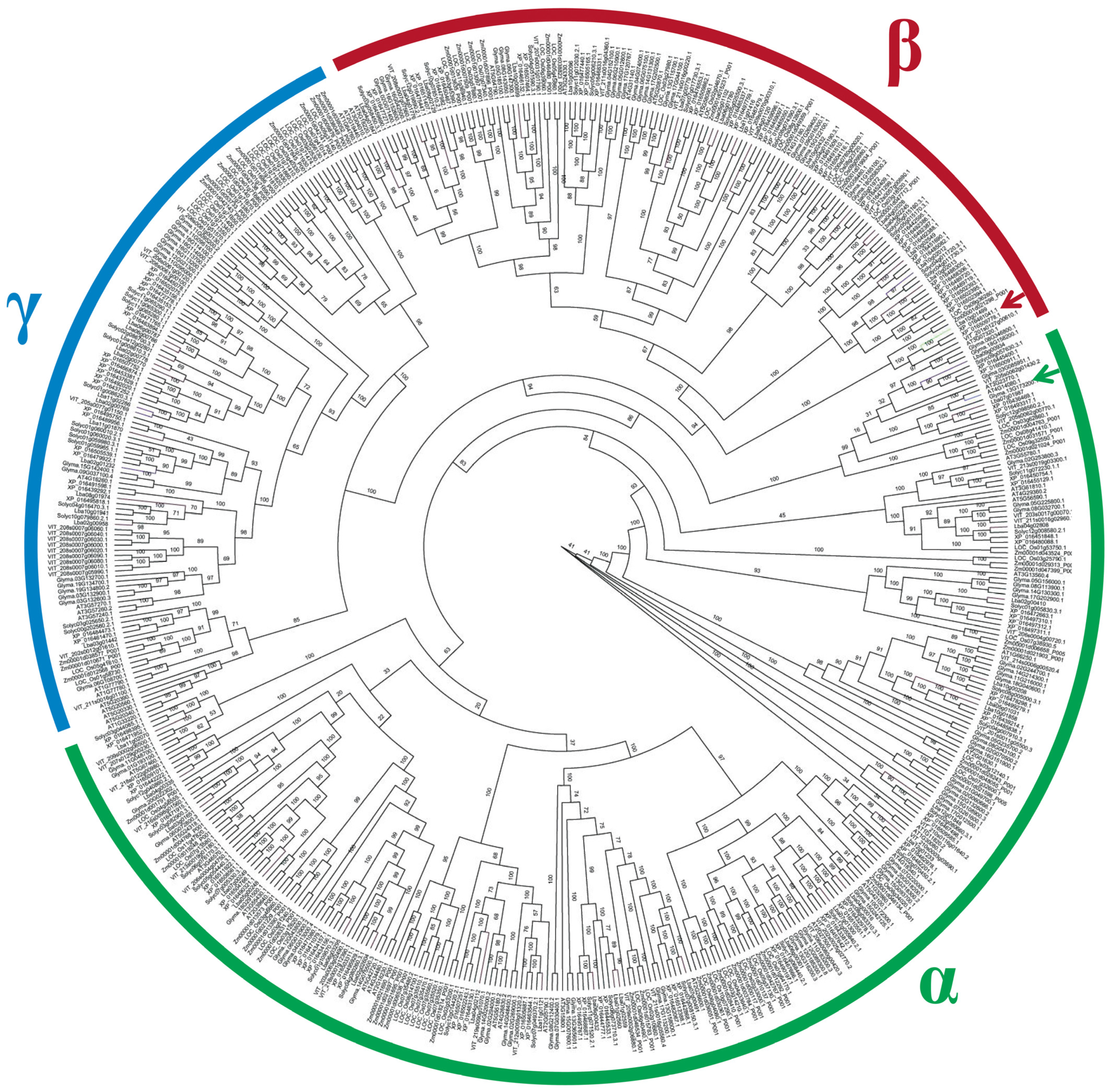
2.7. Identification of Glu Gene Family Related to Callose Metabolism in L. barbarum
2.8. Phylogenetic Analysis of Glu
3. Results
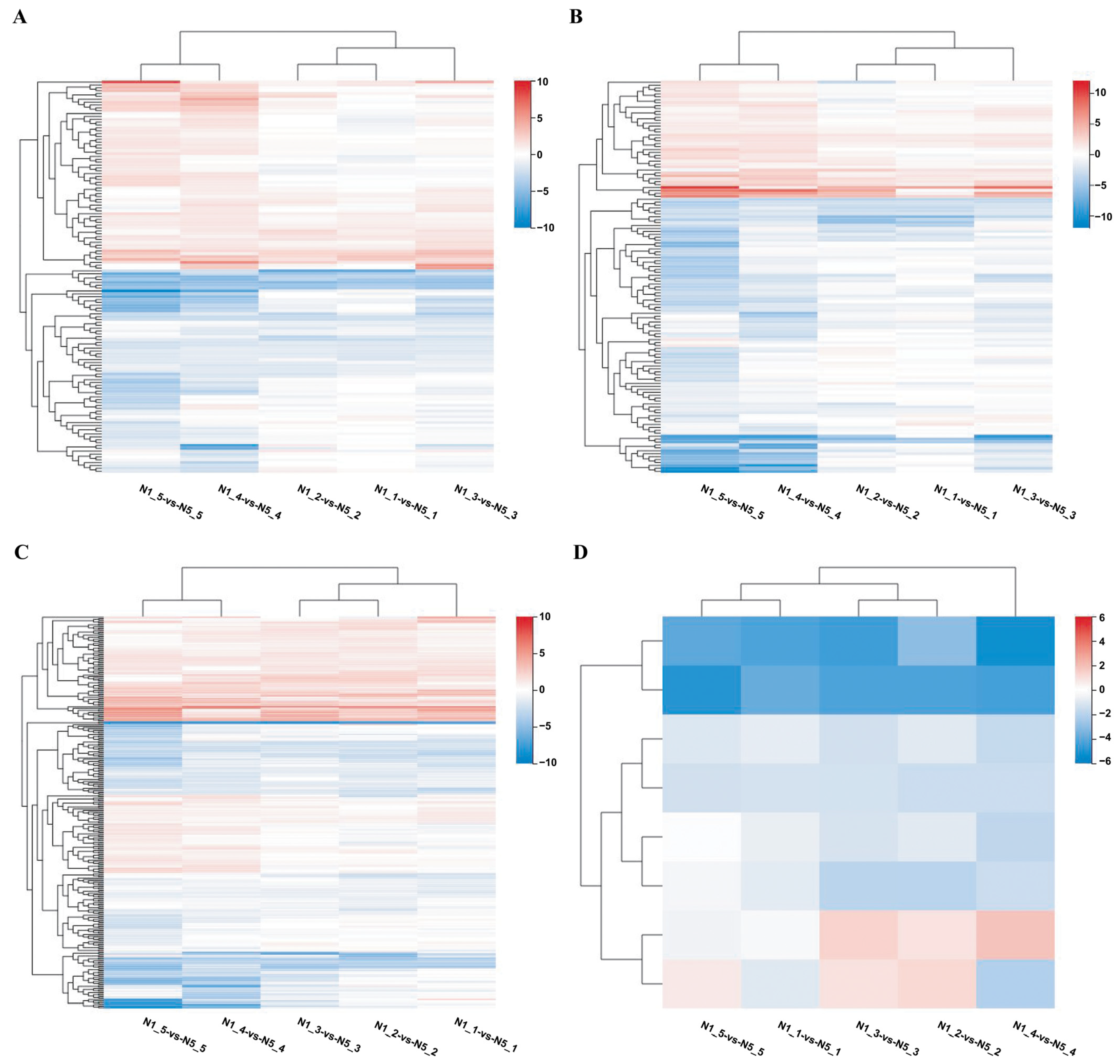
3.1. DEGs in Sterile Line Ningqi No. 5 and Fertile Line Ningqi No. 1
3.2. Anther DEGs in All Control Groups
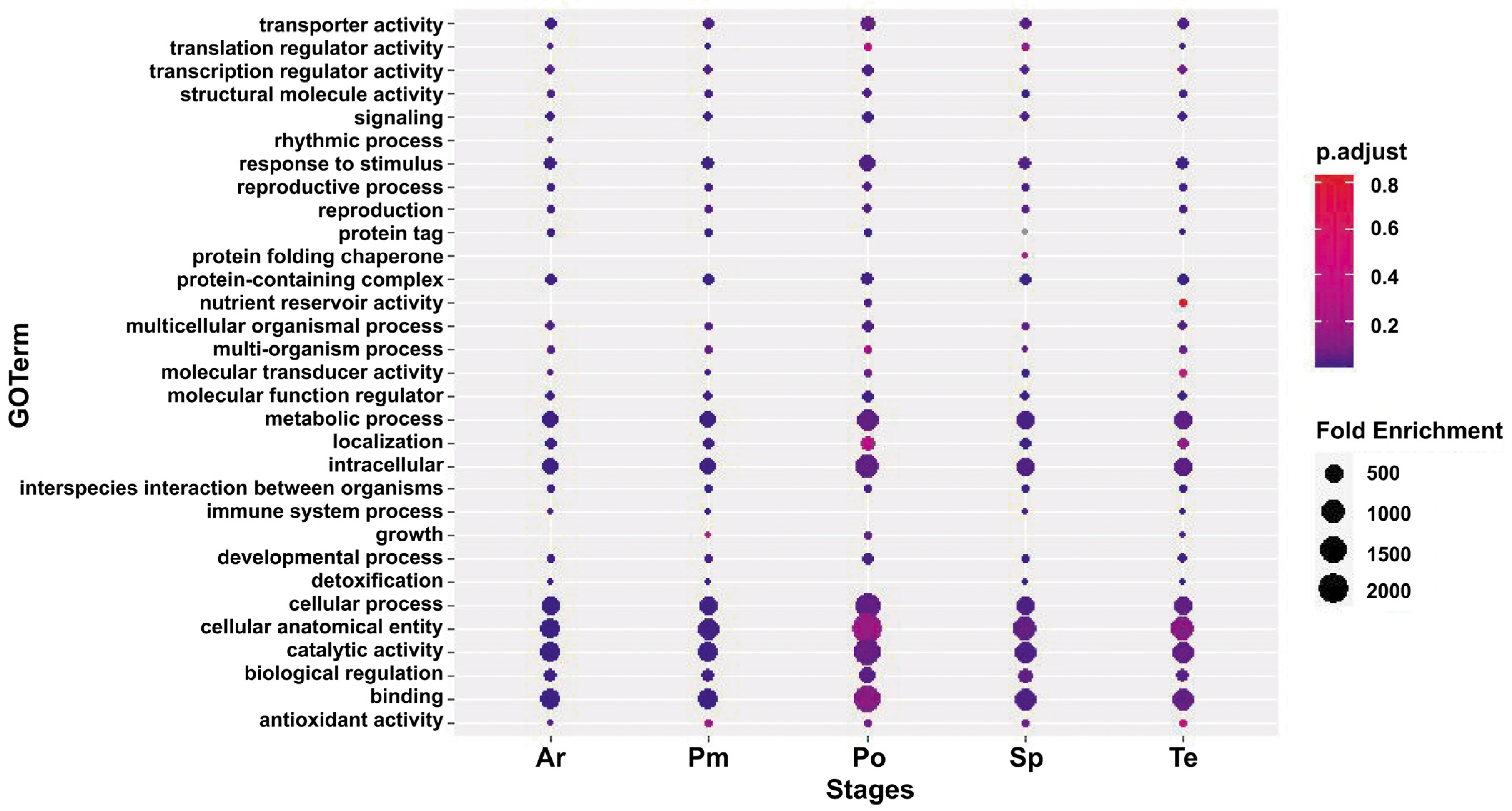
3.3. Functional Annotation and Classification of DEGs
3.4. DEGs Related to Energy Metabolism
3.5. DEGs Related to Pollen Development
3.6. DEGs Related to Hydrolase Activity
3.7. DEGs Related to Active Oxygen Metabolism
3.8. Identification of Hub Genes
3.9. TFs Co-Expressed with Glu
3.10. Analysis of the Phylogenetic Relationship of Glu Protein System
4. Discussion
4.1. Abnormal Glycometabolism Can Affect Pollen Fertility
4.2. Hydrolase Activity Can Regulate the Release of Microspores from Callus
4.3. TFs Can Affect Pollen Fertility by Regulating Gene Expression
4.4. Addressing Alignment Challenges through Enhanced Genome Assembly and Annotation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Longin, C.F.H.; Mühleisen, J.; Maurer, H.P.; Zhang, H.; Gowda, M.; Reif, J.C. Hybrid breeding in autogamous cereals. Theor. Appl. Genet. 2012, 125, 1087–1096. [Google Scholar] [CrossRef]
- Wilson, Z.A.; Zhang, D.-B. From Arabidopsis to rice: Pathways in pollen development. J. Exp. Bot. 2009, 60, 1479–1492. [Google Scholar] [CrossRef]
- Alves-Ferreira, M.; Wellmer, F.; Banhara, A.; Kumar, V.; Riechmann, J.L.; Meyerowitz, E.M. Global expression profiling applied to the analysis of Arabidopsis stamen development. Plant Physiol. 2007, 145, 747–762. [Google Scholar] [CrossRef]
- Wijeratne, A.J.; Zhang, W.; Sun, Y.; Liu, W.; Albert, R.; Zheng, Z.; Oppenheimer, D.G.; Zhao, D.; Ma, H. Differential gene expression in Arabidopsis wild-type and mutant anthers: Insights into anther cell differentiation and regulatory networks. Plant J. 2007, 52, 14–29. [Google Scholar] [CrossRef]
- Chang, H.-S.; Zhang, C.; Chang, Y.-H.; Zhu, J.; Xu, X.-F.; Shi, Z.-H.; Zhang, X.-L.; Xu, L.; Huang, H.; Zhang, S. No primexine and plasma membrane undulation is essential for primexine deposition and plasma membrane undulation during microsporogenesis in Arabidopsis. Plant Physiol. 2012, 158, 264–272. [Google Scholar] [CrossRef]
- Lee, S.-K.; Eom, J.-S.; Hwang, S.-K.; Shin, D.; An, G.; Okita, T.W.; Jeon, J.-S. Plastidic phosphoglucomutase and ADP-glucose pyrophosphorylase mutants impair starch synthesis in rice pollen grains and cause male sterility. J. Exp. Bot. 2016, 67, 5557–5569. [Google Scholar] [CrossRef]
- Park, J.-I.; Ishimizu, T.; Suwabe, K.; Sudo, K.; Masuko, H.; Hakozaki, H.; Nou, I.-S.; Suzuki, G.; Watanabe, M. UDP-glucose pyrophosphorylase is rate limiting in vegetative and reproductive phases in Arabidopsis thaliana. Plant Cell Physiol. 2010, 51, 981–996. [Google Scholar] [CrossRef]
- Hirose, T.; Hashida, Y.; Aoki, N.; Okamura, M.; Yonekura, M.; Ohto, C.; Terao, T.; Ohsugi, R. Analysis of gene-disruption mutants of a sucrose phosphate synthase gene in rice, OsSPS1, shows the importance of sucrose synthesis in pollen germination. Plant Sci. 2014, 225, 102–106. [Google Scholar] [CrossRef]
- Sivitz, A.B.; Reinders, A.; Ward, J.M. Arabidopsis sucrose transporter AtSUC1 is important for pollen germination and sucrose-induced anthocyanin accumulation. Plant Physiol. 2008, 147, 92–100. [Google Scholar] [CrossRef]
- Somashekar, H.; Mimura, M.; Tsuda, K.; Nonomura, K.-I. Rice GLUCAN SYNTHASE-LIKE5 promotes anther callose deposition to maintain meiosis initiation and progression. Plant Physiol. 2023, 191, 400–413. [Google Scholar] [CrossRef]
- Scott, R.J.; Spielman, M.; Dickinson, H. Stamen structure and function. Plant Cell 2004, 16, S46–S60. [Google Scholar] [CrossRef]
- Hird, D.L.; Worrall, D.; Hodge, R.; Smartt, S.; Paul, W.; Scott, R. The anther-specific protein encoded by the Brassica napus and Arabidopsis thaliana A6 gene displays similarity to β-1, 3-glucanases. Plant J. 1993, 4, 1023–1033. [Google Scholar] [CrossRef]
- Wan, L.; Zha, W.; Cheng, X.; Liu, C.; Lv, L.; Liu, C.; Wang, Z.; Du, B.; Chen, R.; Zhu, L. A rice β-1, 3-glucanase gene Osg1 is required for callose degradation in pollen development. Planta 2011, 233, 309–323. [Google Scholar] [CrossRef]
- Shi, X.; Sun, X.; Zhang, Z.; Feng, D.; Zhang, Q.; Han, L.; Wu, J.; Lu, T. GLUCAN SYNTHASE-LIKE 5 (GSL5) plays an essential role in male fertility by regulating callose metabolism during microsporogenesis in rice. Plant Cell Physiol. 2015, 56, 497–509. [Google Scholar] [CrossRef]
- Wang, D.; Li, L.; Yang, H.; Ferguson, S.S.; Baer, M.R.; Gartenhaus, R.B.; Wang, H. The constitutive androstane receptor is a novel therapeutic target facilitating cyclophosphamide-based treatment of hematopoietic malignancies. Blood J. Am. Soc. Hematol. 2013, 121, 329–338. [Google Scholar] [CrossRef]
- Dong, X.; Hong, Z.; Sivaramakrishnan, M.; Mahfouz, M.; Verma, D.P.S. Callose synthase (CalS5) is required for exine formation during microgametogenesis and for pollen viability in Arabidopsis. Plant J. 2005, 42, 315–328. [Google Scholar] [CrossRef]
- Enns, L.C.; Kanaoka, M.M.; Torii, K.U.; Comai, L.; Okada, K.; Cleland, R.E. Two callose synthases, GSL1 and GSL5, play an essential and redundant role in plant and pollen development and in fertility. Plant Mol. Biol. 2005, 58, 333–349. [Google Scholar] [CrossRef]
- Amagase, H.; Farnsworth, N.R. A review of botanical characteristics, phytochemistry, clinical relevance in efficacy and safety of Lycium barbarum fruit (Goji). Food Res. Int. 2011, 44, 1702–1717. [Google Scholar] [CrossRef]
- Chang, R.C.-C.; So, K.-F. Use of anti-aging herbal medicine, Lycium barbarum, against aging-associated diseases. What do we know so far? Cell. Mol. Neurobiol. 2008, 28, 643–652. [Google Scholar] [CrossRef]
- Zhang, X.; Bei, Z.; Ma, H.; Wei, Z.; Zhou, J.; Ren, Y.; Xu, W.; Nan, P.; Wang, Y.; Li, L. Abnormal Programmed Cell Death of Tapetum Leads to the Pollen Abortion of Lycium barbarum Linnaeus. Horticulturae 2022, 8, 1056. [Google Scholar] [CrossRef]
- Portillo, M.; Fenoll, C.; Escobar, C. Evaluation of different RNA extraction methods for small quantities of plant tissue: Combined effects of reagent type and homogenization procedure on RNA quality-integrity and yield. Physiol. Plant. 2006, 128, 1–7. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Wickham, H.; Chang, W.; Henry, L.; Pedersen, T.L.; Takahashi, K.; Wilke, C.; Woo, K.; Yutani, H.; Dunnington, D. ggplot2: Create Elegant Data Visualisations Using the Grammar of Graphics. R Package Version 2016, 2. Available online: https://ggplot2.tidyverse.org/reference/ggplot2-package.html (accessed on 12 March 2024).
- Horvath, S.; Dong, J. Geometric interpretation of gene coexpression network analysis. PLoS Comput. Biol. 2008, 4, e1000117. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Doxey, A.C.; Yaish, M.W.; Moffatt, B.A.; Griffith, M.; McConkey, B.J. Functional divergence in the Arabidopsis β-1, 3-glucanase gene family inferred by phylogenetic reconstruction of expression states. Mol. Biol. Evol. 2007, 24, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.; Thomas, C.; Findlay, K.; Bayer, E.; Maule, A.J. An Arabidopsis GPI-anchor plasmodesmal neck protein with callose binding activity and potential to regulate cell-to-cell trafficking. Plant Cell 2009, 21, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-M.; Lo, S.-F.; Ho, T.-H.D. Source–sink communication: Regulated by hormone, nutrient, and stress cross-signaling. Trends Plant Sci. 2015, 20, 844–857. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, R.; La Camera, S.; Atanassova, R.; Dédaldéchamp, F.; Allario, T.; Pourtau, N.; Bonnemain, J.-L.; Laloi, M.; Coutos-Thévenot, P.; Maurousset, L. Source-to-sink transport of sugar and regulation by environmental factors. Front. Plant Sci. 2013, 4, 53278. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.; Godt, D.E.; Guivarc’h, A.; Kahmann, U.; Chriqui, D.; Roitsch, T. Induction of male sterility in plants by metabolic engineering of the carbohydrate supply. Proc. Natl. Acad. Sci. USA 2001, 98, 6522–6527. [Google Scholar] [CrossRef]
- Suzuki, T.; Narciso, J.O.; Zeng, W.; van de Meene, A.; Yasutomi, M.; Takemura, S.; Lampugnani, E.R.; Doblin, M.S.; Bacic, A.; Ishiguro, S. KNS4/UPEX1: A type II arabinogalactan β-(1, 3)-galactosyltransferase required for pollen exine development. Plant Physiol. 2017, 173, 183–205. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Osborne, E.; Poindexter, P.D.; Somerville, C.R. Microspore separation in the quartet 3 mutants of Arabidopsis is impaired by a defect in a developmentally regulated polygalacturonase required for pollen mother cell wall degradation. Plant Physiol. 2003, 133, 1170–1180. [Google Scholar] [CrossRef] [PubMed]
- Honys, D.; Twell, D. Comparative analysis of the Arabidopsis pollen transcriptome. Plant Physiol. 2003, 132, 640–652. [Google Scholar] [CrossRef]
- Santiago, J.P.; Sharkey, T.D. Pollen development at high temperature and role of carbon and nitrogen metabolites. Plant Cell Environ. 2019, 42, 2759–2775. [Google Scholar] [CrossRef]
- Fujii, S.; Komatsu, S.; Toriyama, K. Retrograde regulation of nuclear gene expression in CW-CMS of rice. Plant Mol. Biol. 2007, 63, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Minic, Z. Physiological roles of plant glycoside hydrolases. Planta 2008, 227, 723–740. [Google Scholar] [CrossRef]
- Tohge, T.; de Souza, L.P.; Fernie, A.R. Current understanding of the pathways of flavonoid biosynthesis in model and crop plants. J. Exp. Bot. 2017, 68, 4013–4028. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.-Y.; Yang, J.-F.; Fan, T.; Ye, N.-H.; Liu, Y.-G. A phylogenetically informed comparison of GH1 hydrolases between Arabidopsis and rice response to stressors. Front. Plant Sci. 2017, 8, 233759. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, T. The Acyl-Glucose-Dependent Anthocyanin Glucosyltransferases of Angiosperm. Ph.D. Thesis, Tokyo University of Agriculture and Technology, Fuchū, Japan, 2013. [Google Scholar]
- Zhang, D.; Yang, L. Specification of tapetum and microsporocyte cells within the anther. Curr. Opin. Plant Biol. 2014, 17, 49–55. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Toriyama, K.; Yoshikawa, M.; Ejiri, S.-i.; Hinata, K. Tapetum-specific expression of the gene for an endo-β-1, 3-glucanase causes male sterility in transgenic tobacco. Plant Cell Physiol. 1995, 36, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Francis, K.E.; Lam, S.Y.; Copenhaver, G.P. Separation of Arabidopsis pollen tetrads is regulated by QUARTET1, a pectin methylesterase gene. Plant Physiol. 2006, 142, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Jopcik, M.; Matusikova, I.; Moravcikova, J.; Durechova, D.; Libantova, J. The expression profile of Arabidopsis thaliana β-1, 3-glucanase promoter in tobacco. Mol. Biol. 2015, 49, 543–549. [Google Scholar] [CrossRef]
- Bucciaglia, P.A.; Smith, A.G. Cloning and characterization of Tag 1, a tobacco anther β-1, 3-glucanase expressed during tetrad dissolution. Plant Mol. Biol. 1994, 24, 903–914. [Google Scholar] [CrossRef]
- Wang, B.; Xue, J.-S.; Yu, Y.-H.; Liu, S.-Q.; Zhang, J.-X.; Yao, X.-Z.; Liu, Z.-X.; Xu, X.-F.; Yang, Z.-N. Fine regulation of ARF17 for anther development and pollen formation. BMC Plant Biol. 2017, 17, 243. [Google Scholar] [CrossRef]
- Lu, P.; Chai, M.; Yang, J.; Ning, G.; Wang, G.; Ma, H. The Arabidopsis CALLOSE DEFECTIVE MICROSPORE1 gene is required for male fertility through regulating callose metabolism during microsporogenesis. Plant Physiol. 2014, 164, 1893–1904. [Google Scholar] [CrossRef]
- Zhang, Z.B.; Zhu, J.; Gao, J.F.; Wang, C.; Li, H.; Li, H.; Zhang, H.Q.; Zhang, S.; Wang, D.M.; Wang, Q.X. Transcription factor AtMYB103 is required for anther development by regulating tapetum development, callose dissolution and exine formation in Arabidopsis. Plant J. 2007, 52, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Zhao, T.; Zhang, L.; Du, G.; Shen, Y.; Tang, D.; Li, Y.; Luo, Q.; Cheng, Z. Defective microspore development 1 is required for microspore cell integrity and pollen wall formation in rice. Plant J. 2020, 103, 1446–1459. [Google Scholar] [CrossRef] [PubMed]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2020, 30, 1083–1096. [Google Scholar] [CrossRef] [PubMed]
- Sarwal, V.; Niehus, S.; Ayyala, R.; Chang, S.; Lu, A.; Darci-Maher, N.; Littman, R.; Chhugani, K.; Soylev, A.; Comarova, Z.; et al. A comprehensive benchmarking of WGS-based structural variant callers. BMC Genom. 2021, 22, 314. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Bei, Z.; Li, J.; Ma, H.; Wang, C.; Xu, W.; Ren, Y.; Zhou, J.; Yan, X. Regulatory Mechanisms of Pollen Development: Transcriptomic and Bioinformatic Insights into the Role of β-1,3 Glucanase Gene (LbGlu1) in Lycium barbarum. Horticulturae 2024, 10, 512. https://doi.org/10.3390/horticulturae10050512
Zhang X, Bei Z, Li J, Ma H, Wang C, Xu W, Ren Y, Zhou J, Yan X. Regulatory Mechanisms of Pollen Development: Transcriptomic and Bioinformatic Insights into the Role of β-1,3 Glucanase Gene (LbGlu1) in Lycium barbarum. Horticulturae. 2024; 10(5):512. https://doi.org/10.3390/horticulturae10050512
Chicago/Turabian StyleZhang, Xin, Zhanlin Bei, Jinglong Li, Haijun Ma, Cuiping Wang, Wendi Xu, Yufeng Ren, Jun Zhou, and Xingfu Yan. 2024. "Regulatory Mechanisms of Pollen Development: Transcriptomic and Bioinformatic Insights into the Role of β-1,3 Glucanase Gene (LbGlu1) in Lycium barbarum" Horticulturae 10, no. 5: 512. https://doi.org/10.3390/horticulturae10050512