Chemical Atherogenesis: Role of Endogenous and Exogenous Poisons in Disease Development

Abstract
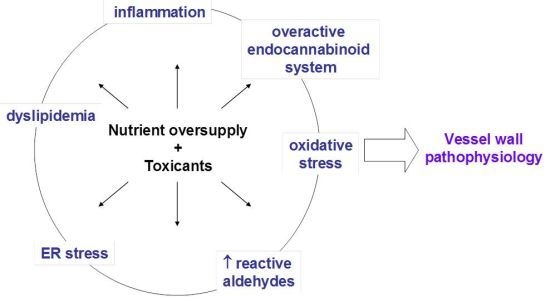
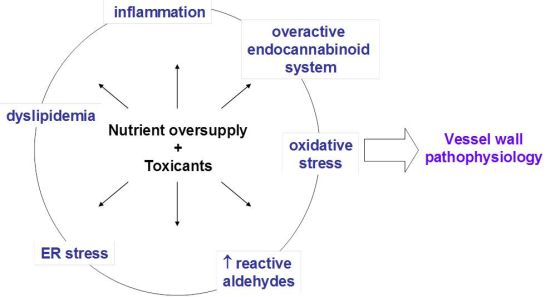
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pollutant | References |
|---|---|
| Acrolein | [3] |
| Allylamine | [4] |
| Arsenic | [5] |
| Benzo(a)pyrene, other PAHs | [6] |
| Bisphenol A | [7] |
| PCBs | [8] |
| Cigarette smoke constituents | [9] |
| Vinyl chloride | [10] |
| Air pollutants (particulate matter, ozone, and NOx) | [11,12] |
| Compound | References |
|---|---|
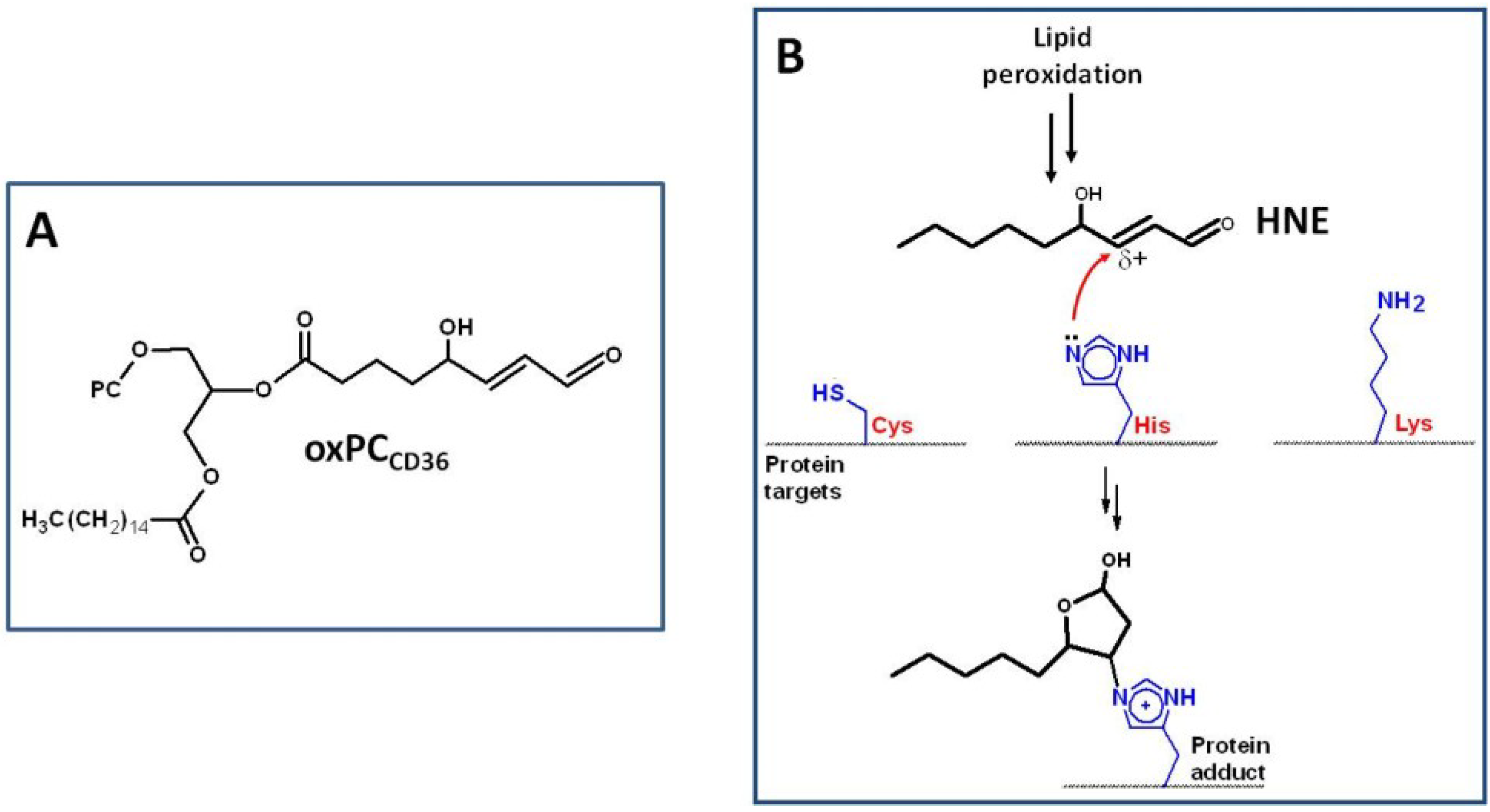
| oxPAPCCD36 | [14] |
| 4-Hydroxynonenal | [15] |
| 4-Oxononenal | [16] |
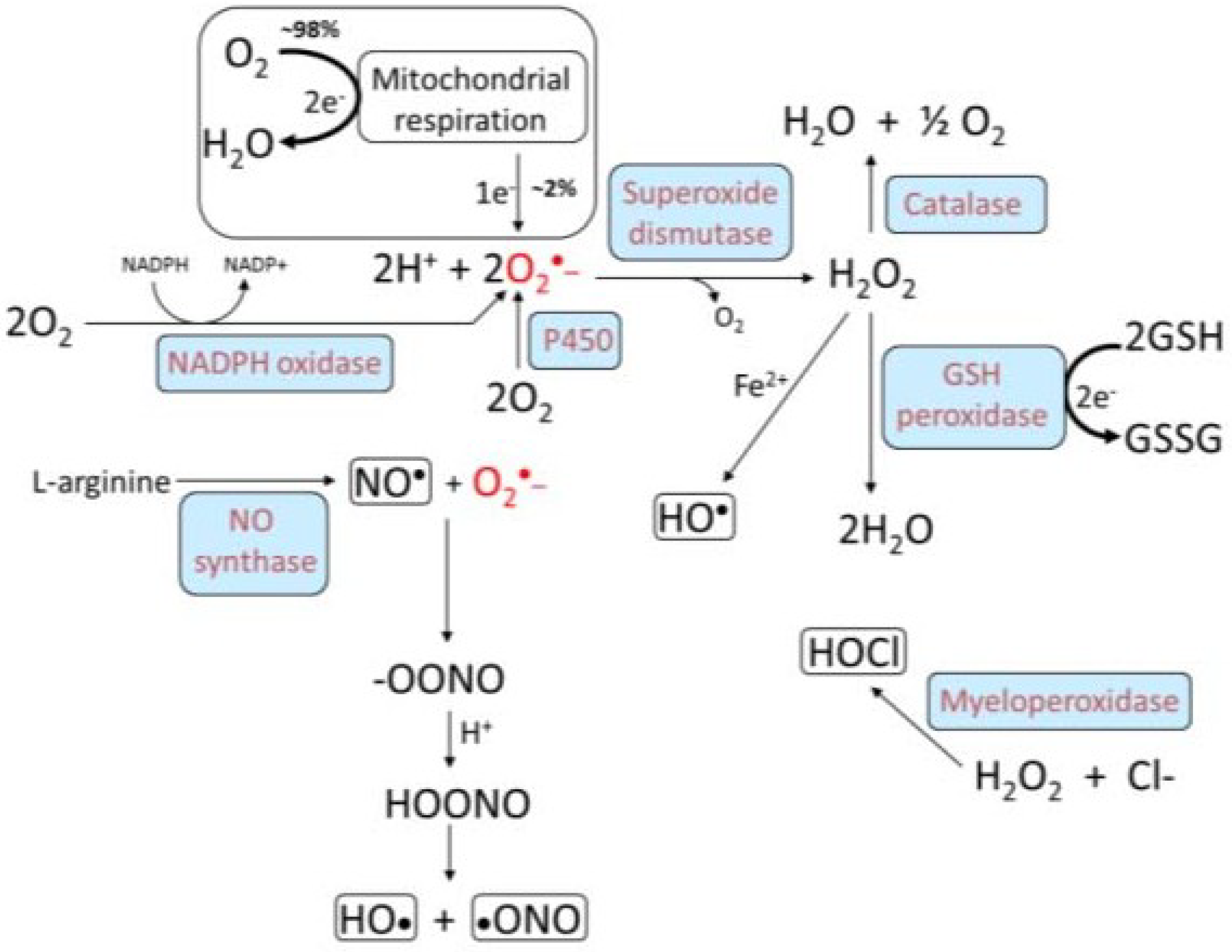
| Reactive oxygen/nitrogen species (O2·−,·OH,·NO, H2O2) | [17] |
| Saturated fatty acids | [18] |
| Cholesterol | [19] |
| Oxysterols | [20] |
| Isoprostanes | [21] |
| Eicosanoids | [22] |
| Lipopolysaccharide (LPS) | [23] |
2. Atherogenesis
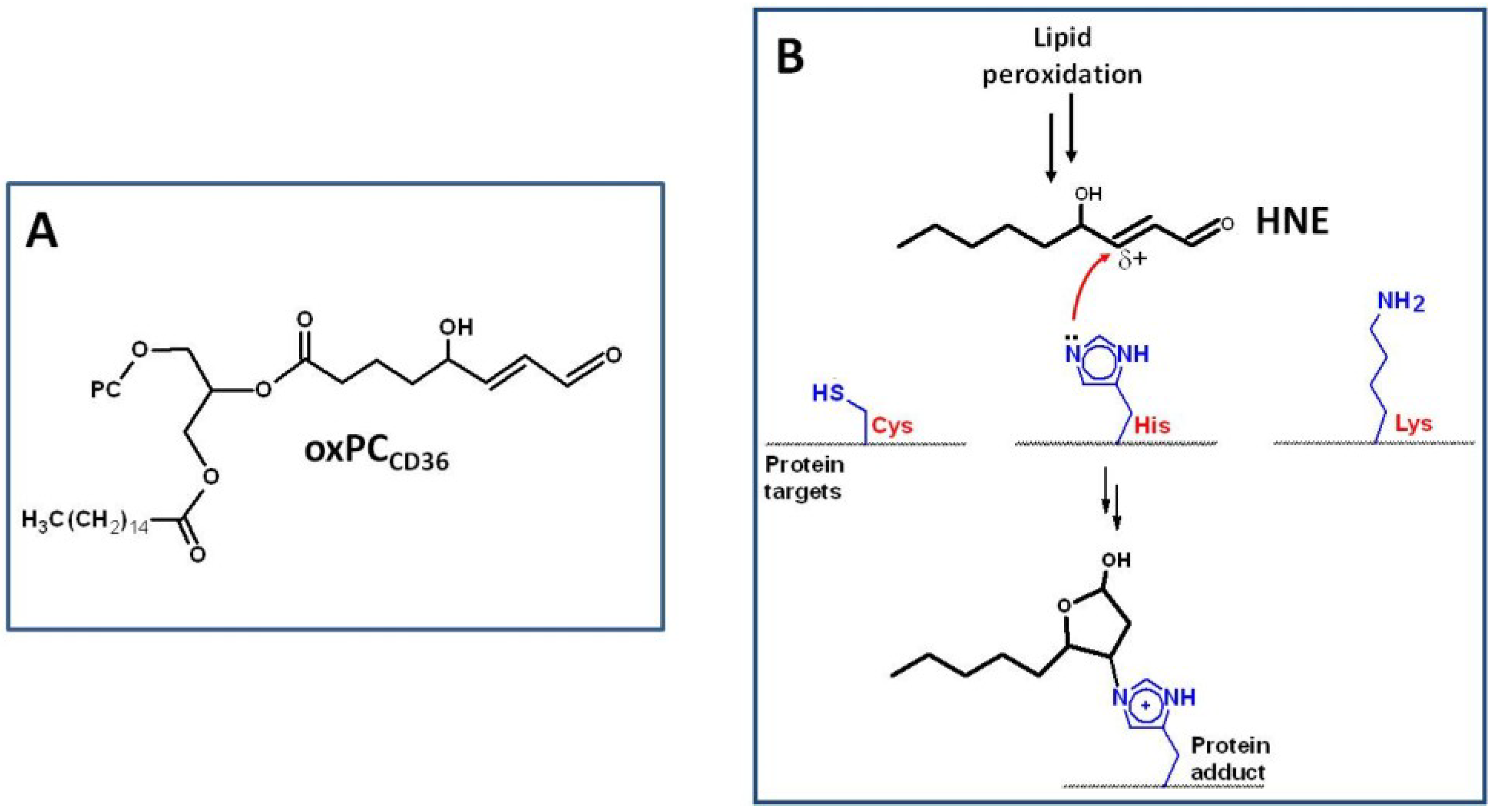
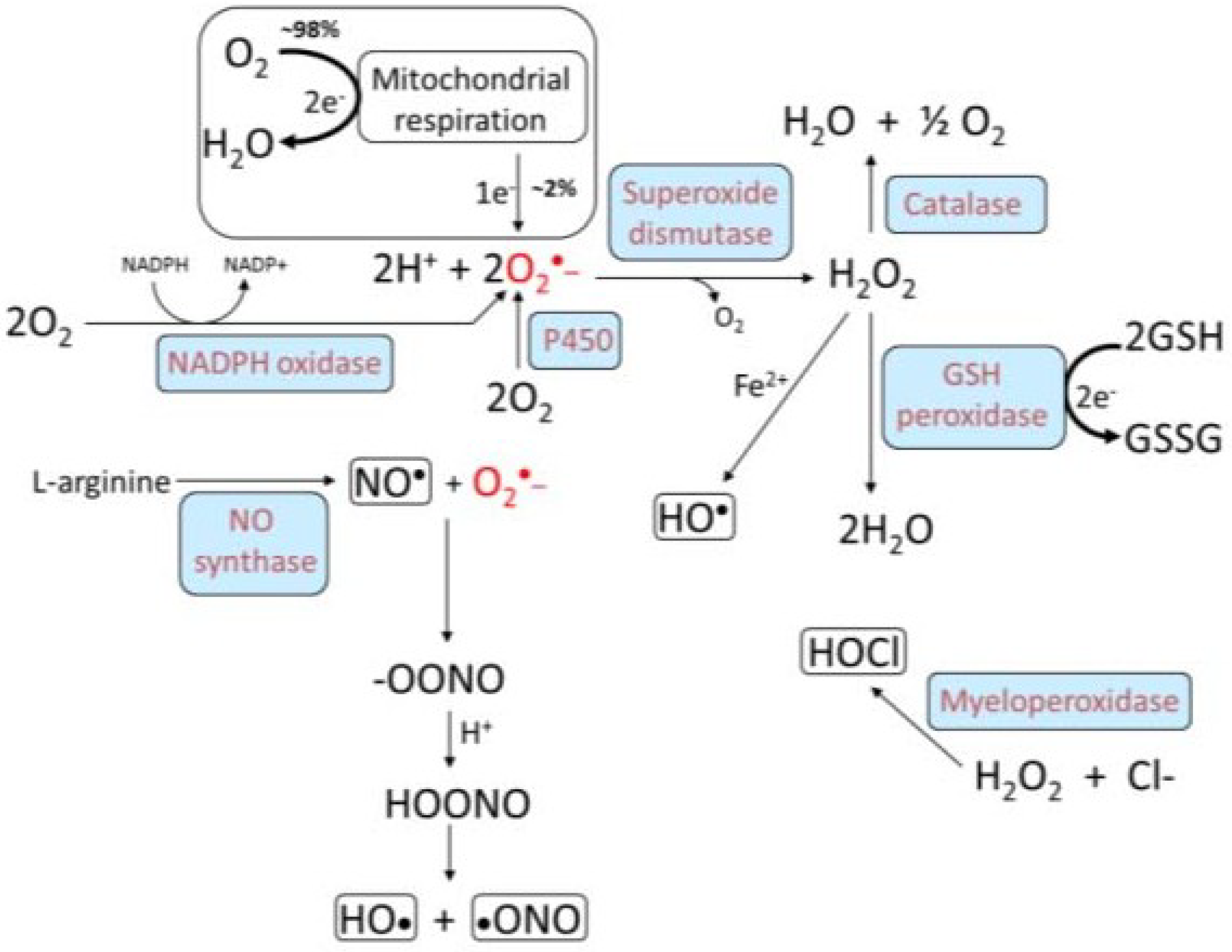
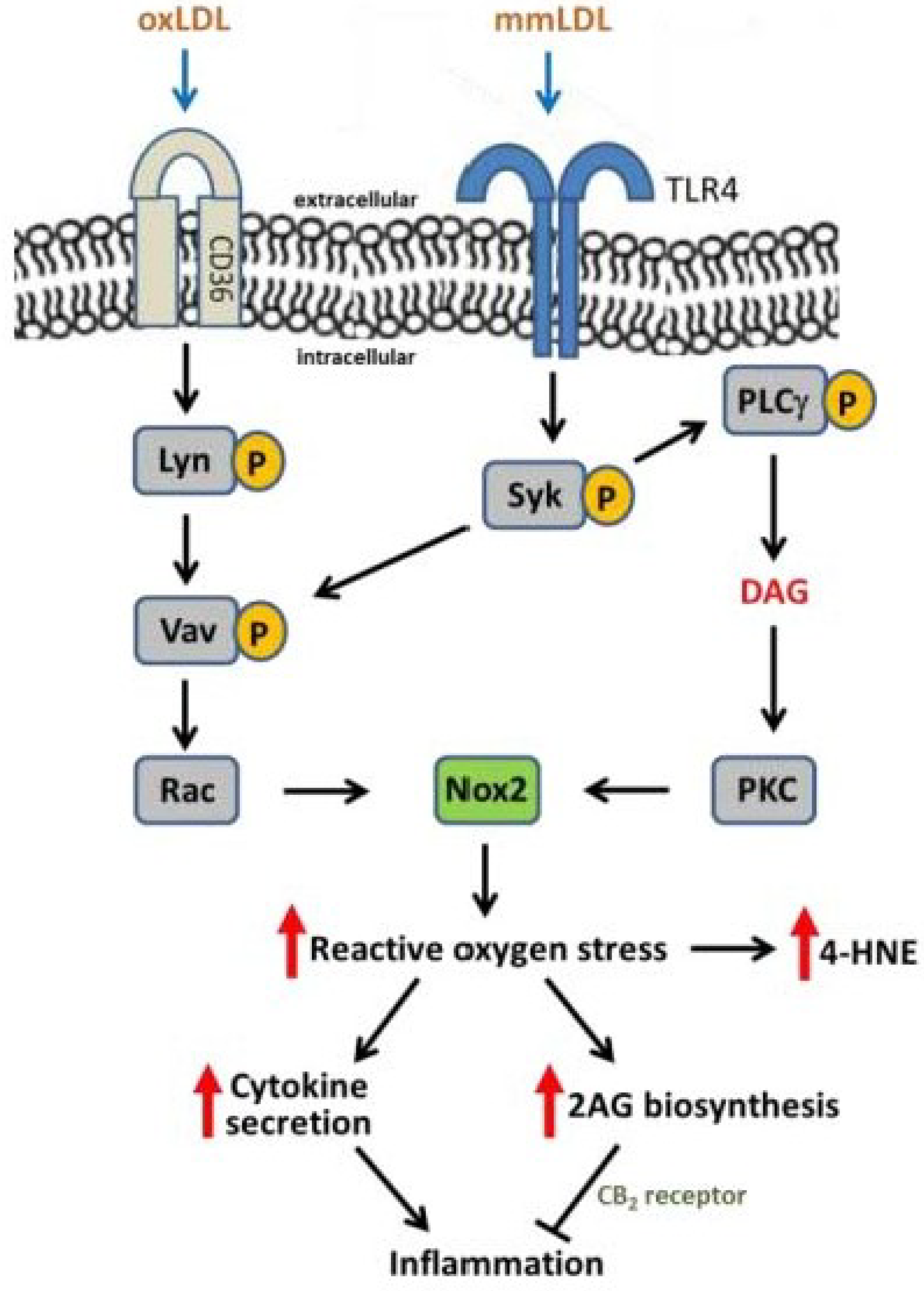
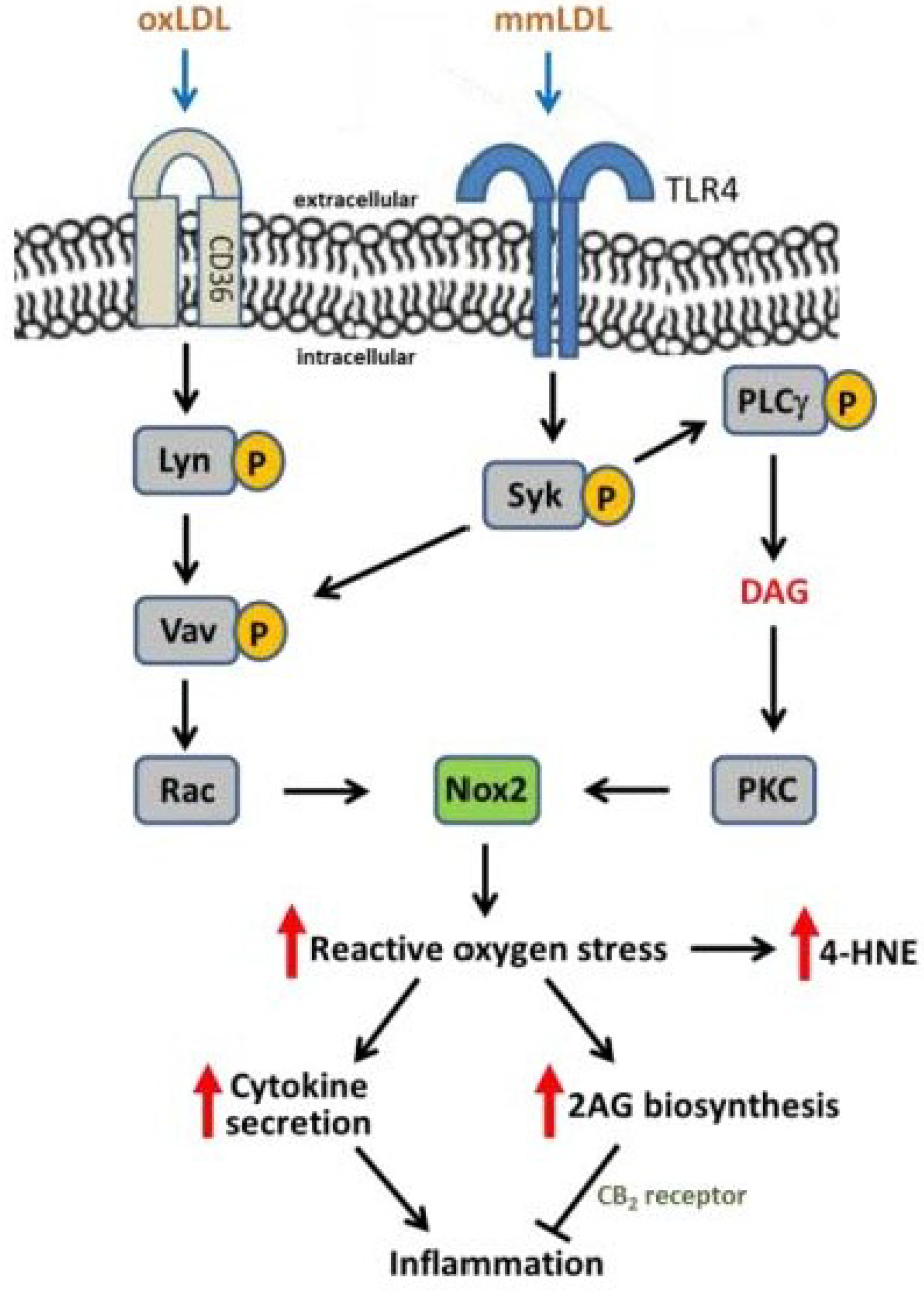
3. Oxidative Stress, NADPH Oxidase, and Atherosclerosis
4. Macrophage Reverse Cholesterol Transport
5. Endocannabinoid System and Atherosclerosis
6. Cigarette Smoke: A Modifiable Risk Factor
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ramos, K.S.; Partridge, C.R.; Teneng, I. Genetic and molecular mechanisms of chemical atherogenesis. Mutat. Res. 2007, 621, 18–30. [Google Scholar]
- Campen, M.J.; Lund, A.; Rosenfeld, M. Mechanisms linking traffic-related air pollution and atherosclerosis. Curr. Opin. Pulm. Med. 2012, 18, 155–160. [Google Scholar] [CrossRef]
- Srivastava, S.; Sithu, S.D.; Vladykovskaya, E.; Haberzettl, P.; Hoetker, D.J.; Siddiqui, M.A.; Conklin, D.J.; D’Souza, S.E.; Bhatnagar, A. Oral exposure to acrolein exacerbates atherosclerosis in apoE-null mice. Atherosclerosis 2011, 215, 301–308. [Google Scholar] [CrossRef]
- Conklin, D.J.; Boor, P.J. Allylamine cardiovascular toxicity: Evidence for aberrant vasoreactivity in rats. Toxicol. Appl. Pharmacol. 1998, 148, 245–251. [Google Scholar] [CrossRef]
- Simeonova, P.P.; Luster, M.I. Arsenic and atherosclerosis. Toxicol. Appl. Pharmacol. 2004, 198, 444–449. [Google Scholar] [CrossRef]
- Curfs, D.M.; Lutgens, E.; Gijbels, M.J.; Kockx, M.M.; Daemen, M.J.; van Schooten, F.J. Chronic exposure to the carcinogenic compound benzo[a]pyrene induces larger and phenotypically different atherosclerotic plaques in ApoE-knockout mice. Am. J. Pathol. 2004, 164, 101–108. [Google Scholar] [CrossRef]
- Kim, M.J.; Moon, M.K.; Kang, G.H.; Lee, K.J.; Choi, S.H.; Lim, S.; Oh, B.C.; Park, D.J.; Park, K.S.; Jang, H.C.; et al. Chronic exposure to Bisphenol A can accelerate atherosclerosis in high-fat-fed apolipoprotein e knockout mice. Cardiovasc. Toxicol. 2013. [Google Scholar] [CrossRef]
- Arsenescu, V.; Arsenescu, R.I.; King, V.; Swanson, H.; Cassis, L.A. Polychlorinated biphenyl-77 induces adipocyte differentiation and proinflammatory adipokines and promotes obesity and atherosclerosis. Environ. Health Perspect. 2008, 116, 761–768. [Google Scholar] [CrossRef]
- Gairola, C.G.; Drawdy, M.L.; Block, A.E.; Daugherty, A. Sidestream cigarette smoke accelerates atherogenesis in apolipoprotein E−/− mice. Atherosclerosis 2001, 156, 49–55. [Google Scholar] [CrossRef]
- Hansen, E.S. International Commission for Protection Against Environmental Mutagens and Carcinogens. ICPEMC Working Paper 7/1/2. Shared risk factors for cancer and atherosclerosis—A review of the epidemiological evidence. Mutat. Res. 1990, 239, 163–179. [Google Scholar] [CrossRef]
- Araujo, J.A.; Barajas, B.; Kleinman, M.; Wang, X.; Bennett, B.J.; Gong, K.W.; Navab, M.; Harkema, J.; Sioutas, C.; Lusis, A.J.; et al. Ambient particulate pollutants in the ultrafine range promote early atherosclerosis and systemic oxidative stress. Circ. Res. 2008, 102, 589–596. [Google Scholar] [CrossRef]
- Yin, F.; Lawal, A.; Ricks, J.; Fox, J.R.; Larson, T.; Navab, M.; Fogelman, A.M.; Rosenfeld, M.E.; Araujo, J.A. Diesel exhaust induces systemic lipid peroxidation and development of dysfunctional pro-oxidant and pro-inflammatory high-density lipoprotein. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1153–1161. [Google Scholar] [CrossRef]
- Liebler, D.C. The poisons within: Application of toxicity mechanisms to fundamental disease processes. Chem. Res. Toxicol. 2006, 19, 610–613. [Google Scholar] [CrossRef]
- Furnkranz, A.; Schober, A.; Bochkov, V.N.; Bashtrykov, P.; Kronke, G.; Kadl, A.; Binder, B.R.; Weber, C.; Leitinger, N. Oxidized phospholipids trigger atherogenic inflammation in murine arteries. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 633–638. [Google Scholar] [CrossRef]
- Yun, M.R.; Im, D.S.; Lee, S.J.; Woo, J.W.; Hong, K.W.; Bae, S.S.; Kim, C.D. 4-Hydroxynonenal contributes to macrophage foam cell formation through increased expression of class A scavenger receptor at the level of translation. Free Radic. Biol. Med. 2008, 45, 177–183. [Google Scholar] [CrossRef]
- Salomon, R.G.; Gu, X. Critical insights into cardiovascular disease from basic research on the oxidation of phospholipids: The gamma-hydroxyalkenal phospholipid hypothesis. Chem. Res. Toxicol. 2011, 24, 1791–1802. [Google Scholar] [CrossRef]
- Lassegue, B.; San Martin, A.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef]
- Imaizumi, K. Diet and atherosclerosis in apolipoprotein E-deficient mice. Biosci. Biotechnol. Biochem. 2011, 75, 1023–1035. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Brewer, H.B., Jr.; Davidson, W.S.; Fayad, Z.A.; Fuster, V.; Goldstein, J.; Hellerstein, M.; Jiang, X.C.; Phillips, M.C.; Rader, D.J.; et al. Cholesterol efflux and atheroprotection: Advancing the concept of reverse cholesterol transport. Circulation 2012, 125, 1905–1919. [Google Scholar] [CrossRef]
- Feldmann, R.; Fischer, C.; Kodelja, V.; Behrens, S.; Haas, S.; Vingron, M.; Timmermann, B.; Geikowski, A.; Sauer, S. Genome-wide analysis of LXRalpha activation reveals new transcriptional networks in human atherosclerotic foam cells. Nucleic Acids Res. 2013, 41, 3518–3531. [Google Scholar] [CrossRef]
- Milne, G.L.; Yin, H.; Hardy, K.D.; Davies, S.S.; Roberts, L.J., 2nd. Isoprostane generation and function. Chem. Rev. 2011, 111, 5973–5996. [Google Scholar] [CrossRef]
- Gleim, S.; Stitham, J.; Tang, W.H.; Martin, K.A.; Hwa, J. An eicosanoid-centric view of atherothrombotic risk factors. Cell. Mol. Life Sci. 2012, 69, 3361–3380. [Google Scholar] [CrossRef]
- Azzam, K.M.; Fessler, M.B. Crosstalk between reverse cholesterol transport and innate immunity. Trends Endocrinol. Metab. 2012, 23, 169–178. [Google Scholar] [CrossRef]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef]
- Murray, C.J.; Lopez, A.D. Evidence-based health policy—Lessons from the Global Burden of Disease Study. Science 1996, 274, 740–743. [Google Scholar] [CrossRef]
- Hopkins, P.N. Molecular biology of atherosclerosis. Physiol. Rev. 2013, 93, 1317–1542. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Li, W.; Park, Y.M.; Rahaman, S.O. Mechanisms of cell signaling by the scavenger receptor CD36: Implications in atherosclerosis and thrombosis. Trans. Am. Clin. Climatol. Assoc. 2010, 121, 206–220. [Google Scholar]
- Szapacs, M.E.; Kim, H.Y.; Porter, N.A.; Liebler, D.C. Identification of proteins adducted by lipid peroxidation products in plasma and modifications of apolipoprotein A1 with a novel biotinylated phospholipid probe. J. Proteome Res. 2008, 7, 4237–4246. [Google Scholar] [CrossRef]
- Shao, B.; Heinecke, J.W. Impact of HDL oxidation by the myeloperoxidase system on sterol efflux by the ABCA1 pathway. J Proteomics 2011, 74, 2289–2299. [Google Scholar] [CrossRef]
- Stemmer, U.; Ramprecht, C.; Zenzmaier, E.; Stojcic, B.; Rechberger, G.; Kollroser, M.; Hermetter, A. Uptake and protein targeting of fluorescent oxidized phospholipids in cultured RAW 264.7 macrophages. Biochim. Biophys. Acta 2012, 1821, 706–718. [Google Scholar] [CrossRef]
- Fritz, K.S.; Petersen, D.R. Exploring the biology of lipid peroxidation-derived protein carbonylation. Chem. Res. Toxicol. 2011, 24, 1411–1419. [Google Scholar] [CrossRef]
- Minekura, H.; Kumagai, T.; Kawamoto, Y.; Nara, F.; Uchida, K. 4-Hydroxy-2-nonenal is a powerful endogenous inhibitor of endothelial response. Biochem. Biophys. Res. Commun. 2001, 282, 557–561. [Google Scholar] [CrossRef]
- Feng, B.; Yao, P.M.; Li, Y.; Devlin, C.M.; Zhang, D.; Harding, H.P.; Sweeney, M.; Rong, J.X.; Kuriakose, G.; Fisher, E.A.; et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 2003, 5, 781–792. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L.; Krieger, M.; Ho, Y.K.; Anderson, R.G. Reversible accumulation of cholesteryl esters in macrophages incubated with acetylated lipoproteins. J. Cell Biol. 1979, 82, 597–613. [Google Scholar] [CrossRef]
- Abid, M.R.; Spokes, K.C.; Shih, S.C.; Aird, W.C. NADPH oxidase activity selectively modulates vascular endothelial growth factor signaling pathways. J. Biol. Chem. 2007, 282, 35373–35385. [Google Scholar] [CrossRef]
- Mao, H.; Fang, X.; Floyd, K.M.; Polcz, J.E.; Zhang, P.; Liu, B. Induction of microglial reactive oxygen species production by the organochlorinated pesticide dieldrin. Brain Res. 2007, 1186, 267–274. [Google Scholar]
- Tithof, P.K.; Olivero, J.; Ruehle, K.; Ganey, P.E. Activation of neutrophil calcium-dependent and -independent phospholipases A2 by organochlorine compounds. Toxicol. Sci. 2000, 53, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Shiose, A.; Sumimoto, H. Arachidonic acid and phosphorylation synergistically induce a conformational change of p47phox to activate the phagocyte NADPH oxidase. J. Biol. Chem. 2000, 275, 13793–13801. [Google Scholar] [CrossRef]
- Miller, Y.I.; Choi, S.H.; Wiesner, P.; Fang, L.; Harkewicz, R.; Hartvigsen, K.; Boullier, A.; Gonen, A.; Diehl, C.J.; Que, X.; et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ. Res. 2011, 108, 235–248. [Google Scholar] [CrossRef]
- Park, Y.M.; Febbraio, M.; Silverstein, R.L. CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima. J. Clin. Investig. 2009, 119, 136–145. [Google Scholar]
- Nguyen-Khoa, T.; Massy, Z.A.; Witko-Sarsat, V.; Canteloup, S.; Kebede, M.; Lacour, B.; Drueke, T.; Descamps-Latscha, B. Oxidized low-density lipoprotein induces macrophage respiratory burst via its protein moiety: A novel pathway in atherogenesis? Biochem. Biophys. Res. Commun. 1999, 263, 804–809. [Google Scholar] [CrossRef]
- Judkins, C.P.; Diep, H.; Broughton, B.R.; Mast, A.E.; Hooker, E.U.; Miller, A.A.; Selemidis, S.; Dusting, G.J.; Sobey, C.G.; Drummond, G.R. Direct evidence of a role for Nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in ApoE−/− mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H24–H32. [Google Scholar] [CrossRef]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef]
- Libby, P.; Aikawa, M.; Jain, M.K. Vascular endothelium and atherosclerosis. Handb. Exp. Pharmacol. 2006, 176, 285–306. [Google Scholar] [CrossRef]
- Pratico, D.; O’Mahony, D.; Lawson, J.; Kinsella, T.; Fitzgerald, G.A. Cellular activation by thromboxane A2 and 8-EPI-PGF2 alpha. Adv. Exp. Med. Biol. 1997, 400A, 229–233. [Google Scholar]
- Kinsella, B.T.; O’Mahony, D.J.; Fitzgerald, G.A. The human thromboxane A2 receptor alpha isoform (TP alpha) functionally couples to the G proteins Gq and G11 in vivo and is activated by the isoprostane 8-epi prostaglandin F2 alpha. J. Pharmacol. Exp. Ther. 1997, 281, 957–964. [Google Scholar]
- Leitinger, N.; Huber, J.; Rizza, C.; Mechtcheriakova, D.; Bochkov, V.; Koshelnick, Y.; Berliner, J.A.; Binder, B.R. The isoprostane 8-iso-PGF(2alpha) stimulates endothelial cells to bind monocytes: Differences from thromboxane-mediated endothelial activation. FASEB J. 2001, 15, 1254–1256. [Google Scholar]
- Barbieri, S.S.; Amadio, P.; Gianellini, S.; Zacchi, E.; Weksler, B.B.; Tremoli, E. Tobacco smoke regulates the expression and activity of microsomal prostaglandin E synthase-1: Role of prostacyclin and NADPH-oxidase. FASEB J. 2011, 25, 3731–3740. [Google Scholar] [CrossRef]
- Janowski, B.A.; Grogan, M.J.; Jones, S.A.; Wisely, G.B.; Kliewer, S.A.; Corey, E.J.; Mangelsdorf, D.J. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc. Natl. Acad. Sci. USA 1999, 96, 266–271. [Google Scholar] [CrossRef]
- Russell, D.W. Oxysterol biosynthetic enzymes. Biochim. Biophys. Acta 2000, 1529, 126–135. [Google Scholar]
- Ghosh, S. Cholesteryl ester hydrolase in human monocyte/macrophage: Cloning, sequencing, and expression of full-length cDNA. Physiol. Genomics 2000, 2, 1–8. [Google Scholar]
- Ghosh, S.; Zhao, B.; Bie, J.; Song, J. Macrophage cholesteryl ester mobilization and atherosclerosis. Vasc. Pharmacol. 2010, 52, 1–10. [Google Scholar] [CrossRef]
- Zhao, B.; Song, J.; St Clair, R.W.; Ghosh, S. Stable overexpression of human macrophage cholesteryl ester hydrolase results in enhanced free cholesterol efflux from human THP1 macrophages. Am. J. Physiol. Cell Physiol. 2007, 292, C405–C412. [Google Scholar]
- Zhao, B.; Song, J.; Chow, W.N.; St Clair, R.W.; Rudel, L.L.; Ghosh, S. Macrophage-specific transgenic expression of cholesteryl ester hydrolase significantly reduces atherosclerosis and lesion necrosis in Ldlr mice. J. Clin. Investig. 2007, 117, 2983–2992. [Google Scholar] [CrossRef]
- Crow, J.A.; Middleton, B.L.; Borazjani, A.; Hatfield, M.J.; Potter, P.M.; Ross, M.K. Inhibition of carboxylesterase 1 is associated with cholesteryl ester retention in human THP-1 monocyte/macrophages. Biochim. Biophys. Acta 2008, 1781, 643–654. [Google Scholar]
- Crow, J.A.; Bittles, V.; Herring, K.L.; Borazjani, A.; Potter, P.M.; Ross, M.K. Inhibition of recombinant human carboxylesterase 1 and 2 and monoacylglycerol lipase by chlorpyrifos oxon, paraoxon and methyl paraoxon. Toxicol. Appl. Pharmacol. 2012, 258, 145–150. [Google Scholar] [CrossRef]
- Bie, J.; Zhao, B.; Song, J.; Ghosh, S. Improved insulin sensitivity in high fat- and high cholesterol-fed Ldlr−/− mice with macrophage-specific transgenic expression of cholesteryl ester hydrolase: Role of macrophage inflammation and infiltration into adipose tissue. J. Biol. Chem. 2010, 285, 13630–13637. [Google Scholar] [CrossRef]
- Zhu, X.; Lee, J.Y.; Timmins, J.M.; Brown, J.M.; Boudyguina, E.; Mulya, A.; Gebre, A.K.; Willingham, M.C.; Hiltbold, E.M.; Mishra, N.; et al. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J. Biol. Chem. 2008, 283, 22930–22941. [Google Scholar] [CrossRef]
- Pacher, P.; Steffens, S. The emerging role of the endocannabinoid system in cardiovascular disease. Semin. Immunopathol. 2009, 31, 63–77. [Google Scholar] [CrossRef]
- Rajesh, M.; Mukhopadhyay, P.; Hasko, G.; Liaudet, L.; Mackie, K.; Pacher, P. Cannabinoid-1 receptor activation induces reactive oxygen species-dependent and -independent mitogen-activated protein kinase activation and cell death in human coronary artery endothelial cells. Br. J. Pharmacol. 2010, 160, 688–700. [Google Scholar] [CrossRef]
- Dol-Gleizes, F.; Paumelle, R.; Visentin, V.; Mares, A.M.; Desitter, P.; Hennuyer, N.; Gilde, A.; Staels, B.; Schaeffer, P.; Bono, F. Rimonabant, a selective cannabinoid CB1 receptor antagonist, inhibits atherosclerosis in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 12–18. [Google Scholar] [CrossRef]
- Sugamura, K.; Sugiyama, S.; Fujiwara, Y.; Matsubara, J.; Akiyama, E.; Maeda, H.; Ohba, K.; Matsuzawa, Y.; Konishi, M.; Nozaki, T.; et al. Cannabinoid 1 receptor blockade reduces atherosclerosis with enhances reverse cholesterol transport. J. Atheroscler. Thromb. 2010, 17, 141–147. [Google Scholar] [CrossRef]
- Steffens, S.; Veillard, N.R.; Arnaud, C.; Pelli, G.; Burger, F.; Staub, C.; Karsak, M.; Zimmer, A.; Frossard, J.L.; Mach, F. Low dose oral cannabinoid therapy reduces progression of atherosclerosis in mice. Nature 2005, 434, 782–786. [Google Scholar] [CrossRef]
- Booz, G.W. Cannabidiol as an emergent therapeutic strategy for lessening the impact of inflammation on oxidative stress. Free Radic. Biol. Med. 2011, 51, 1054–1061. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, Y.; Zhang, W.; Xue, J.; Wu, Y.Z.; Xu, W.; Liang, X.; Chen, T.; Kishimoto, C.; Yuan, Z. WIN55212-2 ameliorates atherosclerosis associated with suppression of pro-inflammatory responses in ApoE-knockout mice. Eur. J. Pharmacol. 2010, 649, 285–292. [Google Scholar] [CrossRef]
- Tsubakio-Yamamoto, K.; Matsuura, F.; Koseki, M.; Oku, H.; Sandoval, J.C.; Inagaki, M.; Nakatani, K.; Nakaoka, H.; Kawase, R.; Yuasa-Kawase, M.; et al. Adiponectin prevents atherosclerosis by increasing cholesterol efflux from macrophages. Biochem. Biophys. Res. Commun. 2008, 375, 390–394. [Google Scholar] [CrossRef]
- Jiang, L.S.; Pu, J.; Han, Z.H.; Hu, L.H.; He, B. Role of activated endocannabinoid system in regulation of cellular cholesterol metabolism in macrophages. Cardiovasc. Res. 2009, 81, 805–813. [Google Scholar] [CrossRef]
- Astarita, G.; Geaga, J.; Ahmed, F.; Piomelli, D. Targeted lipidomics as a tool to investigate endocannabinoid function. Int. Rev. Neurobiol. 2009, 85, 35–55. [Google Scholar]
- Gao, Y.; Vasilyev, D.V.; Goncalves, M.B.; Howell, F.V.; Hobbs, C.; Reisenberg, M.; Shen, R.; Zhang, M.Y.; Strassle, B.W.; Lu, P.; et al. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J. Neurosci. 2010, 30, 2017–2024. [Google Scholar] [CrossRef]
- Xie, S.; Borazjani, A.; Hatfield, M.J.; Edwards, C.C.; Potter, P.M.; Ross, M.K. Inactivation of lipid glyceryl ester metabolism in human THP1 monocytes/macrophages by activated organophosphorus insecticides: Role of carboxylesterases 1 and 2. Chem. Res. Toxicol. 2010, 23, 1890–1904. [Google Scholar] [CrossRef]
- Wang, R.; Borazjani, A.; Matthews, A.T.; Mangum, L.C.; Edelmann, M.J.; Ross, M.K. Identification of palmitoyl protein thioesterase 1 in human THP1 monocytes and macrophages and characterization of unique biochemical activities for this enzyme. Biochemistry 2013, 52, 7559–7574. [Google Scholar] [CrossRef]
- Csordas, A.; Bernhard, D. The biology behind the atherothrombotic effects of cigarette smoke. Nat. Rev. Cardiol. 2013, 10, 219–230. [Google Scholar] [CrossRef]
- Miyaura, S.; Eguchi, H.; Johnston, J.M. Effect of a cigarette smoke extract on the metabolism of the proinflammatory autacoid, platelet-activating factor. Circ. Res. 1992, 70, 341–347. [Google Scholar] [CrossRef]
- Knight-Lozano, C.A.; Young, C.G.; Burow, D.L.; Hu, Z.Y.; Uyeminami, D.; Pinkerton, K.E.; Ischiropoulos, H.; Ballinger, S.W. Cigarette smoke exposure and hypercholesterolemia increase mitochondrial damage in cardiovascular tissues. Circulation 2002, 105, 849–854. [Google Scholar] [CrossRef]
- Atochin, D.N.; Huang, P.L. Endothelial nitric oxide synthase transgenic models of endothelial dysfunction. Pflugers Arch. 2010, 460, 965–974. [Google Scholar] [CrossRef]
- Uchida, K. Current status of acrolein as a lipid peroxidation product. Trends Cardiovasc. Med. 1999, 9, 109–113. [Google Scholar] [CrossRef]
- Uchida, K. Role of reactive aldehyde in cardiovascular diseases. Free Radic. Biol. Med. 2000, 28, 1685–1696. [Google Scholar] [CrossRef]
- Watanabe, K.; Nakazato, Y.; Saiki, R.; Igarashi, K.; Kitada, M.; Ishii, I. Acrolein-conjugated low-density lipoprotein induces macrophage foam cell formation. Atherosclerosis 2013, 227, 51–57. [Google Scholar] [CrossRef]
- O’Toole, T.E.; Zheng, Y.T.; Hellmann, J.; Conklin, D.J.; Barski, O.; Bhatnagar, A. Acrolein activates matrix metalloproteinases by increasing reactive oxygen species in macrophages. Toxicol. Appl. Pharmacol. 2009, 236, 194–201. [Google Scholar] [CrossRef]
- Lemaitre, V.; Dabo, A.J.; D’Armiento, J. Cigarette smoke components induce matrix metalloproteinase-1 in aortic endothelial cells through inhibition of mTOR signaling. Toxicol. Sci. 2011, 123, 542–549. [Google Scholar] [CrossRef]
- Seo, K.W.; Lee, S.J.; Kim, C.E.; Yun, M.R.; Park, H.M.; Yun, J.W.; Bae, S.S.; Kim, C.D. Participation of 5-lipoxygenase-derived LTB(4) in 4-hydroxynonenal-enhanced MMP-2 production in vascular smooth muscle cells. Atherosclerosis 2010, 208, 56–61. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, C.E.; Seo, K.W.; Kim, C.D. HNE-induced 5-LO expression is regulated by NF-κB/ERK and Sp1/p38 MAPK pathways via EGF receptor in murine macrophages. Cardiovasc. Res. 2010, 88, 352–359. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, C.E.; Yun, M.R.; Seo, K.W.; Park, H.M.; Yun, J.W.; Shin, H.K.; Bae, S.S.; Kim, C.D. 4-Hydroxynonenal enhances MMP-9 production in murine macrophages via 5-lipoxygenase-mediated activation of ERK and p38 MAPK. Toxicol. Appl. Pharmacol. 2010, 242, 191–198. [Google Scholar] [CrossRef]
- Akiba, S.; Kumazawa, S.; Yamaguchi, H.; Hontani, N.; Matsumoto, T.; Ikeda, T.; Oka, M.; Sato, T. Acceleration of matrix metalloproteinase-1 production and activation of platelet-derived growth factor receptor beta in human coronary smooth muscle cells by oxidized LDL and 4-hydroxynonenal. Biochim. Biophysi. Acta 2006, 1763, 797–804. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ross, M.K.; Matthews, A.T.; Mangum, L.C. Chemical Atherogenesis: Role of Endogenous and Exogenous Poisons in Disease Development. Toxics 2014, 2, 17-34. https://doi.org/10.3390/toxics2010017
Ross MK, Matthews AT, Mangum LC. Chemical Atherogenesis: Role of Endogenous and Exogenous Poisons in Disease Development. Toxics. 2014; 2(1):17-34. https://doi.org/10.3390/toxics2010017
Chicago/Turabian StyleRoss, Matthew K., Anberitha T. Matthews, and Lee C. Mangum. 2014. "Chemical Atherogenesis: Role of Endogenous and Exogenous Poisons in Disease Development" Toxics 2, no. 1: 17-34. https://doi.org/10.3390/toxics2010017
APA StyleRoss, M. K., Matthews, A. T., & Mangum, L. C. (2014). Chemical Atherogenesis: Role of Endogenous and Exogenous Poisons in Disease Development. Toxics, 2(1), 17-34. https://doi.org/10.3390/toxics2010017